

Abstract
Metabolism of sphingolipids into downstream lipid mediators followed by signaling modulates tumor microenvironment and the cancer cells to influence tumor progression. As such, sphingolipid signaling represents a novel way to modulate tumor biology. Neuroblastoma (NB), the most common extracranial solid tumor of childhood, is highly angiogenic and often displays poor prognosis. However, the role of sphingolipid mediators is not known in NB. We found that NB expresses high levels of sphingosine kinase-2 (SphK2), which is essential for the formation of sphingosine-1-phosphate (S1P). S1P induced vascular endothelial growth factor (VEGF) expression in SK-N-AS NB cells. The effect occurred at the transcriptional level. Hypoxia in combination with S1P had a synergistic effect on VEGF expression. Strong correlation was detected between S1P receptor-2 (S1P2) and VEGF mRNAs in 11 different cell lines and 17 NB tissues. Blockade of S1P2 with the selective antagonist JTE-013 significantly inhibited S1P-induced VEGF expression. Overexpression and knockdown of S1P2 in SK-N-AS cells increased or inhibited S1P-induced VEGF secretion, respectively. Interestingly, JTE-013 significantly inhibited tumor growth, VEGF mRNA expression and induced apoptosis in the NB tumor xenografts. Taken together, our data suggest that enhanced formation of sphingolipid mediator S1P in NB profoundly influences tumor microenvironment by inducing VEGF expression via S1P2. Modulation of sphingolipid signaling by inhibiting S1P2 may constitute a novel strategy to control NB.
Keywords: vascular endothelial growth factor, sphingosine-1-phosphate, sphingosine-1-phosphate receptor 2, sphingosine kinase 2, neuroblastoma
Introduction
Bioactive lipid sphingosine-1-phosphate (S1P), an important sphingolipid mediator, has been implicated in many pathological processes via binding to its five specific G-protein-coupled receptors (S1P1–5), which are differentially expressed and coupled to a variety of G-proteins (1). Sphingosine kinase (SphK) 1 and 2, which are overexpressed in many pathological conditions including cancer (2), are the two key enzymes responsible for catalyzing the formation of S1P. Recent studies have shown that S1P signaling may have an important role in cancer (3). Blockade of S1P signaling by using S1P-specific antibody (4), SphK inhibitors (5, 6) and S1P receptor modulator such as FTY720 (7, 8) successfully reduced tumor growth, blocked tumor angiogenesis and inhibited tumor progression in various cancers, suggesting that modulation of sphingolipid signaling may be an effective cancer preventive and/or therapeutic strategy.
Neuroblastoma (NB), derived from cells of the primitive neural crest, is the most common extracranial solid tumor in childhood and the most frequently diagnosed neoplasm during infancy. It has a broad spectrum of clinical behavior which can range from spontaneous regression to dissemination and death (9). Despite improvements in outcome for those with low-risk NB, the outcome for children with high-risk NB has improved only modestly, with long-term survival seen in less than 40% (10). Therefore, novel therapeutic strategies against NB are desperately needed. Interestingly, Meitar et al. found that tumor vascularity correlated well with an aggressive phenotype in NB (11), which made angiogenesis inhibitors an attractive therapeutic option.
Angiogenesis, the formation of new blood vessels, is a prerequisite for tumor growth and metastasis. Among many pro- and anti-angiogenic growth factors, vascular endothelial growth factor (VEGF) is a key mediator of angiogenesis. Overexpression of VEGF has been observed in a variety of solid tumors including NB and is associated with NB tumor progression (12).
However, little is known concerning the role of sphingolipid mediators in NB. Herein we show that NB produces the bioactive lipid S1P and S1P regulates angiogenesis in NB.
Materials and Methods
Materials
S1P was purchased from Biomol (Plymouth Meeting, PA) and JTE-013 was from Tocris (Ellisville, MO). V5 antibody R960-25 (1:5000) was purchased from Invitrogen (Carlsbad, CA) and β-actin antibody sc-8432 (1:1000) was from Santa Cruz (Santa Cruz, CA). FTY720-P and VPC44116 were kindly provided by Dr. Volker Brinkmann (Novartis, Basel) and Dr. Kevin Lynch (University of Virginia), respectively. Seventeen human NB frozen tissues were obtained from Children’s Oncology Group (COG).
Cell culture, adenoviral transduction and siRNA transfection
SK-N-AS and SK-N-FI cell lines were obtained from the American Type Culture Collection and cultured in DMEM (Sigma, Saint Louis, MO) supplemented with 10% FBS (Hyclone, Logan, UT), 1x MEM NEAA (Gibco, Grand Island, NY) and penicillin-streptomycin (Gibco) at 37 °C in a humidified chamber of 5% CO2 (referred to as normoxic conditions). SK-N-BE(2), CHP-134 and LAN-5 cell lines were kindly provided by Dr. Nehal Parikh (Connecticut Children’s Medical Center). SK-N-BE(2) was cultured in 1:1 MEM/F12 (Gibco) contained with 10% FBS and antibiotics while CHP-134 and LAN-5 were cultured in RPMI-1640 (Sigma) supplemented with 10% FBS and antibiotics. All the above-mentioned cell lines have not been authenticated in our laboratory. Hypoxia treatments (1% O2), adenoviral transduction and siRNA transfection (Dharmacon Lafayette, CO) were performed as previously described (13, 14).
Quantitative real-time PCR
SYBR Green-based quantitative real-time PCR was carried out as described (13). Primers were designed using Primer Express™ 2.0 (Applied Biosystems) and the sequences for VEGF gene were 5′-CGAGGGCCTGGAGTGTGT-3′ (forward) and 5′-GGCCTTGGTGAGGTTTGATC -3′ (reverse).
VEGF ELISA analysis of conditioned media
NB cells (1×106 cells/well) were seeded into 6-well plates. After attachment, they were serum starved for 24 h and treated with or without S1P in 1ml/well serum-free (SF) media under different conditions for another 24 h. The conditioned media was collected, spin down at 1000 g for 5 min at 4°C and analyzed for VEGF production using Human VEGF ELISA Development Kit (PeproTech, Rocky Hill, NJ) according to the manufacturer’s instructions. Simultaneously, MTT assay was also performed to determine the effects of different treatments on cell survival and the final VEGF protein concentration was corrected by the MTT data.
S1P measurement
SK-N-AS cells were transfected with pcDNA3 or SphK2 plasmid by Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions, serum starved for 24 h and cultured in SF DMEM for another 24 h. Cells were then washed with cold PBS, scraped and spin down to yield the cell pellets. The measurement of S1P in those pellets was performed by the analytical LC/MS/MS core facility at the Medical University of South Carolina.
Transient transfection and luciferase assay
A human 2.6-Kb (bp −2361 to +298 relative to the transcription start site) VEGF promoter-luciferase construct, which was kindly provided by Dr. Debabrata Mukhopadhyay (Mayo Clinic, Minnesota) was used in this assay (15). Briefly, SK-N-AS cells were transfected with targeted DNA construct or control vector using Lipofectamine 2000, serum-starved and stimulated with 1 μM of S1P for another 24 h. Cell extracts were prepared by using luciferase cell culture lysis buffer (Promega, Madison, WI) and assessed for luciferase activities. Final luciferase activity was normalized by the total amount of cellular protein as assayed by the BCA protein assay Kit (Pierce).
Western blot analysis
Treated SK-N-AS cells were washed with ice-cold PBS and homogenized in RIPA buffer followed by western blot analysis as previously described (16).
Subcutaneous tumor model
Six-week-old male athymic NCr-nu/nu nude mice (National Cancer Institute, Frederick, MD) were used in this study. The study was conducted according to our institution’s and the National Research Council’s guide for the care and use of laboratory animals. Briefly, each mouse received a subcutaneous flank injection containing 1 × 107 SK-N-AS cells in 0.1 ml PBS. Five days later, the mice were randomized into two groups: JTE-013-treated group receiving 30mg/Kg JTE-013 in 2% (2-hydroxypropyl)-β-cyclodextrin (Sigma) in PBS by gavage daily and the control group receiving the vehicle only. Treatments started on the day of randomization. Tumors were measured every other day with a caliper and tumor volumes were calculated using the following formula: TV = length × width2 × 0.52. Two weeks later, the mice were euthanized; tumor masses were collected followed by different assays.
PECAM-1 staining
Fresh tumor tissues derived from subcutaneous tumor model were fixed, paraffin-embedded and cut for 5μm sections followed by PECAM-1 staining (sc-1506, 1:100, Santa Cruz) as previously described (13). Sections were examined under microscope and the area containing the maximum number of discrete microvessels (“hot spot”) was identified. Five hot spots per sample were selected, photos were taken under x20 field, vessels were counted and the mean of the counts was calculated.
Terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assay
TUNEL assay was performed on the paraffin sections using ApopTag® Peroxidase In Situ Apoptosis Detection Kit (Millipore, Billerica, MA) according to the manufacturer’s instructions. Sections were examined under light microscope and six random areas in viable tumor area per sample were chosen and photos were taken under 20x field. Data were presented as the number of apoptotic cells per field.
Statistical Analysis
All experiments on cells were performed at least twice on separate occasions. Data were presented as means ± SE from representative experiments. Statistical significance of differences between two groups was determined by two-tailed homoscedastic Student’s t-test using Microsoft Excel software.
Results
SphK2 was abundantly expressed in NB cells and tissues
To investigate the role of sphingolipid mediators in NB, we determined the expression of SphK, the key rate-limiting enzyme in S1P synthesis, in NB cell lines and tumor tissues by quantitative real-time PCR. Both SphK1 and SphK2 were expressed in all 5 NB cell lines tested while SphK2 was more abundantly expressed (Fig. 1A). This phenomenon was also observed in 17 NB tumor tissues, in which the mRNA expression of SphK2 was at least two orders of magnitude higher than that of SphK1 (Fig. 1B). We also measured S1P level in NB cells by LC/MS/MS. NB cells synthesized significant levels of S1P and transfection of SphK2 into SK-N-AS cells further increased its level (Fig. 1C). These data suggest that S1P produced by SphK might play a functional role in NB.
FIGURE 1.
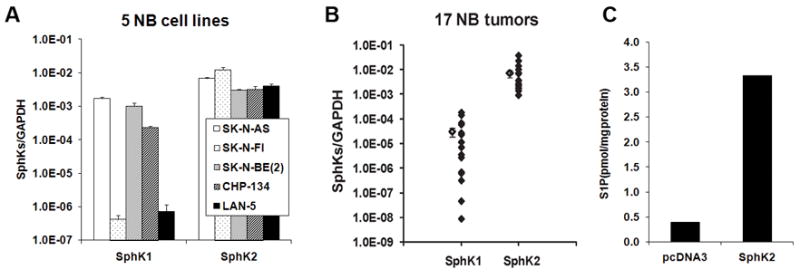
SphK2 was abundantly expressed in NB cells and tissues. A and B, The mRNA expression of SphK1 and SphK2 in 5 NB cell lines (A) and 17 human NB tissues (B) by quantitative real-time PCR. The mean values for SphK1 and SphK2 in B were shown at the left side of each column. C, The S1P synthesis in SK-N-AS cells transfected with pcDNA3 or SphK2 plasmid. Representative data from two independent experiments was shown.
S1P induced VEGF expression in NB cells, distinct from hypoxia
S1P treatment of NB cells in vitro did not induce cell proliferation significantly (data not shown). However, to determine whether S1P, the product of SphK, regulates the tumor microenvironment, we measured the expression of VEGF, a major regulator of angiogenesis. S1P concentration-dependently increased VEGF mRNA levels (Fig. 2A). Therefore, conditioned media of SK-N-AS cells with S1P treatment was also collected followed by VEGF ELISA analysis. Similarly, S1P induced VEGF protein secretion concentration-dependently (Fig. 2A). Moreover, the induction of VEGF secretion by S1P was observed in other NB cell lines (Fig. 2B), indicating the effect of S1P on VEGF induction is a general phenomenon in NB. Because hypoxia is a major stimulator of VEGF expression in tumors (17), we also determined the effect of hypoxia in conjunction with S1P stimulation on VEGF induction. S1P stimulation and hypoxia both upregulated VEGF mRNA levels and protein secretion. And the two stimuli together had a synergistic effect (Fig. 2C), suggesting that VEGF induced by S1P may play a role distinct from that of hypoxia in NB.
FIGURE 2.

S1P induced VEGF expression in NB cells, distinct from hypoxia. A, SK-N-AS cells were serum starved and treated with S1P for 2 h or 24 h followed by quantitative real-time PCR (2h) and VEGF ELISA (24h). B, NB cells were serum starved and treated with S1P (1μM) for 24 h followed by VEGF ELISA. C, Serum-starved SK-N-AS cells were incubated with or without S1P (1μM) under normoxia or hypoxia for 2 h or 24 h followed by quantitative real-time PCR (2h) and VEGF ELISA (24h). Results were means ± SE. n=3. *, P < 0.05, **, P < 0.01 versus without S1P treatment; ##, P < 0.01 versus normoxia.
S1P induced VEGF mRNA transcription, not mRNA stability
To determine whether S1P-induced VEGF mRNA expression occurred on the transcriptional level or post-transcriptional level, SK-N-AS cells were treated with or without S1P for 2 h followed by the addition of actinomycin D to inhibit transcription. It showed that in control and S1P-stimulated cells, VEGF mRNA levels decreased with almost the same degradation rate, suggesting that S1P did not alter VEGF mRNA stability (Fig. 3A). Stimulation with S1P was not able to induce VEGF mRNA expression in cells pretreated with actinomycin D (Fig. 3B), suggesting the effect of S1P on VEGF mRNA expression occurred on the transcriptional level. To confirm this hypothesis, human VEGF promoter luciferase construct was introduced into SK-N-AS cells, and luciferase activity was measured in cell lysates after S1P stimulation. S1P increased VEGF promoter activity significantly by 1.7 fold (Fig. 3C). Taken together, these data demonstrate that S1P increased VEGF mRNA transcription but not its mRNA stability.
FIGURE 3.
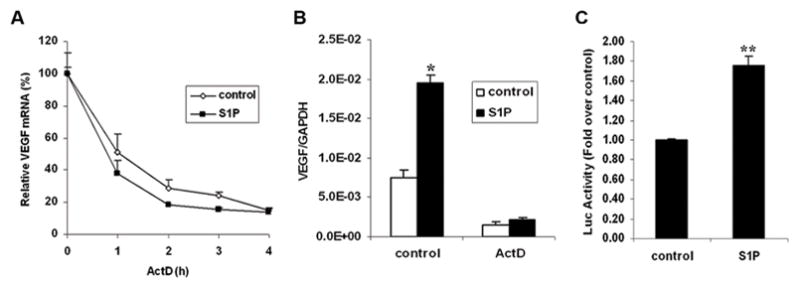
S1P induced VEGF mRNA transcription, not mRNA stability. A, Serum-starved SK-N-AS cells were treated with or without S1P (1 μM) for 2 h followed by incubation with ActD (10μg/ml) for different times (0, 1, 2, 3, 4 h) before quantitative real-time PCR was performed. Data are expressed as relative to control, assigning a value of 100% to the control cells. B, Serum-starved SK-N-AS cells were pretreated with ActD (10μg/ml) for 0.5 h and stimulated with S1P (1μM) for another 2 h followed by quantitative real-time PCR. C, SK-N-AS cells were transfected with the VEGF promoter-luciferase construct by Lipofectamine 2000, serum starved and stimulated with S1P (1μM) for another 24 h followed by the assessment of luciferase activity. Data are expressed as fold over control. Results were means ± SE. n=3. *, P < 0.05, **, P < 0.01 versus non S1P-treated control.
S1P2 was responsible for S1P-induced VEGF expression in NB cells
Five S1P receptors (S1P1–5) have been identified to bind specifically to S1P (1). To determine which S1P receptor was responsible for S1P-induced VEGF expression, we detected the expression profile of S1P receptors in NB cells and NB tissues. S1P1, S1P2 and S1P3 were mainly expressed whereas S1P4 and S1P5 were barely detectable (Fig. 4A and 4B). There was a good correlation between S1P2 mRNA and VEGF mRNA expression in 5 NB cell lines (R=0.7). Interestingly, introduction of more cell lines into this statistical analysis revealed the existence of strong correlation between S1P2 and VEGF (R=0.875, P <0.001; Fig. 4C). More importantly, this strong correlation was also seen in 17 NB tissues (R=0.884, P<0.001; Fig. 4D), suggesting that S1P2 might be responsible for VEGF expression.
FIGURE 4.
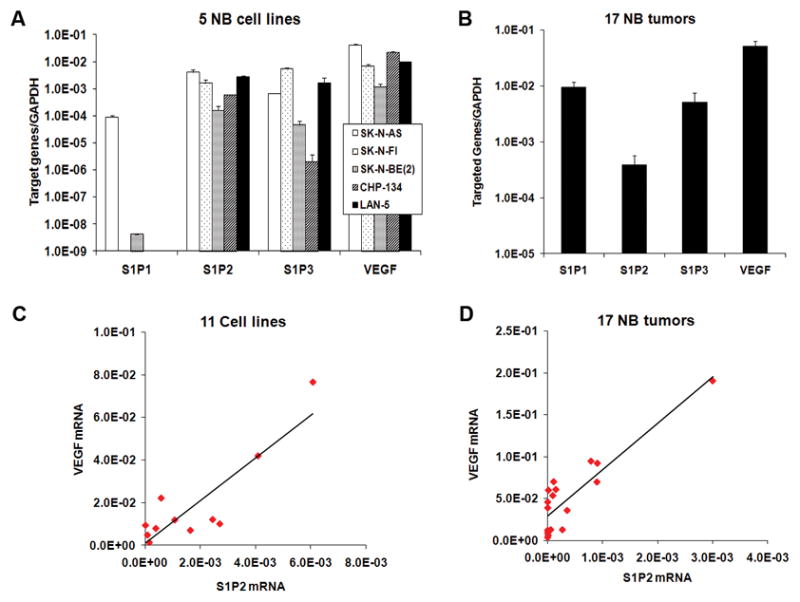
VEGF mRNA level correlated well with S1P2 mRNA level in different cells and NB tissues. A and B, The mRNA expression of S1P receptors and VEGF in 5 NB cell lines (A) and 17 human NB tissues (B) by quantitative real-time PCR. C and D, Correlation analysis between S1P2 and VEGF mRNA was performed by Origin 7.0 in 11 cell lines including 5 NB cell lines, WiT49, G401, ACHN, HEY, SKOV-3 and IOSE (C) and 17 human NB tissues (D).
Using FTY720-phosphate (FTY720-P), the agonist for all S1P receptors except S1P2 (18), we did not see the same degree of induction as we observed with S1P (Fig. 5A), which further suggested that S1P-induced VEGF expression might be mediated by S1P2. To substantiate this notion, we used the selective S1P2 antagonist JTE-013 (16, 19). It showed that JTE-013 significantly inhibited S1P-induced VEGF mRNA expression and protein secretion. In contrast, inhibition of S1P1 signaling by S1P1 antagonist VPC44116 (13, 20) did not affect S1P-induced VEGF expression (Fig. 5B). Next, we altered S1P2 expression level in SK-N-AS cells by adenoviral transduction and siRNA transfection. In agreement with our hypothesis, overexpression and downregualtion of S1P2 either increased or inhibited S1P-induced VEGF protein expression (Fig. 5C and 5D). Taken together, these data strongly suggest that S1P-induced VEGF expression is mediated by S1P2 in NB cells.
FIGURE 5.

S1P2 was responsible for S1P-induced VEGF expression in NB cells. A, SK-N-AS cells were serum starved and stimulated with FTY720-P (10, 100nM) for 2 h or 24 h followed by quantitative real-time PCR (2h) and VEGF ELISA (24h). B, Serum-starved SK-N-AS cells were pretreated with JTE-013 (1μM) or VPC-44116 (1μM) for 0.5 h and stimulated with S1P (1μM) for another 2 h or 24 h followed by quantitative real-time PCR (2h) and VEGF ELISA (24h). C, SK-N-AS cells were infected with adenovirus overexpressing S1P2 or GFP, serum-starved and stimulated with S1P (1μM) for 24 h followed by western blot (left) and VEGF ELISA (right). D, SK-N-AS cells were transfected with 100 nM S1P2 siRNA or non-specific (NS) siRNA, serum-starved and treated with S1P (1μM) for 2 h or 24 h followed by quantitative real-time PCR (left, middle) and VEGF ELISA(right). Results were means ± SE. n=2. *, P < 0.05, **, P < 0.01 versus without S1P treatment or NS siRNA(4D, left); #, P < 0.05, ##, P < 0.01 versus indicated control.
S1P2 antagonist JTE-013 inhibited growth of NB xenografts
Next we were interested in what the role of this pathway was on tumor growth. Therefore, a subcutaneous xenograft tumor model was used. After treatment of the mice with S1P2 antagonist JTE-013 for two weeks, we found that JTE-013, at its expected maximal dose comparing to that of S1P receptor modulator FTY720 (7), was able to significantly inhibit tumor growth from day 10 compared to the control group (Fig. 6A). Quantitative real-time PCR confirmed that VEGF mRNA expression was significantly decreased in JTE-013-treated tumors (Fig. 6B). Histological studies utilizing PECAM-1 staining did not find a significant decrease in number of vessels in JTE-013-treated tumors, but a trend in this direction was observed (Fig. 6C). However, more apoptotic cells were observed in JTE-013-treated tumors by TUNEL assay (Fig. 6D).
FIGURE 6.
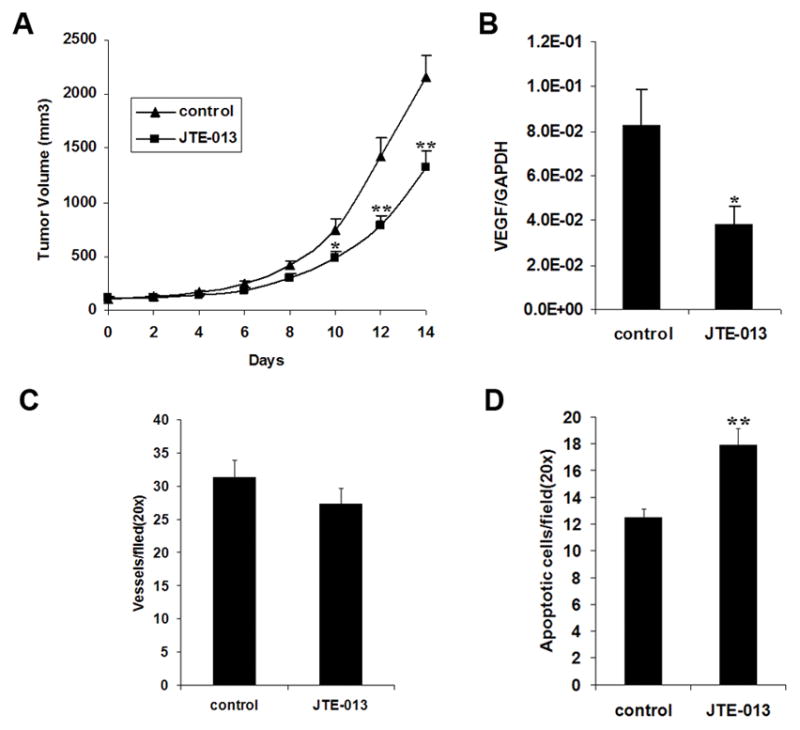
S1P2 antagonist JTE-013 inhibited growth of NB xenografts. A, The tumor growth curves of control and JTE-013-treated mice (n=7). B, VEGF mRNA levels in control and JTE-013-treated tumors by quantitative real-time PCR. C, Vessel density in control and JTE-013-treated tumors by PECAM-1 staining. D, Apoptotic cells in control and JTE-013-treated tumors labeled by TUNEL method. *, P < 0.05, **, P < 0.01 versus the corresponding control.
Discussion
VEGF has been found to be overexpressed in NB and it has been determined a marker of poor prognosis (12). Therefore, VEGF inhibition has potential therapeutic implications for NB treatment. On the other hand, accumulating evidence shows that sphingolipid signaling plays an important role in human cancers, indicating that modulation of this pathway might be one of the potential cancer therapeutic strategies. To date, cross-talk between S1P signaling and VEGF signaling has been identified (21, 22). However, it is unclear whether these two important molecules might directly affect each other and elicit biological functions in human cancers. Interestingly, in this study, we found that SphKs, especially SphK2, was abundantly expressed in NB cells and tissues (Fig. 1). As the product of these enzymes, S1P induced VEGF expression in different NB cells via hypoxia-independent pathway (Fig. 2). And it occurred on the transcriptional level (Fig. 3), in a HIF-1-independent manner (data not shown). These data suggest that SphK/S1P/VEGF signaling might play an important role in NB, which prompted us to further examine this pathway in NB.
As an exogenous stimulator, S1P exerts its biological functions by interaction with its specific receptors. Among them, S1P1 and S1P2 usually display opposite effects. Typically, S1P1 stimulates while S1P2 inhibits cell migration (23, 24). We have expanded the known functions of S1P2 in our lab to include induction of anti-proliferative CTGF expression and induction of the inflammatory factor COX-2 expression in Wilms tumor (16, 25). Recently, more complex functions of S1P2 have been revealed in the tumor-related immune system and vascular system (24, 26). However, the function of S1P2 on tumor cells is unclear. Interestingly in our study, correlation analysis showed a good linear relationship exists between S1P2 mRNA and VEGF mRNA in different cancer cells and NB tissues (Fig. 4), implying that S1P2 might be responsible for S1P-induced VEGF expression in tumor cells. To confirm this hypothesis, several techniques including the employment of S1P analogue FTY720-P, S1P2 antagonist JTE-013, adenoviral transduction and siRNA transfection were used. All our findings suggest that S1P-induced VEGF expression is mediated by S1P2 in NB cells (Fig. 5). It has been shown that S1P2 can activate RhoA and three MAPKs signaling pathways (27). Further investigation of downstream signaling using different pharmacological inhibitors revealed that RhoA/ROCK and ERK pathways might be involved in this process (data not shown).
In 2009, Cattoretti et al. reported that targeted disruption of S1P2 resulted in alterations in the immune system which led to diffuse large B-cell lymphoma formation (26). However, the authors did not propose a mechanism for the potential lymphoma suppressive action of S1P2. More recently, Du et al. implanted mouse tumor cells into S1P2-deficient mice and found that S1P2 on host endothelial cells (ECs) negatively regulated tumor angiogenesis and tumor growth (24). Arguably, their model focused on the function of S1P2 from ECs. In fact, the S1P2 status and function of these tumor cells were not described. Interestingly, we report for the first time that S1P2 antagonist JTE-013 significantly inhibited growth of NB xenografts. Further analysis demonstrated lower human VEGF mRNA levels and more apoptotic cells in the JTE-013-treated tumors (Fig. 6). Although the difference in microvessel density did not reach statistical significance, the trend was towards lower vessels in JTE-013-treated tumors and this may lead to alterations in blood flow and tumor apoptosis, and finally lead to reduced tumor growth. Taken together, these data validated our in vitro findings and further suggest that S1P/S1P2/VEGF signaling in tumor cells may promote NB growth by affecting angiogenesis and the relevant tumor cell apoptosis. Cumulatively, the available data suggests that S1P2 from different sources might play distinct roles on tumor angiogenesis and growth through different mechanism.
In summary, our findings for the first time demonstrate that SphK2 was abundantly expressed in NB and its product S1P can induce VEGF expression in cancer via S1P2, which further extends the biological functions of S1P2 in human cancers. Moreover, on the basis of the finding that S1P2 antagonist JTE-013 significantly inhibited NB tumor growth, it strongly suggests that modulation of sphingolipid signaling by inhibiting S1P2 may constitute a novel strategy to control NB.
Acknowledgments
Grant support: NIH grants k08DK070468A (F.F.), P01CA77839 (T.H.), R01HL89934 (T.H.) and the Seraph Foundation.
Footnotes
T.H. consulted the following commercial entities – Novartis Pharma, Sidley Austin LLC and Wilmer Hale LLC.
References
- 1.Sanchez T, Hla T. Structural and functional characteristics of S1P receptors. J Cell Biochem. 2004;92:913–22. doi: 10.1002/jcb.20127. [DOI] [PubMed] [Google Scholar]
- 2.French KJ, Schrecengost RS, Lee BD, et al. Discovery and evaluation of inhibitors of human sphingosine kinase. Cancer Res. 2003;63:5962–9. [PubMed] [Google Scholar]
- 3.Pyne NJ, Pyne S. Sphingosine 1-phosphate and cancer. Nat Rev Cancer. 2010;10:489–503. doi: 10.1038/nrc2875. [DOI] [PubMed] [Google Scholar]
- 4.Visentin B, Vekich JA, Sibbald BJ, et al. Validation of an anti-sphingosine-1-phosphate antibody as a potential therapeutic in reducing growth, invasion, and angiogenesis in multiple tumor lineages. Cancer cell. 2006;9:225–38. doi: 10.1016/j.ccr.2006.02.023. [DOI] [PubMed] [Google Scholar]
- 5.Chumanevich AA, Poudyal D, Cui X, et al. Suppression of colitis-driven colon cancer in mice by a novel small molecule inhibitor of sphingosine kinase. Carcinogenesis. 2010;31:1787–93. doi: 10.1093/carcin/bgq158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.French KJ, Zhuang Y, Maines LW, et al. Pharmacology and antitumor activity of ABC294640, a selective inhibitor of sphingosine kinase-2. J Pharmacol Exp Ther. 2010;333:129–39. doi: 10.1124/jpet.109.163444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.LaMontagne K, Littlewood-Evans A, Schnell C, et al. Antagonism of sphingosine-1-phosphate receptors by FTY720 inhibits angiogenesis and tumor vascularization. Cancer Res. 2006;66:221–31. doi: 10.1158/0008-5472.CAN-05-2001. [DOI] [PubMed] [Google Scholar]
- 8.Pchejetski D, Bohler T, Brizuela L, et al. FTY720 (fingolimod) sensitizes prostate cancer cells to radiotherapy by inhibition of sphingosine kinase-1. Cancer Res. 2010;70:8651–61. doi: 10.1158/0008-5472.CAN-10-1388. [DOI] [PubMed] [Google Scholar]
- 9.Rossler J, Taylor M, Geoerger B, et al. Angiogenesis as a target in neuroblastoma. Eur J Cancer. 2008;44:1645–56. doi: 10.1016/j.ejca.2008.05.015. [DOI] [PubMed] [Google Scholar]
- 10.Maris JM, Hogarty MD, Bagatell R, Cohn SL. Neuroblastoma. Lancet. 2007;369:2106–20. doi: 10.1016/S0140-6736(07)60983-0. [DOI] [PubMed] [Google Scholar]
- 11.Meitar D, Crawford SE, Rademaker AW, Cohn SL. Tumor angiogenesis correlates with metastatic disease, N-myc amplification, and poor outcome in human neuroblastoma. J Clin Oncol. 1996;14:405–14. doi: 10.1200/JCO.1996.14.2.405. [DOI] [PubMed] [Google Scholar]
- 12.Eggert A, Ikegaki N, Kwiatkowski J, Zhao H, Brodeur GM, Himelstein BP. High-level expression of angiogenic factors is associated with advanced tumor stage in human neuroblastomas. Clin Cancer Res. 2000;6:1900–8. [PubMed] [Google Scholar]
- 13.Li MH, Sanchez T, Yamase H, et al. S1P/S1P1 signaling stimulates cell migration and invasion in Wilms tumor. Cancer Lett. 2009;276:171–9. doi: 10.1016/j.canlet.2008.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vumbaca F, Phoenix KN, Rodriguez-Pinto D, Han DK, Claffey KP. Double-stranded RNA-binding protein regulates vascular endothelial growth factor mRNA stability, translation, and breast cancer angiogenesis. Mol Cell Biol. 2008;28:772–83. doi: 10.1128/MCB.02078-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mukhopadhyay D, Knebelmann B, Cohen HT, Ananth S, Sukhatme VP. The von Hippel-Lindau tumor suppressor gene product interacts with Sp1 to repress vascular endothelial growth factor promoter activity. Mol Cell Biol. 1997;17:5629–39. doi: 10.1128/mcb.17.9.5629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li MH, Sanchez T, Pappalardo A, Lynch KR, Hla T, Ferrer F. Induction of antiproliferative connective tissue growth factor expression in Wilms’ tumor cells by sphingosine-1-phosphate receptor 2. Mol Cancer Res. 2008;6:1649–56. doi: 10.1158/1541-7786.MCR-07-2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Minchenko A, Bauer T, Salceda S, Caro J. Hypoxic stimulation of vascular endothelial growth factor expression in vitro and in vivo. Lab Invest. 1994;71:374–9. [PubMed] [Google Scholar]
- 18.Brinkmann V, Davis MD, Heise CE, et al. The immune modulator FTY720 targets sphingosine 1-phosphate receptors. J Biol Chem. 2002;277:21453–7. doi: 10.1074/jbc.C200176200. [DOI] [PubMed] [Google Scholar]
- 19.Sanchez T, Skoura A, Wu MT, Casserly B, Harrington EO, Hla T. Induction of vascular permeability by the sphingosine-1-phosphate receptor-2 (S1P2R) and its downstream effectors ROCK and PTEN. Arterioscler Thromb Vasc Biol. 2007;27:1312–8. doi: 10.1161/ATVBAHA.107.143735. [DOI] [PubMed] [Google Scholar]
- 20.Oo ML, Thangada S, Wu MT, et al. Immunosuppressive and anti-angiogenic sphingosine 1-phosphate receptor-1 agonists induce ubiquitinylation and proteasomal degradation of the receptor. J Biol Chem. 2007;282:9082–9. doi: 10.1074/jbc.M610318200. [DOI] [PubMed] [Google Scholar]
- 21.Igarashi J, Erwin PA, Dantas AP, Chen H, Michel T. VEGF induces S1P1 receptors in endothelial cells: Implications for cross-talk between sphingolipid and growth factor receptors. Proc Natl Acad Sci U S A. 2003;100:10664–9. doi: 10.1073/pnas.1934494100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tanimoto T, Jin ZG, Berk BC. Transactivation of vascular endothelial growth factor (VEGF) receptor Flk-1/KDR is involved in sphingosine 1-phosphate-stimulated phosphorylation of Akt and endothelial nitric-oxide synthase (eNOS) J Biol Chem. 2002;277:42997–3001. doi: 10.1074/jbc.M204764200. [DOI] [PubMed] [Google Scholar]
- 23.Anelli V, Gault CR, Snider AJ, Obeid LM. Role of sphingosine kinase-1 in paracrine/transcellular angiogenesis and lymphangiogenesis in vitro. FASEB J. 2010 doi: 10.1096/fj.09-150540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Du W, Takuwa N, Yoshioka K, et al. S1P(2), the G protein-coupled receptor for sphingosine-1-phosphate, negatively regulates tumor angiogenesis and tumor growth in vivo in mice. Cancer Res. 2010;70:772–81. doi: 10.1158/0008-5472.CAN-09-2722. [DOI] [PubMed] [Google Scholar]
- 25.Li MH, Sanchez T, Milne GL, Morrow JD, Hla T, Ferrer F. S1P/S1P2 signaling induces cyclooxygenase-2 expression in Wilms tumor. J Urol. 2009;181:1347–52. doi: 10.1016/j.juro.2008.10.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cattoretti G, Mandelbaum J, Lee N, et al. Targeted disruption of the S1P2 sphingosine 1-phosphate receptor gene leads to diffuse large B-cell lymphoma formation. Cancer Res. 2009;69:8686–92. doi: 10.1158/0008-5472.CAN-09-1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taha TA, Argraves KM, Obeid LM. Sphingosine-1-phosphate receptors: receptor specificity versus functional redundancy. Biochim Biophys Acta. 2004;1682:48–55. doi: 10.1016/j.bbalip.2004.01.006. [DOI] [PubMed] [Google Scholar]