

Abstract
Kinetic Target-Guided Synthesis (TGS) and in situ click chemistry are among unconventional discovery strategies having the potential to streamline the development of protein-protein interaction modulators (PPIMs). In kinetic TGS and in situ click chemistry, the target is directly involved in the assembly of its own potent, bidentate ligand from a pool of reactive fragments. Herein, we report the use and validation of kinetic TGS based on the sulfo-click reaction between thio acids and sulfonyl azides as a screening and synthesis platform for the identification of high-quality PPIMs. Starting from a randomly designed library consisting of 9 thio acids and 9 sulfonyl azides leading to 81 potential acylsulfonamides, the target protein, Bcl-XL selectively assembled four PPIMs, acylsulfonamides SZ4TA2, SZ7TA2, SZ9TA1, and SZ9TA5, which have been shown to modulate Bcl-XL/BH3 interactions. To further investigate the Bcl-XL templation effect, control experiments were carried out using two mutants of Bcl-XL. In one mutant, phenylalanine Phe131 and aspartic acid Asp133, which are critical for the BH3 domain binding, have been substituted by alanines, while arginine Arg139, a residue identified to play a crucial role in the binding of ABT-737, a BH3 mimetic, has been replaced by an alanine in the other mutant. Incubation of these mutants with the reactive fragments and subsequent LC/MS-SIM analysis confirmed that these building block combinations yield the corresponding acylsulfonamides at the BH3 binding site, the actual “hot spot” of Bcl-XL. These results validate kinetic TGS using the sulfo-click reaction as a valuable tool for the straightforward identification of high-quality PPIMs.
Keywords: Kinetic Target-Guided Synthesis, Sulfo-Click Chemistry, Protein-Protein Interactions, Bcl-2 Family
Introduction
Protein-protein interactions (PPIs) are central to a large number of vital biological processes and thus represent attractive targets for the development of novel therapies for a variety of diseases.(1-4) Although scientists recognized the tremendous potential in targeting PPIs over the last two decades, the development of small molecules, which specifically modulate or disrupt a particular PPI, remains a challenging and risky undertaking.(1) Commonly, protein-protein interfaces are large and flat, and they lack deep cavities that might serve as good binding sites for small molecules.(5, 6) Moreover, amino acids at the interfaces of PPIs are flexible and thus pose challenges at conducting computer-guided compound design.(7-9)
Although protein-protein interfaces bury 500–3000 Å2 of total surface area, which exceeds the potential binding area of low-molecular-weight compounds,(10, 11) Wells and co-workers demonstrated that only a fraction of the amino acid residues at the protein-protein interface contributes to the major portion of the binding free energy.(12-14) These key amino acids, defined as recognition patches or hot spots, therefore provide the theoretical and experimental evidence that PPIs can be disrupted or modulated by low-molecular-weight compounds. In the last 15 years, numerous approaches have been developed for the discovery of small molecules modulating or disrupting PPIs. Often, small molecule design is aimed at mimicking a peptide or a protein secondary structure in a truncated form.(15, 16) Alternatively, fragment-based drug discovery strategies using biomolecular NMR, X-ray crystallography, or surface plasmon resonance (SPR) lead to the identification of fragments with good ligand efficiencies, which are further developed into potent protein-protein interaction modulators (PPIMs). Herein we report the expansion and utilization of kinetic Target-Guided Synthesis (TGS) as a screening platform for the identification of PPIMs.
In the last two decades, several TGS approaches have been described, in which the target biomolecule assembles its inhibitory ligand from a collection of reactive fragments. Depending on the nature of the assembly step, TGS approaches can be classified into (a) dynamic combinatorial chemistry (DCC), (b) reagent-accelerated TGS, and (c) kinetic TGS.(17-20) In dynamic combinatorial chemistry, the assembly process is reversible, whereas reagent-accelerated TGS uses building blocks, which combine in an irreversible fashion only in presence of an external reagent or a catalyst upon binding to the biological target. In kinetic TGS, a biological target accelerates the irreversible covalent bond formation only between complementary reacting fragments binding to adjacent binding sites of the target (Figure 1A). Kinetic TGS(16) and in situ click chemistry(17, 18) have been exclusively applied for the identification of inhibitors of enzymatic targets with well defined binding pockets. In a recent proof-of-concept study with the anti-apoptotic protein Bcl-XL as the biological target, we demonstrated that kinetic TGS can also be used for the “rediscovery” of a PPIM previously reported by the Abbott Laboratories starting from smaller fragments bearing a thio acid or a sulfonyl azide functional group.(20) Williams and coworkers described that the amidation reaction between thio acids and sulfonyl azides,(21, 22) which in the meantime has been named as the sulfo-click reaction,(23) proceeds in aqueous media.
Figure 1.
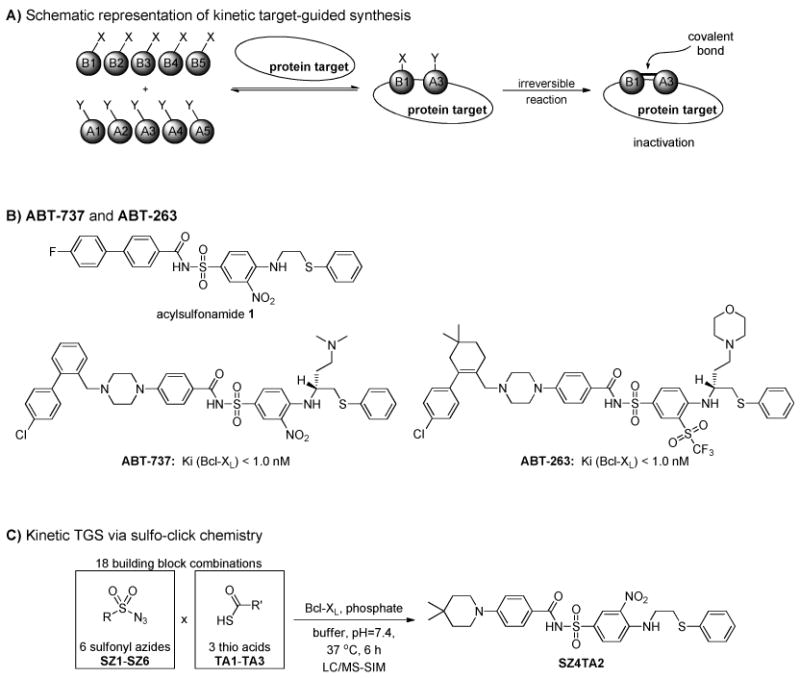
Kinetic TGS approach targeting PPIs. A) TGS approaches are based on the principle that multidentate interactions between a ligand and a biological target are collectively much stronger than the corresponding monovalent interactions of each of the fragments.(60) Thus, target-assembled compound most likely will have a stronger interaction with the biological target as compared to the individual building blocks.(60) In kinetic TGS, fragments decorated with complementary reactive groups are incubated with the target biomolecule. If two fragments reside simultaneously in close proximity in binding pockets of the target, the two reactive functionalities react with each other forming a covalent linkage between the two fragments. B) Acylsulfonamide 1, ABT-737 and ABT-263 compounds targeting Bcl-XL. C) Proof-of-concept study to demonstrate that the amidation between thio acids and sulfonyl azides is suited for kinetic TGS targeting PPIs.
The proteins of the Bcl-2 family have been validated as attractive PPI targets for cancer therapy.(24) The Bcl-2 family of proteins, which consists of both anti- and pro-apoptotic molecules, plays a pivotal role in the regulation of the intrinsic pathway of apoptosis. The anti-apoptotic Bcl-2 family proteins Bcl-2, Bcl-XL, and Mcl-1 inhibit the release of certain pro-apoptotic factors from mitochondria. In contrast, pro-apoptotic Bcl-2 family members, which can be further separated into two subgroups, the multidomain BH1–3 proteins (i.e., Bax and Bak) and the BH3-only proteins (e.g., Bad, Bim, and Noxa), induce the release of mitochondrial apoptogenic molecules into the cytosol.(25, 26) Evidence has been accumulated that the majority of human cancers overexpress the pro-survival Bcl-2 family proteins, which not only contribute to cancer progression by preventing normal cell turnover, but also render cancer cells resistant to current cancer treatments.(27, 28) Although there is a controversy over how anti-apoptotic Bcl-2 family proteins function,(29, 30) it is generally accepted that apoptosis is initiated by the binding of pro-apoptotic BH3-only proteins to anti-apoptotic Bcl-2 family molecules in cancer cells. These interactions are mediated by the insertion of the BH3 domain of pro-death proteins into the hydrophobic groove on the surface of anti-apoptotic proteins Bcl-2, Bcl-XL, or Mcl-1.(31, 32) Therefore, small molecules that mimic the BH3 domains of pro-apoptotic Bcl-2 family proteins have potential as anti-cancer therapeutics.
Previously, Abbott Laboratories developed acylsulfonamide 1, ABT-737, ABT-263, and other structurally related acylsulfonamides, which efficiently disrupt Bcl-XL-Bad interaction (Figure 1B).(33-35) Based on these reports, we designed reactive fragments structurally related to ABT-737 and ABT-263 (SZ1–SZ6 and TA1–TA3), and incubated these as binary fragment mixtures in presence of Bcl-XL (Figure 1C). Analysis of each incubation sample by liquid chromatography combined with mass spectrometry detection in the selected ion mode (LC/MS-SIM) showed that of all 18 possible products only compound SZ4TA2, which was developed by Abbott Laboratories, has been detected. In comparison, incubations of fragments in the absence of Bcl-XL or in presence of Bcl-XL and various BH3-containing peptides failed to yield detectable amounts of acylsulfonamide products. In addition, IC50 inhibitory constants in the nM range have been determined for SZ4TA2, while their corresponding thio acid or sulfonyl azide fragments did not show any inhibition up to 100 μM concentrations.
Herein, we successfully employed and validated the sulfo-click kinetic TGS approach as a straightforward yet reliable PPIM screening platform for the identification of Bcl-XL-protein modulators. The design of kinetic TGS incubations with wildtype and mutant Bcl-XL proteins provided an additional layer of confirmatory experiments for the delivery of high-quality PPIMs. Furthermore, experimental evidence has been accumulated indicating that kinetic TGS is a PPIM screening and synthesis method generating only active compounds.
Results and Discussion
Screening of an extended reactive fragment library
The proof-of-concept study motivated us to investigate whether kinetic TGS is also successful at generating hit compounds which have not been previously reported. Two sublibraries of reactive fragments, one consisting of thio acids and the other of sulfonyl azides, have been synthesized. The thio acids were generated from the corresponding acid chlorides and sodium hydrosulfide, while the sulfonyl azides were prepared by alkylation of amines with 4-(bromomethyl)benzenesulfonyl azide (Figure 2A-C). A selection of acylsulfonamides has been synthesized mainly by: a) EDCI coupling of corresponding carboxylic acids and sulfonamides, or b) the previsouly reported reaction between sulfonyl azides and selenocarboxylates which were generated from corresponding carboxylic acids and the selenating reagent, LiAlHSeH (Figure 2D).(36)
Figure 2.
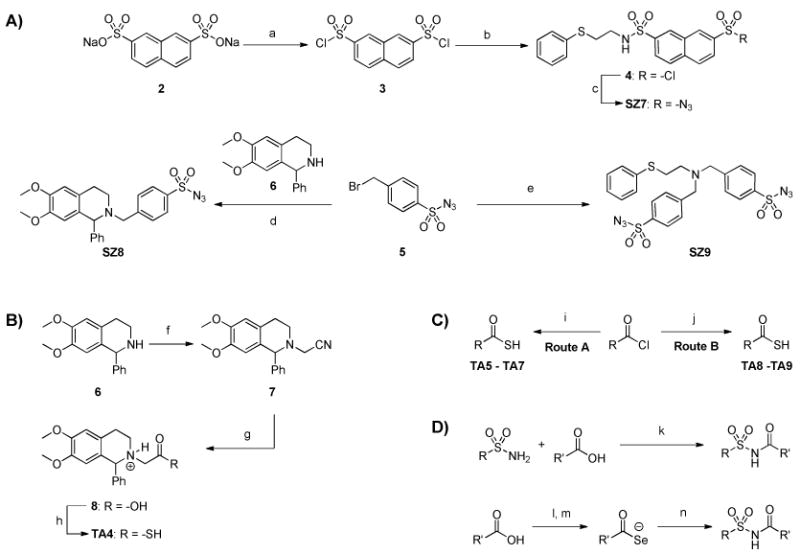
Synthesis of sulfonyl azides, thio acids and acylsulfonamides.
Reaction conditions: (a) SOCl2, DMF, reflux, 2 h (b) 2-(phenylthio)ethanamine (0.5 eq), K2CO3, CHCl3, 12 h, RT (c) NaN3, acetone, H2O, 0 °C, 3 h, 70% (over 3 steps) (d) K2CO3, CH3CN:H2O (9:1), 12 h, RT, 87% (e) 2-(phenylthio)ethanamine (0.5 eq), K2CO3, CH3CN:H2O (9:1), 12 h, RT, 60% (f) ICH2CN, K2CO3, CH3CN:H2O (10:1), 2 d, 60 °C, 79% (g) 12 N HCl, 90 °C, 3 h, 66% (h) i) (COCl)2, CH2Cl2, 0 °C to RT, 8 h; ii) dimethylthioformamide, H2S, 15 min, 25% (i) NaSH, acetone, H2O, 2 h, RT (j) NaSH, neat, 0 °C to RT, 1 h (k) EDCI, DMAP, CH2Cl2, RT, 24 - 48 h (l) (CH3)2CHOCOCl, N-methyl piperidine, THF, 0 °C, 30 min (m) LiAlHSeH, THF, 0 °C, 30 min (n) RSO2N3, THF, 0 °C to RT, 3 h.
The majority of the reactive fragments have been randomly selected, while a small fraction of the reactive fragments has been designed to be structurally related to ABT-737 or ABT-263. Eighty one binary mixtures containing one thio acid (TA1–TA9) and one sulfonyl azide (SZ1–SZ9) were incubated with the target protein Bcl-XL for 6 hours at 37 °C (Figure 3). In parallel, identical binary fragment mixtures were incubated in buffer without Bcl-XL. Similar to in situ click chemistry,(17, 18) all incubations were directly subjected to HPLC analysis with acylsulfonamide product detection by electrospray ionization in the positive selected ion mode (LC/MS-SIM).(37) Comparison of the LC/MS-SIM traces of identical fragment combinations with or without protein Bcl-XL, led to the identification of the previously reported fragment combination SZ4TA2(20) and three new combinations SZ7TA2, SZ9TA1, and SZ9TA5 with increased amounts of acylsulfonamide products in the incubations containing Bcl-XL (Figure 4A-B and supporting information).
Figure 3.
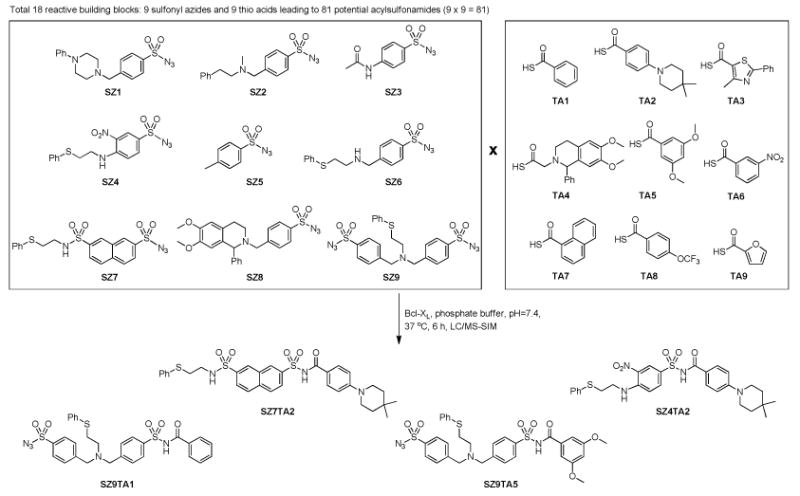
Kinetic TGS screening of Bcl-XL via sulfo-click chemistry.
Figure 4.

LC/MS-SIM analysis of kinetic TGS incubations with fragments SZ7 and TA2 targeting Bcl-XL. The samples were incubated at 37 °C for 6 hours and subjected to LC/MS-SIM analysis with gradient system 1 (see supporting information). A) Incubation sample containing fragments SZ7 and TA2 in absence of Bcl-XL; B) Incubation sample containing fragments SZ7 and TA2 in presence of 2 μM Bcl-XL; C) Incubation sample containing fragments SZ7 and TA2 in presence of 2 μM Bcl-XL and 20 μM Bim BH3 peptide; D) Incubation sample containing fragments SZ7 and TA2 in presence of 2 μM Bcl-XL and 20 μM mutant Bim BH3 peptide; E) Synthetic SZ7TA2 as the reference compound.
Prior to synthesizing the new TGS hit compounds SZ7TA2, SZ9TA1, and SZ9TA5, control incubations with wildtype and mutant pro-apoptotic Bim BH3 peptides were conducted to assess whether the hit combinations assemble at the targeted binding sites of Bcl-XL or randomly elsewhere on the protein surface (Figure 4C-D and supporting information). These control experiments with Bak BH3 peptide have been previously introduced to confirm the kinetic TGS assembly of compound SZ4TA2.(20) Wildtype Bim BH3 peptide (Bim sequence CEIWIAQELRRIGDEFNAYYAR), the natural Bcl-XL ligand, outcompetes the reactive fragments for binding at the BH3 binding site of Bcl-XL and thus suppresses the Bcl-XL-templated assembly of acylsulfonamides SZ7TA2, SZ9TA1, and SZ9TA5. Contrarily, mutant of the Bim BH3 peptide (mutant Bim sequence CEIWIAQEARRIGAEFNAYYAR) exhibits low affinity towards Bcl-XL and therefore does not significantly affect the Bcl-XL-templated assembly of SZ7TA2, SZ9TA1, and SZ9TA5. Since these co-incubations with wildtype and mutant BH3 peptides strongly suggest that the formation of acylsulfonamides SZ7TA2, SZ9TA1, and SZ9TA5 takes place at the BH3 binding site of Bcl-XL, compounds SZ7TA2, SZ9TA1, and SZ9TA5 have been synthesized and subjected to LC/MS-SIM analysis. Comparison of the LC/MS-SIM traces of the Bcl-XL-templated reactions with the ones of the synthetic compounds clearly confirmed that Bcl-XL templates the formation of hit compounds SZ7TA2, SZ9TA1, and SZ9TA5 (Figure 4E and supporting information).
Kinetic TGS with mutant Bcl-XL
Experiments were designed, in which mutated Bcl-XL proteins are incubated with reactive fragments. Alterations of the BH3 binding site directly affect the binding of reactive fragments SZ4, SZ7, SZ9, TA1, TA2, and TA5 to the protein, which in turn will influence the rate of the protein-templated acylsulfonamide formation. The purpose of these mutant Bcl-XL proteins is to expand the repertoire of controls with Bim BH3 peptides with complementary experiments indicating whether the TGS reaction occurs with the help of the target protein Bcl-XL and specifically at the binding site of interest. The known mutant of Bcl-XL, in which phenylalanine Phe131 and aspartic acid Asp133 have been substituted by alanines, has been prepared since it fails at interacting with Bak or Bim BH3 peptides.(38) In addition, a second mutant Bcl-XL has been prepared, in which arginine Arg139 has been replaced by alanine. Arginine Arg139 has been identified to be a key residue interacting with ABT-737 and analogues thereof.(33) As a proof-of-concept, incubations of the mutant Bcl-XL with building blocks SZ4 and TA2 were first undertaken at various reactive fragment concentrations (Figures 5,6 and supporting information). In comparison to the incubation with wildtype Bcl-XL, a reduction in the templation activity by approximately 40% or more has been observed in both mutant Bcl-XL-templated reactions (Table 1). This observation can be explained by closer examination of a reported NMR-structure of Bcl-XL complexed with acylsulfonamide 1, whose structure is closely related to the kinetic TGS product SZ4TA2.(33) Comparison of the location of Phe131 and Asp133 with the position of compound 1 in the wildtype Bcl-XL binding site reveals that the residues Phe131 and Asp133, although important for the binding to Bak or Bim BH3 peptides, are relatively distant from the acylsulfonamide 1, while Arg139 appears to be closer to compound 1. Surprisingly, mutant R139ABcl-XL displays a slightly increased templation reaction in comparison to F131A,D133ABcl-XL. Conformational changes induced by seemingly distant amino acid residues are difficult to trace and may probably influence the templation effect observed during the incubations with wildtype and mutant Bcl-XL proteins.
Figure 5.

LC/MS-SIM analysis of kinetic TGS incubations with fragments SZ4 and TA2 targeting the wildtype and mutant of Bcl-XL. The samples were incubated at 37 °C for 6 hours and subjected to LC/MS-SIM analysis with gradient system 1 (see supporting information). A) Incubation sample containing fragments SZ4 and TA2 in absence of wildtype Bcl-XL; B) Incubation sample containing fragments SZ4 and TA2 in presence of 2 μM wildtype Bcl-XL; C) Incubation sample containing fragments SZ4 and TA2 in presence of 2 μM single mutant R139ABcl-XL; D) Synthetic SZ4TA2 as the reference compound.
Figure 6.

LC/MS-SIM analysis of kinetic TGS incubations with fragments SZ4 and TA2 targeting the wildtype and double mutant of Bcl-XL. The samples were incubated at 37 °C for 6 hours and subjected to LC/MS-SIM analysis with gradient system 2 (see supporting information). A) Incubation sample containing fragments SZ4 and TA2 in absence of wildtype Bcl-XL; B) Incubation sample containing fragments SZ4 and TA2 in presence of 2 μM wildtype Bcl-XL; C) Incubation sample containing fragments SZ4 and TA2 in presence of 2 μM double mutant F131A,D133ABcl-XL; D) Synthetic SZ4TA2 as the reference compound.
Table 1.
Kinetic TGS incubations.
| Incubation | Fragment Combinations | |||||||
|---|---|---|---|---|---|---|---|---|
| SZ4TA2 | SZ7TA2 | SZ9TA1 | SZ9TA5 | |||||
| Peak Area | % Signal | Peak Area | % Signal | Peak Area | % Signal | Peak Area | % Signal | |
| Buffer alone | 26,794 | 7.4 | 3,594 | 6.8 | 313 | 35.3 | 466 | 10.9 |
| WT Bcl-XL | 363,187 | 100.0 | 52,920 | 100.0 | 887 | 100.0 | 4,275 | 100.0 |
| WT Bcl-XL and WT Bak BH3 | 59,437 | 16.3 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| WT Bcl-XL and mutant Bak BH3 | 181,156 | 49.8 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| WT Bcl-XL and WT Bim BH3 | 51,773 | 14.3 | 28,911 | 54.6 | 552 | 62.2 | 944 | 22.1 |
| WT Bcl-XL and mutant Bim BH3 | 217,813 | 59.9 | 47,728 | 90.2 | 761 | 85.8 | 2,557 | 59.8 |
|
| ||||||||
| Buffer alone | 44195 | 9.6 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| WT Bcl-XL | 460532 | 100.0 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
| F131A,D133ABcl-XL | 196429 | 42.7 | n.d. | n.d. | n.d. | n.d. | n.d. | n.d. |
|
| ||||||||
| Buffer alone | 4,733 | 11.0 | 2,046 | 7.2 | 939 | 25.0 | 726 | 11.4 |
| WT Bcl-XL | 43,210 | 100.0 | 28,600 | 100.0 | 3,750 | 100.0 | 6,370 | 100.0 |
| R139ABcl-XL | 25,959 | 60.1 | 16,965 | 59.3 | 2,637 | 70.3 | 4,406 | 69.2 |
n.d. = not determined; WT = wildtype
For TGS hit combinations SZ7TA2, SZ9TA1, and SZ9TA5, confirmatory experiments have been conducted with single mutant R139ABcl-XL only, since the preparation of double mutant F131A,D133ABcl-XL has been cumbersome. Similar to the incubations of fragments SZ4 and TA2, experiments with the mutant protein leading to acylsulfonamides SZ7TA2, SZ9TA1, and SZ9TA5 displayed a reduction in acylsulfonamide formation compared to the incubations with wildtype Bcl-XL. These experiments suggest that the acylsulfonamide genesis occurs in proximity to key amino acid residue Arg139.
PPIM activity of kinetic TGS hits and additional acylsulfonamides
The kinetic TGS hits were subjected to dose response studies to obtain IC50s and to investigate if the hit compounds are also modulating or disrupting the interaction between Bcl-XL and a native BH3 peptide ligand. Previously, Abbott Laboratories determined by their assay, that SZ4TA2 is a good PPIM with a Ki constant of 19 nM.(34, 35) Abbott determined the dissociation constants by a competitive fluorescence polarization assay using a fluorescein-labeled Bad-BH3 peptide. In order to precisely compare the inhibitory properties of our kinetic TGS hits with the compounds reported by Abbott, we decided to perform binding studies by a fluorescence polarization assay implemented in our laboratories, which uses GST-Bcl-XL and fluorescein-labeled Bak-BH3 peptide. Consistently, compound SZ4TA2 has been validated by our assay as a Bcl-XL inhibitor against Bak-BH3 with an IC50 constant of 106 nM (Table 2). Kinetic TGS hit compounds SZ7TA2, SZ9TA1, and SZ9TA5 showed IC50s in the low μM range (Figure 3 and supporting information). Taken together, these results indicate that the hit compounds SZ4TA2, SZ7TA2, SZ9TA1, and SZ9TA5 identified through the kinetic TGS screening are indeed respectable ligands of the biological target, which underscores the utility of kinetic TGS as a valuable approach to PPIM discovery.
Table 2.
PPIM activity of kinetic TGS hit compounds.
| Compound | IC50 | Ki |
|---|---|---|
| SZ4TA2 | 106 ± 12 nM | 37.5 ± 5.0 nM |
| SZ7TA2 | 28.4 ± 3.5 μM | 11.5 ± 1.4 μM |
| SZ9TA1 | 28.7 ± 4.1 μM | 11.6 ± 1.6 μM |
| SZ9TA5 | 36.0 ± 2.5 μM | 14.6 ± 1.0 μM |
To assess whether the kinetic TGS hits are more potent than acylsulfonamides, which were not identified in the kinetic TGS screening, 33 randomly selected acylsulfonamides were synthesized. All compounds, as well as TGS hit compounds SZ4TA2, SZ7TA2, SZ9TA1, and SZ9TA5 were tested at a 50 μM concentration for PPI disruption in the Bcl-XL/Bak-BH3 fluorescence polarization assay. The 37 acylsulfonamides tested corresponds to 45.7% of the 81 member library. Strikingly, the four kinetic TGS hits were the most potent compounds tested, disrupting the Bcl-XL/BH3 interaction with 60% inhibition or more, while the randomly selected acylsulfonamides demonstrated an average of 15% inhibition (Table 3). Only four of the 33 randomly selected acylsulfonamides demonstrated moderate inhibition (35–45%). In contrast, all reactive fragments SZ1–SZ9 and TA1–TA9 have been tested in the fluorescence polarization assay at 100 μM concentration and less than 5% inhibition was detected. These measurements indicate that the dissociation constants for the corresponding reactive building blocks SZ1–SZ9 and TA1–TA9 have to be higher than 100 μM. These important results suggest that the amidation reaction between thio acids and sulfonyl azides is suitable for kinetic TGS using building blocks displaying weak binding affinities. In addition, this study strongly suggests that the kinetic TGS screening identified the more active members of the library of potential acylsulfonamides arising from reactive fragments SZ1–SZ9 and TA1–TA9.
Table 3.
Percentage Inhibition displayed by an acylsulfonamide at 50 μM concentration. Of the 37 compounds tested, the four most potent compounds were identified by kinetic TGS.
| Fragments | SZ1 | SZ2 | SZ3 | SZ4 | SZ5 | SZ6 | SZ7 | SZ8 | SZ9 |
|---|---|---|---|---|---|---|---|---|---|
| TA1 | n.d. | 2 | 0 | 14 | 29 | n.d. | n.d. | 19 | 80 |
| TA2 | n.d. | 8 | n.d. | 100 | 28 | 26 | 76 | n.d. | 38 |
| TA3 | 6 | 7 | n.d. | n.d. | n.d. | n.d. | n.d. | 30 | 22 |
| TA4 | n.d. | 25 | n.d. | n.d. | n.d. | n.d. | n.d. | 8 | n.d. |
| TA5 | 5 | n.d. | n.d. | n.d. | 0 | n.d. | 15 | 11 | 60 |
| TA6 | 4 | n.d. | 0 | n.d. | 0 | n.d. | 20 | n.d. | n.d. |
| TA7 | n.d. | n.d. | 0 | n.d. | n.d. | n.d. | 47 | 30 | 45 |
| TA8 | n.d. | n.d. | 0 | n.d. | n.d. | n.d. | n.d. | 38 | n.d. |
| TA9 | 3 | n.d. | 0 | n.d. | 1 | n.d. | n.d. | 24 | n.d. |
n.d. = not determined
Discussion
Generally, cell-permeable small modulators of PPIs have been considered to be desirable tools with great implications for drug discovery and development.(3, 4) Nevertheless, reliable yet straightforward techniques or approaches for the development of potent and effective PPIMs are currently unavailable. Over the past 15 years, a variety of fragment-based lead discovery approaches have been developed and successfully applied for the development of potent PPIMs.(39-41) These approaches are commonly based on the detection of fragments binding to the target protein followed by the study of their binding to the protein target at atomic level resolution using X-ray crystallography or NMR spectroscopy. The initial hits are further optimized via fragment growing, in which fragments are extended into identified binding sites step-by-step, or via fragment linking, in which fragments identified to bind to adjacent binding sites are covalently linked together.(41-44) Even though fragment-based lead discovery strategies have been very successful for the development of PPIMs, they are mainly limited by two constraints. Detection and quantification of fragment binding requires specially designed methodology due to the weak binding typically observed for fragments. Furthermore, the optimization of fragments into potent and selective compounds is not straightforward and not rapidly achievable, even though structural information is available.(43, 45) For example, though high quality NMR structures were available, the development of Bcl-XL PPIMs by Abbott(33, 34) required several design iterations, and the preparation and testing of more than thousand compounds in order to yield ABT-737 and ABT-263.(46) Furthermore, of the very first design consisting of 21 different structures containing the structural motifs of the initial fragments identified by NMR, most compounds bound to Bcl-XL with a dissociation constant greater than 10 μM.(35) Thus, though the hit compounds SZ7TA2, SZ9TA1, and SZ9TA5 display IC50 constants of 28 to 37 μM in the Bak-BH3 fluorescence polarization assay, the herein reported kinetic TGS approach suggests that the high-quality PPIMs will be identified early on in the screening process. This outcome is consistent with previously reported kinetic TGS studies, in which the enzyme carbonic anhydrase II preferably accelerates the formation of the better inhibitory compounds from a pool of reactive fragments.(47, 48) Other kinetic TGS examples using exclusively in situ click chemistry also suggest that the triazoles generated in the protein-templated reactions are the more potent inhibitors.(37, 48-54)
Recently, fragment-based discovery strategies have been reported which involve the protein target directly to select and assemble its own inhibitory compounds from a pool of reactive fragments. These approaches, also termed as in situ click chemistry or kinetic TGS approaches,(16, 18) were conceptually described in the 1980s(55) and are still relatively unexplored compared to dynamic combinatorial chemistry. Thus far, kinetic TGS has mainly been applied to the identification of potent enzyme inhibitors. Nevertheless, the herein reported kinetic TGS offers an attractive approach to PPIM lead discovery because it allows the protein to select and combine building blocks that fit best into its binding sites, thus assembling larger compounds.(16, 18) The screening method can be as simple as determining whether or not the PPIM product has been formed in a given test mixture. This is especially advantageous over a conventional high-throughput screening of difficult targets such as protein interfaces requiring cumbersome and time-consuming experiments to confirm whether screening hits are true or false positives.
Finally, considering that the flexible nature of protein interfaces complicates the development of PPIMs by conventional means, kinetic TGS has the potential to target the protein in a conformation, which is short-lived, undetectable or easily missed with present techniques. A small number of in situ click chemistry approaches targeting enzymatic systems lead to the identification of triazole inhibitors stabilizing the protein in an unprecedented and less abundant conformation.(56-58) Thus, we speculate that the herein reported sulfo-click chemistry kinetic TGS approach provides medicinal chemists a straightforward search strategy to stabilize conformations of dynamic protein targets such as PPIs.
Conclusions
Herein, we demonstrate that the sulfo-click kinetic TGS approach exhibits great promise in fragment-based PPIM discovery since it combines synthesis and screening of libraries of low-molecular-weight PPIMs into a single step. Samples containing the protein target Bcl-XL and reacting fragments leading to 81 structurally different acylsulfonamides have been incubated and analyzed by LC/MS-SIM for acylsulfonamide formation. Of the 81 possible fragment combinations, only combinations SZ4TA2, SZ7TA2, SZ9TA1, and SZ9TA5 yielded acylsulfonamides in the Bcl-XL-templated reactions. Control incubations with the four hit fragment combinations, in which the BH3 binding site of the wildtype Bcl-XL has been competitively occupied by a Bim BH3 peptide, generated decreased amounts of acylsulfonamides. Furthermore, control incubations with mutants R139ABcl-XL or F131A,D133ABcl-XL, in which amino acid residues at the BH3 binding site have been replaced by alanines, also failed at generating the hit acylsulfonamides suggesting that the protein-templated assembly of SZ4TA2, SZ7TA2, SZ9TA1, and SZ9TA5 occurs at the desired BH3 binding site of Bcl-XL. Subsequent testing of synthesized kinetic TGS hit acylsulfonamides in a fluorescence-based competitive binding assay demonstrated that the kinetic TGS hit compounds indeed display PPIM activity. These findings have been supported by a set of 33 additional acylsulfonamides randomly selected from the 81-member library, which have been shown to fail at demonstrating potent PPIM activity in the fluorescence-based competitive binding assay. These results provide a general test case for the sulfo-click kinetic TGS approach to generate hits targeting the proteins of the Bcl-2 family and further validate the kinetic TGS approach to be suitable for PPIM discovery. In contrast to conventional screening approaches, experimental data suggests that PPIM screening via kinetic TGS reduces the number of false positives, cutting down the number of screening hits to be validated in confirmatory assays. We speculate that the herein reported PPIM discovery strategy for the family of the Bcl-2 proteins is general and can easily be implemented to lead development targeting other PPIs such as MDM2/p53, IAP/caspase, and others.(1, 4, 59)
Methods
Synthesis of selected compounds
The synthesis of reactive fragments and acylsulfonamides has been reported in the supporting information.
Expression and purification of wildtype and mutant Bcl-XL fusion proteins
The protocols have been reported in the supporting information.
General protocol for incubations of Bcl-XL with reactive fragments
In a 96-well plate, one thio acid building block (1 μL of a 2 mM solution in methanol) and one sulfonyl azide building block (1 μL of a 2 mM solution in methanol) were added to a solution of Bcl-XL (98 μL of a 2 μM Bcl-XL solution in buffer (58 mM Na2HPO4, 17 mM NaH2PO4, 68 mM NaCl, 1 mM NaN3, pH = 7.40)). The 96-well plate was sealed and incubated at 37 °C for six hours. The incubation samples were then subjected to liquid chromatography combined with mass spectrometry analysis in the selected ion mode (LC/MS-SIM, Zorbax SB-C18 preceded by a Phenomenex C18 guard column, electrospray ionization and mass spectrometric detection in the positive selected ion mode, tuned to the expected molecular mass of the product). The TGS hit compound was identified by the mass and the retention time. As a control, identical building block combinations were incubated in buffer without Bcl-XL and subjected to LC/MS-SIM analysis. Comparison of the LC/MS-SIM chromatograms of these control incubations with the chromatograms of the Bcl-XL containing incubations allows us to determine whether the protein is templating the corresponding amidation reaction. Furthermore, synthetically prepared acylsulfonamide was subjected to LC/MS-SIM analysis and the retention time was compared with the one identified in the Bcl-XL containing incubation.
Fluorescence polarization-based competitive binding assay
The detailed protocol to conduct fluorescence polarization-based competitive binding assays has been previously reported.(20)
Supplementary Material
Acknowledgments
We are grateful to the James and Esther King Biomedical Research Program (NIR Grant 07KN-08 to R.M.) and the National Cancer Institute, National Institutes of Health (Grant P01CA118210 to H-G.W.) for financial support.
Footnotes
Supporting Information. Synthetic procedures, LC/MS-SIM traces, 1H and 13C NMR spectra and determination of IC50 values. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Wells JA, McClendon CL. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature (London, United Kingdom) 2007;450:1001–1009. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- 2.Arkin M. Protein-protein interactions and cancer: small molecules going in for the kill. Current Opinion in Chemical Biology. 2005;9:317–324. doi: 10.1016/j.cbpa.2005.03.001. [DOI] [PubMed] [Google Scholar]
- 3.Arkin MR, Wells JA. Small-molecule inhibitors of protein-protein interactions: Progressing towards the dream. Nature Reviews Drug Discovery. 2004;3:301–317. doi: 10.1038/nrd1343. [DOI] [PubMed] [Google Scholar]
- 4.Berg T. Modulation of protein-protein interactions with small organic molecules. Angewandte Chemie-International Edition. 2003;42:2462–2481. doi: 10.1002/anie.200200558. [DOI] [PubMed] [Google Scholar]
- 5.Preissner R, Goede A, Frommel C. Dictionary of interfaces in proteins (DIP). Data bank of complementary molecular surface patches. Journal of Molecular Biology. 1998;280:535–550. doi: 10.1006/jmbi.1998.1878. [DOI] [PubMed] [Google Scholar]
- 6.McCoy AJ, Epa VC, Colman PM. Electrostatic complementarity at protein/protein interfaces. Journal of Molecular Biology. 1997;268:570–584. doi: 10.1006/jmbi.1997.0987. [DOI] [PubMed] [Google Scholar]
- 7.DeLano WL, Ultsch MH, de Vos AM, Wells JA. Convergent solutions to binding at a protein-protein interface. Science. 2000;287:1279–1283. doi: 10.1126/science.287.5456.1279. [DOI] [PubMed] [Google Scholar]
- 8.Janin J, Henrick K, Moult J, Eyck Ten L, Sternberg MJE, Vajda S, Vasker I, Wodak SJ. CAPRI: A Critical Assessment of PRedicted Interactions. Proteins-Structure Function and Genetics. 2003;52:2–9. doi: 10.1002/prot.10381. [DOI] [PubMed] [Google Scholar]
- 9.Nooren IMA, Thornton JM. Structural characterisation and functional significance of transient protein-protein interactions. Journal of Molecular Biology. 2003;325:991–1018. doi: 10.1016/s0022-2836(02)01281-0. [DOI] [PubMed] [Google Scholar]
- 10.Lo Conte L, Chothia C, Janin J. The atomic structure of protein-protein recognition sites. Journal of Molecular Biology. 1999;285:2177–2198. doi: 10.1006/jmbi.1998.2439. [DOI] [PubMed] [Google Scholar]
- 11.Nooren IMA, Thornton JM. Diversity of protein-protein interactions. Embo Journal. 2003;22:3486–3492. doi: 10.1093/emboj/cdg359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Clackson T, Wells JA. A Hot-Spot of Binding-Energy in a Hormone-Receptor Interface. Science. 1995;267:383–386. doi: 10.1126/science.7529940. [DOI] [PubMed] [Google Scholar]
- 13.Bogan AA, Thorn KS. Anatomy of hot spots in protein interfaces. Journal of Molecular Biology. 1998;280:1–9. doi: 10.1006/jmbi.1998.1843. [DOI] [PubMed] [Google Scholar]
- 14.DeLano WL. Current Opinion in Structural Biology. Vol. 12. 2002. Unraveling hot spots in binding interfaces: progress and challenges; pp. 14–20. [DOI] [PubMed] [Google Scholar]
- 15.Stigers KD, Soth MJ, Nowick JS. Designed molecules that fold to mimic protein secondary structures. Current Opinion in Chemical Biology. 1999;3:714–723. doi: 10.1016/s1367-5931(99)00030-7. [DOI] [PubMed] [Google Scholar]
- 16.Hu XD, Manetsch R. Kinetic target-guided synthesis. Chemical Society Reviews. 2010;39:1316–1324. doi: 10.1039/b904092g. [DOI] [PubMed] [Google Scholar]
- 17.Mamidyala SK, Finn MG. In situ click chemistry: probing the binding landscapes of biological molecules. Chemical Society Reviews. 2010;39:1252–1261. doi: 10.1039/b901969n. [DOI] [PubMed] [Google Scholar]
- 18.Sharpless KB, Manetsch R. In situ click chemistry: a powerful means for lead discovery. Expert Opin Drug Discovery. 2006;1:525–538. doi: 10.1517/17460441.1.6.525. [DOI] [PubMed] [Google Scholar]
- 19.Corbett PT, Leclaire J, Vial L, West KR, Wietor JL, Sanders JKM, Otto S. Dynamic Combinatorial Chemistry. Chemical Reviews (Washington, DC, United States) 2006;106:3652–3711. doi: 10.1021/cr020452p. [DOI] [PubMed] [Google Scholar]
- 20.Hu X, Sun J, Wang HG, Manetsch R. Bcl-XL-Templated Assembly of Its Own Protein-Protein Interaction Modulator from Fragments Decorated with Thio Acids and Sulfonyl Azides. Journal of the American Chemical Society. 2008;130:13820–13821. doi: 10.1021/ja802683u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shangguan N, Katukojvala S, Greenburg R, Williams LJ. The reaction of thio acids with azides: A new mechanism and new synthetic applications. Journal of the American Chemical Society. 2003;125:7754–7755. doi: 10.1021/ja0294919. [DOI] [PubMed] [Google Scholar]
- 22.Kolakowski RV, Shangguan N, Sauers RR, Williams LJ. Mechanism of thio acid/azide amidation. Journal of the American Chemical Society. 2006;128:5695–5702. doi: 10.1021/ja057533y. [DOI] [PubMed] [Google Scholar]
- 23.Rijkers DTS, Merkx R, Yim CB, Brouwer AJ, Liskamp RMJ. Sulfo-click' for ligation as well as for site-specific conjugation with peptides, fluorophores, and metal chelators. J Pept Sci. 16:1–5. doi: 10.1002/psc.1197. [DOI] [PubMed] [Google Scholar]
- 24.Danial NN, Korsmeyer SJ. Cell death: Critical control points. Cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 25.Green DR, Evan GI. A matter of life and death. Cancer Cell. 2002;1:19–30. doi: 10.1016/s1535-6108(02)00024-7. [DOI] [PubMed] [Google Scholar]
- 26.Reed JC. Apoptosis-regulating proteins as targets for drug discovery. Trends in Molecular Medicine. 2001;7:314–319. doi: 10.1016/s1471-4914(01)02026-3. [DOI] [PubMed] [Google Scholar]
- 27.Reed JC, Miyashita T, Takayama S, Wang HG, Sato T, Krajewski S, AimeSempe C, Bodrug S, Kitada S, Hanada M. BCL-2 family proteins: Regulators of cell death involved in the pathogenesis of cancer and resistance to therapy. Journal of Cellular Biochemistry. 1996;60:23–32. doi: 10.1002/(SICI)1097-4644(19960101)60:1%3C23::AID-JCB5%3E3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 28.Reed JC. Bcl-2 family proteins: strategies for overcoming chemoresistance in cancer. Adv Pharmacol (San Diego) 1997;41:501–532. doi: 10.1016/s1054-3589(08)61070-4. [DOI] [PubMed] [Google Scholar]
- 29.Green DR. At the gates of death. Cancer Cell. 2006;9:328–330. doi: 10.1016/j.ccr.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 30.Youle RJ, Strasser A. The BCL-2 protein family: opposing activities that mediate cell death. Nat Rev Mol Cell Biol. 2008;9:47–59. doi: 10.1038/nrm2308. [DOI] [PubMed] [Google Scholar]
- 31.Sattler M, Liang H, Nettesheim D, Meadows RP, Harlan JE, Eberstadt M, Yoon HS, Shuker SB, Chang BS, Minn AJ, Thompson CB, Fesik SW. Structure of Bcl-xL-Bak peptide complex: recognition between regulators of apoptosis. Science. 1997;275:983–986. doi: 10.1126/science.275.5302.983. [DOI] [PubMed] [Google Scholar]
- 32.Czabotar PE, Lee EF, van Delft MF, Day CL, Smith BJ, Huang DCS, Fairlie WD, Hinds MG, Colman PM. Structural insights into the degradation of Mcl-1 induced by BH3 domains. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:6217–6222. doi: 10.1073/pnas.0701297104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, Kunzer AR, Letai A, Li C, Mitten MJ, Nettesheim DG, Ng S, Nimmer PM, O'Connor JM, Oleksijew A, Petros AM, Reed JC, Shen W, Tahir SK, Thompson CB, Tomaselli KJ, Wang BL, Wendt MD, Zhang HC, Fesik SW, Rosenberg SH. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005;435:677–681. doi: 10.1038/nature03579. [DOI] [PubMed] [Google Scholar]
- 34.Wendt MD, Shen W, Kunzer A, McClellan WJ, Bruncko M, Oost TK, Ding H, Joseph MK, Zhang HC, Nimmer PM, Ng SC, Shoemaker AR, Petros AM, Oleksijew A, Marsh K, Bauch J, Oltersdorf T, Belli BA, Martineau D, Fesik SW, Rosenberg SH, Elmore SW. Discovery and structure-activity relationship of antagonists of B-cell lymphoma 2 family proteins with chemopotentiation activity in vitro and in vivo. Journal of Medicinal Chemistry. 2006;49:1165–1181. doi: 10.1021/jm050754u. [DOI] [PubMed] [Google Scholar]
- 35.Petros AM, Dinges J, Augeri DJ, Baumeister SA, Betebenner DA, Bures MG, Elmore SW, Hajduk PJ, Joseph MK, Landis SK, Nettesheim DG, Rosenberg SH, Shen W, Thomas S, Wang XL, Zanze I, Zhang HC, Fesik SW. Discovery of a potent inhibitor of the antiapoptotic protein Bcl-x(L) from NMR and parallel synthesis. Journal of Medicinal Chemistry. 2006;49:656–663. doi: 10.1021/jm0507532. [DOI] [PubMed] [Google Scholar]
- 36.Wu XH, Hu LQ. Efficient amidation from carboxylic acids and azides via selenocarboxylates: Application to the coupling of amino acids and peptides with azides. Journal of Organic Chemistry. 2007;72:765–774. doi: 10.1021/jo061703n. [DOI] [PubMed] [Google Scholar]
- 37.Manetsch R, Krasinski A, Radic Z, Raushel J, Taylor P, Sharpless KB, Kolb HC. In situ click chemistry: Enzyme inhibitors made to their own specifications. Journal of the American Chemical Society. 2004;126:12809–12818. doi: 10.1021/ja046382g. [DOI] [PubMed] [Google Scholar]
- 38.Yamaguchi H, Wang HG. Bcl-XL protects BimEL-induced Bax conformational change and cytochrome c release independent of interacting with Bax or BimEL. Journal of Biological Chemistry. 2002;277:41604–41612. doi: 10.1074/jbc.M207516200. [DOI] [PubMed] [Google Scholar]
- 39.Albert JS, Blomberg N, Breeze AL, Brown AJH, Burrows JN, Edwards PD, Folmer RHA, Geschwindner S, Griffen EJ, Kenny PW, Nowak T, Olsson LL, Sanganee H, Shapiro AB. An integrated approach to fragment-based lead generation: Philosophy, strategy and case studies from AstraZeneca's drug discovery programmes. Current Topics in Medicinal Chemistry. 2007;7:1600–1629. doi: 10.2174/156802607782341091. [DOI] [PubMed] [Google Scholar]
- 40.Erlanson DA. Fragment-based lead discovery: a chemical update. Current Opinion in Biotechnology. 2006;17:643–652. doi: 10.1016/j.copbio.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 41.Carr RAE, Congreve M, Murray CW, Rees DC. Fragment-based lead discovery: leads by design. Drug Discovery Today. 2005;10:987–992. doi: 10.1016/S1359-6446(05)03511-7. [DOI] [PubMed] [Google Scholar]
- 42.Poulsen SA, Bornaghi LF. Fragment-based drug discovery of carbonic anhydrase II inhibitors by dynamic combinatorial chemistry utilizing alkene cross metathesis. Bioorganic & Medicinal Chemistry. 2006;14:3275–3284. doi: 10.1016/j.bmc.2005.12.054. [DOI] [PubMed] [Google Scholar]
- 43.Schulz MN, Hubbard RE. Current Opinion in Pharmacology. Vol. 9. 2009. Recent progress in fragment-based lead discovery; pp. 615–621. [DOI] [PubMed] [Google Scholar]
- 44.Congreve M, Chessari G, Tisi D, Woodhead AJ. Recent developments in fragment-based drug discovery. Journal of Medicinal Chemistry. 2008;51:3661–3680. doi: 10.1021/jm8000373. [DOI] [PubMed] [Google Scholar]
- 45.Murray CW, Rees DC. The rise of fragment-based drug discovery. Nature Chemistry. 2009;1:187–192. doi: 10.1038/nchem.217. [DOI] [PubMed] [Google Scholar]
- 46.Hajduk PJ. Fragment-Based Drug Design: How Big Is Too Big? Journal of Medicinal Chemistry. 2006;49:6972–6976. doi: 10.1021/jm060511h. [DOI] [PubMed] [Google Scholar]
- 47.Nguyen R, Huc I. Using an enzyme's active site to template inhibitors. Angewandte Chemie-International Edition. 2001;40:1774–1776. [PubMed] [Google Scholar]
- 48.Mocharla VP, Colasson B, Lee LV, Roper S, Sharpless KB, Wong CH, Kolb HC. In situ click chemistry: Enzyme-generated inhibitors of carbonic anhydrase II. Angewandte Chemie-International Edition. 2005;44:116–120. doi: 10.1002/anie.200461580. [DOI] [PubMed] [Google Scholar]
- 49.Krasinski A, Radic Z, Manetsch R, Raushel J, Taylor P, Sharpless KB, Kolb HC. In situ selection of lead compounds by click chemistry: Target-guided optimization of acetylcholinesterase inhibitors. Journal of the American Chemical Society. 2005;127:6686–6692. doi: 10.1021/ja043031t. [DOI] [PubMed] [Google Scholar]
- 50.Whiting M, Muldoon J, Lin YC, Silverman SM, Lindstrom W, Olson AJ, Kolb HC, Finn MG, Sharpless KB, Elder JH, Fokin VV. Inhibitors of HIV-1 protease by using in situ click chemistry. Angewandte Chemie-International Edition. 2006;45:1435–1439. doi: 10.1002/anie.200502161. [DOI] [PubMed] [Google Scholar]
- 51.Wang J, Sui G, Mocharla VP, Lin RJ, Phelps ME, Kolb HC, Tseng HR. Integrated microfluidics for parallel screening of an in situ click chemistry library. Angewandte Chemie, International Edition. 2006;45:5276–5281. doi: 10.1002/anie.200601677. [DOI] [PubMed] [Google Scholar]
- 52.Hirose T, Sunazuka T, Sugawara A, Endo A, Iguchi K, Yamamoto T, Ui H, Shiomi K, Watanabe T, Sharpless KB, Omura S. Chitinase inhibitors: extraction of the active framework from natural argifin and use of in situ click chemistry. Journal of Antibiotics. 2009;62:277–282. doi: 10.1038/ja.2009.28. [DOI] [PubMed] [Google Scholar]
- 53.Agnew HD, Rohde RD, Millward SW, Nag A, Yeo WS, Hein JE, Pitram SM, Tariq AA, Burns VM, Krom RJ, Fokin VV, Sharpless KB, Heath JR. Iterative In Situ Click Chemistry Creates Antibody-like Protein-Capture Agents. Angewandte Chemie, International Edition. 2009;48:4944–4948. S4944/4941–S4944/4929. doi: 10.1002/anie.200900488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wang Y, Lin WY, Liu K, Lin RJ, Selke M, Kolb HC, Zhang N, Zhao XZ, Phelps ME, Shen CKF, Faull KF, Tseng HR. An integrated microfluidic device for large-scale in situ click chemistry screening. Lab on a Chip. 2009;9:2281–2285. doi: 10.1039/b907430a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jencks WP. On the Attribution and Additivity of Binding-Energies. Proceedings of the National Academy of Sciences of the United States of America-Biological Sciences. 1981;78:4046–4050. doi: 10.1073/pnas.78.7.4046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bourne Y, Kolb HC, Radic Z, Sharpless KB, Taylor P, Marchot P. Freeze-frame inhibitor captures acetylcholinesterase in a unique conformation. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:1449–1454. doi: 10.1073/pnas.0308206100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bourne Y, Radic Z, Kolb HC, Sharpless KB, Taylor P, Marchot P. Structural insights into conformational flexibility at the peripheral site and within the active center gorge of AChE. Chemico-Biological Interactions. 2005;157:159–165. doi: 10.1016/j.cbi.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 58.Willand N, Desroses M, Toto P, Dirie B, Lens Z, Villeret V, Rucktooa P, Locht C, Baulard A, Deprez B. Exploring Drug Target Flexibility Using in Situ Click Chemistry: Application to a Mycobacterial Transcriptional Regulator. ACS Chemical Biology. 5:1007–1013. doi: 10.1021/cb100177g. [DOI] [PubMed] [Google Scholar]
- 59.Bohacek RS, McMartin C, Guida WC. The art and practice of structure-based drug design: a molecular modeling perspective. Med Res Rev. 1996;16:3–50. doi: 10.1002/(SICI)1098-1128(199601)16:1<3::AID-MED1>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 60.Mammen M, Choi SK, Whitesides GM. Polyvalent interactions in biological systems: Implications for design and use of multivalent ligands and inhibitors. Angewandte Chemie-International Edition. 1998;37:2755–2794. doi: 10.1002/(SICI)1521-3773(19981102)37:20<2754::AID-ANIE2754>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.