

Abstract
Arsenic is a cancer chemotherapeutic but hepatotoxicity can be a limiting side effect. O2-Vinyl 1-[2-(carboxylato)pyrrolidin-1-yl]diazen-1-ium-1,2-diolate (V-PROLI/NO) is a nitric oxide (NO) donor prodrug and metabolized by liver cytochromes P450 (CYP450) to release NO. The effects of V-PROLI/NO pretreatment on the toxicity of arsenic (as NaAsO2) were studied in a rat liver cell line (TRL 1215). The cells acted upon the prodrug to release NO, as assessed by nitrite levels, in a time-dependent fashion to maximal levels of 8-fold above basal levels. Pretreatment with V-PROLI/NO markedly reduced arsenic cytolethality which was directly related to the level of NO produced by V-PROLI/NO treatment. Cyp1a1 expression was directly related to the level of NO production and to reduced arsenic cytotoxicity. V-PROLI/NO pretreatment markedly reduced arsenic-induced apoptosis and suppressed phosphorylation of JNK1/2. V-PROLI/NO pretreatment facilitated additional increases in arsenic-induced metallothionein, a metal-binding protein important in arsenic tolerance. Thus, V-PROLI/NO protects against arsenic toxicity in rat liver cells, reducing cytolethality, apoptosis and dysregulation of MAPKs, through generation of NO formed after metabolism by liver cell enzymes, possibly including Cyp1a1. CYP450 required for NO production from V-PROLI/NO treatment in the rat and human appears to differ as we have previously studied the ability of V-PROLI/NO to prevent arsenic toxicity in human liver cells where it reduced toxicity apparently through a CYP2E1-mediated metabolic mechanism. None-the-less, it appears that both rat and human liver cells act upon V-PROLI/NO via a CYP450-related mechanism to produce NO and subsequently reduce arsenic toxicity.
Keywords: V-PROLI/NO, arsenic, rat liver cell, Cyp1a1
1. Introduction
Inorganic arsenic is now a first choice cancer chemotherapeutic against certain leukemias and is thought to have potential against a variety of other cancers, including solid tumors [1,2]. Specifically, arsenic trioxide is used in the treatment of acute promyelocytic leukemia and greatly improves the clinical outcome even in refractory or multiple relapsed cases [3,4]. However, toxic side effects of arsenicals are often a major concern, including the potential for fatal hepatotoxicity [5]. The liver is a major target organ for both arsenic metabolism and toxicity [6]. Thus, an agent able to reduce the toxic potential of arsenic in liver cells would clearly be a useful adjuvant for arsenical chemotherapy, regardless of where the primary tumor of concern was located.
Nitric oxide (NO) is a simple chemical signaling mediator molecule produced by various cells [7]. The study of NO biochemistry and physiology has provided promising avenues for the development of numerous pharmaceuticals based on their ability to release NO [8]. Development of site-specific NO prodrugs is an important trend in production of agents with pharmacologic potential [9]. O2-Vinyl 1-[2-(carboxylato)pyrrolidin-1-yl]diazen-1-ium-1,2-diolate (V-PROLI/NO) is a NO donating prodrug that appears to have liver specificity, as it is metabolized to release NO by liver cytochrome P450s (CYP450) [10]. Emerging evidence indicates that cytochrome P450 1a1 (Cyp1a1), a hepatic and extrahepatic enzyme, is regulated by metal exposure [11] and may play an important role in the detoxication of environmental carcinogens [12]. Recently, we have demonstrated that V-PROLI/NO protects liver cells from arsenic-induced toxicity in a human liver cell line (HepG2) through generation of NO likely formed after metabolism by liver cell enzyme cytochrome P450 2E1 (CYP2E1) [13]. HepG2 is a human heptocarcinoma cell line commonly used as a liver model and it is of interest to see if the same phenomenon occurs in non-tumor cell lines or liver cell lines from non-human species. Whether V-PROLI/NO is metabolized by CYP450 and can be protective against arsenic toxicity is universal in mammalian cells has not been demonstrated.
Mitogen-activated protein kinases (MAPKs) are a family of serine/threonine kinases that include extracellular signal-regulated kinases (ERKs) and c-Jun NH2-terminal kinases/stress-activated protein kinases (JNKs/SAPKs) [14]. ERKs transduce growth factor signals inducing cell proliferation or differentiation. In contrast, JNKs/SAPKs are strongly activated in responses to various stresses, growth arrest, DNA-damaging agents and apoptosis [15]. The levels of phosphorylated JNK1/2 and JNK kinase activity are markedly decreased in cells chronically exposed to arsenic [16]. Activation of the JNK pathway can be critical to apoptosis, and pretreatment with V-PROLI/NO negatively regulates arsenic-induced JNK activation in human HepG2 liver cells [13], indicating that NO has the capacity to alter the adverse effects of arsenic with regard to apoptotic signaling.
Metallothionein (MT) is a small, cysteine-rich, metal-sequestering protein that detoxicates various inorganics [17]. For instance, arsenic enhances MT expression [18] and binds to MT [19]. MT appears to prevent arsenic toxicity on several levels [20]. A recent study showed that human populations poorly expressing MT may be more sensitive to chronic arsenic intoxication [21]. NO released from NO-donating compounds interacts with MT indirectly by releasing bound metal and inducing the protein [22]. Indeed, MT, heme oxygenase-1 (HO-1) and glutathione S-transferase (GST) are well-known markers of acute arsenic-induced oxidative stress. GST plays a key role in phase II metabolism typically observed with detoxification. Polymorphisms in GST genes may affect the behavior of several enzymes involved with the maintenance of cellular glutathione (GSH) levels [23], which protect against cellular reactive oxygen species (ROS) and are thought to be generated in cells that methylate inorganic arsenic [24].
Thus, the purpose of this study was to determine if V-PROLI/NO acts as a potential hepatoprotectant prodrug by blocking arsenic-induced toxicity and apoptosis at the cellular level in the rat liver cell line TRL 1215. We found that although V-PROLI/NO reduced arsenic toxicity in both rat (present work) and human liver cells [13], the rat cells required Cyp1a1 for NO release while the human cells required CYP2E1, an important species difference reported in this present work.
2. Materials and methods
2.1. Chemicals
Sodium arsenite and α-naphthoflavone were purchased from Sigma Chemical Company (St. Louis, MO). V-PROLI/NO was synthesized as previously described [25]. Chemical structure of V-PROLI/NO and its metabolic pathway are shown in Figure 1. Anti-phospho-JNK, anti-phospho-ERK, and anti-JNK1/2 antibodies were purchased from New England Biolabs, Inc. (Beverly, MA).
Fig. 1.
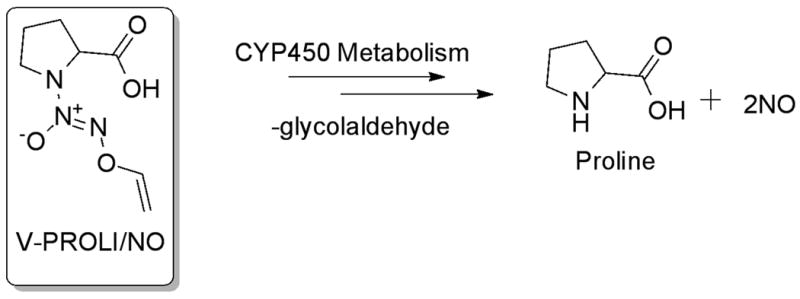
Chemical structure of V-PROLI/NO and its metabolism to produce proline and NO.
2.2. Cell culture
The TRL 1215 rat liver cells were cultured as monolayers in William's E medium containing 10% fetal bovine serum at 37°C in a humidified 5% CO2 atmosphere. These cells metabolically resemble hepatocytes, are diploid and nontumorigenic.
2.3. Metabolic integrity assay
The Promega Cell Titer 96 Non-Radioactive Cell Proliferation Assay kit was used to determine acute cytotoxicity of arsenic (as sodium arsenite) in cells as defined by metabolic integrity. This assay measures the amount of formazan produced by metabolic conversion of Owen's reagent [(3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt, MTS] by dehydrogenase enzymes found in the mitochondria of metabolically active cells. The quantity of formazan product, as measured by absorbance at 490 nm, is directly proportional to the number of living cells. A minimum of 4 replicates of 10,000 cells per well were plated in 96-well plates and allowed to adhere to the plate for 24 h, at which time the media were removed and replaced with fresh media with or without V-PROLI/NO (100 or 200 μM). At the end of this period arsenic (as sodium arsenite) was added in fresh media. Cells were then incubated for an additional 24 h and cell viability was determined. The lethal concentration 50% (LC50) values were determined from analysis of the linear portion of four separately derived metabolic integrity curves.
2.4. Nitrite measurement
A Griess reagent-based system (Promega, Madison, WI) was used to determine nitrite concentration in cell culture media as an indication of NO generated from V-PROLI/NO. A minimum of 4 replicates of 10,000 cells per well were plated in 96-well plates and allowed to adhere to the plate for 24 h. Cells were pretreated with V-PROLI/NO for 24 h. Extracellular media were collected and nitrite was measured.
2.5. Quantification of apoptosis
DNA fragmentation, as an indication of apoptotic cell death, was assessed by determination of cytoplasmic histone-DNA fragments using the Cell Death Detection ELISA kit (Roche, Indianapolis, IN). In all cases, cells were seeded in 96-well plates at 10,000 cells per well in 200 μl medium and treatments were initiated 24 h after plating. To examine the effects of V-PROLI/NO on arsenic-induced apoptosis, cells were pretreated with V-PROLI/NO for 24 h and then incubated with arsenic (as sodium arsenite) for an additional 24 h. Apoptosis was evaluated in both floating and adherent cells.
2.6. Real-time reverse transcriptase polymerase chain reaction analysis (RT-PCR)
Total RNA was isolated from rat liver TRL 1215 cells using TRIzol® (GIBCO/BRL Life Technologies) followed by the cleanup using RNeasy Mini kit (Qiagen, Valencia, CA). The resultant DNA-free RNA was quantitated by UV spectroscopy at 260 nm and stored in RNase-free H2O at −80°C. Quantitative real-time reverse transcription polymerase chain reaction (real time RT-PCR) was conducted. Briefly, total RNA from each sample was reverse transcribed with MuLV reverse transcriptase (Applied Biosystems, Foster City, CA) and Oligo d(T) primers. The SYBR Green PCR master mix (Applied Biosystem) was used for quantitative real-time RT-PCR analysis. The rat primers were designed using Primer Express software (Applied Biosystems) and listed here: Cyp1a1, Forward 5′-GCA GGC CCT GGT GAA ACA-3′; Reverse, 5′-GCT CTG GCC ATT AGC GAT AAG T′; Gst-π, Forward 5′-GGG TCG CTC TTT AGG GCT TTA-3′; Reverse, 5′-GCA GGG CCT TCA CAT AGT CAT C-3′; HO-1, Forward 5′-ATC ATG GCT TGG CCT ACA TTG-3′; Reverse, 5′-CAC GGA TGT GCA CCT CCT T-3′; β-actin, Forward 5′-GGC CAA CCG TGA AAA GAT GA-3′; Reverse 5′-GCC TGG ATG GCT ACG TAC ATG-3′. Relative differences in gene expression between groups were expressed using cycle time (Ct) values; these values were first normalized with that of β-actin in the same sample and expressed as arbitrary units. Real time fluorescence detection was carried out using a MyiQ™ singleColor Real-Time PCR Detection System (Bio-Rad, Hercules, CA).
2.7. Western blot analysis
Protein samples (30 μg) derived from the various cell preparations were subjected to SDS-polyacrylamide gel electrophoresis and transferred onto nitrocellulose membranes. The membranes were blocked with 5% non-fat dry milk in Tris-buffered saline containing 0.05% Tween 20 (TBST) and probed with phospho-specific antibodies against JNK1/2, ERK1/2 and p38 MAPK [16]. After incubation with secondary antibodies, immunoblots were visualized with the LumiGlo detection method (New England Biolabs).
2.8. Metallothionein (MT) quantitation
Cellular MT concentrations were measured by the Cd-hemoglobin method [26]. Values were adjusted to cell number and are expressed as ng MT per 106 cells.
2.9. Statistical analysis
Data are expressed as mean ± statistical error of the mean (SEM). Student's t-test or ANOVA with subsequent Dunnett's test were used as appropriate. Linear (Pearson) correlations were used to determine statistical significance of correlations between V-PROLI/NO concentrations or nitrite production and LC50 for arsenic, correlations between V-PROLI/NO concentration or nitrite production and Cpy1a1 transcript and correlations between Cpy1a1 transcript and LC50 for arsenic. Values are derived from 3 or more replications. Differences were considered significant at a level of p < 0.05.
3. Results
3.1. V-PROLI/NO Toxicity
Initially, we tested the toxicity of V-PROLI/NO in TRL 1215 cells (Figure 2). The results showed V-PROLI/NO had a very low toxicity and 200 μM was non-toxic at 24 h (Figure 2A). This was true for the entire 24 h treatment period (Figure 2B). Thus, concentrations of 100 and 200 μM and a 24 h treatment period for V-PROLI/NO were used for the remainder of this study.
Fig. 2.
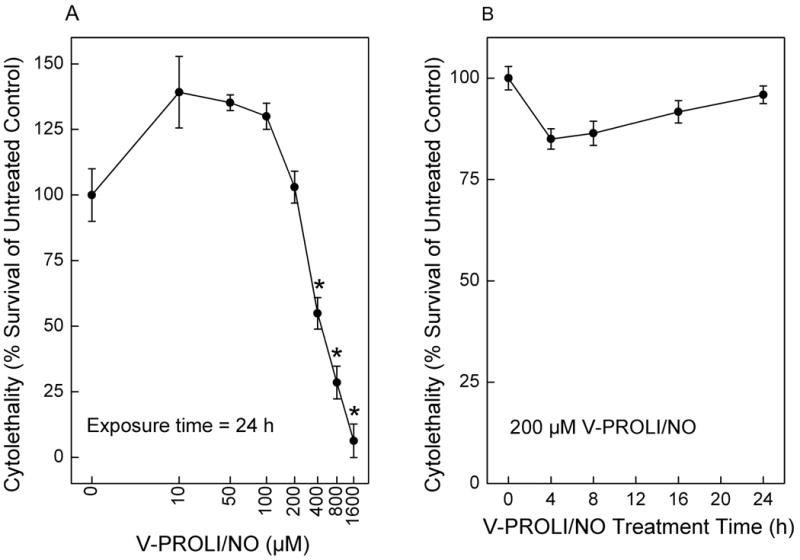
Concentration- and time-dependent effects of V-PROLI/NO treatment on cytolethality.
TRL 1215 rat liver cells were exposed to V-PROLI/NO for 24 h at the levels indicated for the concentration-response curve (A) or at 200 μM for 0 to 24 h for the time course response (B). Results are presented as the mean ± SEM of four separate determinations. Note log scale in (A). *, significantly different (p < 0.05) from untreated control.
3.2. Nitrite formation after V-PROLI/NO exposure and V-PROLI/NO-induced arsenic tolerance
To show that V-PROLI/NO was acted upon by rat liver cells to release NO, cells were exposed to V-PROLI/NO for 24 h at 200 μM for the time indicated and extracellular nitrite was measured by Griess assay as an index of NO production (Figure 3A). Cells clearly acted upon the prodrug to release NO, producing nitrite from V-PROLI/NO in a time-dependent manner. Addition of V-PROLI/NO to medium in the absence of cells did not cause generation of nitrite (not shown). Cells were pretreated with V-PROLI/NO for 24 h then incubated with various levels of arsenic (as sodium arsenite) for an additional 24 h and LC50 values for arsenic were determined (Figure 3B). The V-PROLI/NO pretreatment clearly reduced arsenic-induced cytolethality. The LC50 value for arsenic increased with V-PROLI/NO pretreatment over 2-fold at 100 μM and 3-fold at 200 μM of V-PROLI/NO compared to the control cells. The V-PROLI/NO pretreatment alone was not cytotoxic at the levels used (see Figure 2). Analysis revealed a highly significant correlation between increasing arsenic LC50 and the concentration of V-PROLI/NO (Figure 3C). Also, increasing nitrite production was highly correlated with increases in LC50 for arsenic (Figure 3D). These strong correlations suggest mechanistic significance indicating that when exposed to V-PROLI/NO, TRL 1215 cells metabolize V-PROLI/NO to release NO, which in turn protects against arsenic-induced cytolethality.
Fig. 3.
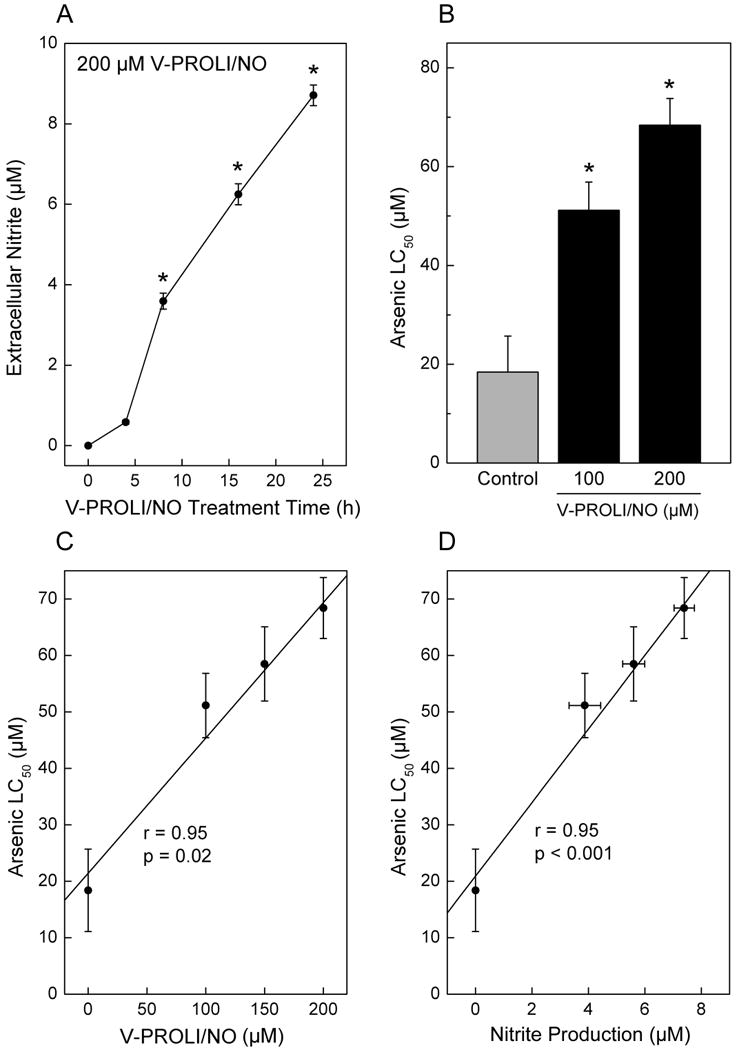
Time-dependent nitrite formation after V-PROLI/NO treatment and V-PROLI/NO–induced tolerance to arsenic-induced cytotoxicity.
Cells were exposed to V-PROLI/NO for 24 h at 200 μM for the times indicated and assessed for NO formation. Extracellular medium was collected and nitrite was measured by Griess assay as an indirect index of NO production (A). Cells were pretreated with V-PROLI/NO for 24 h or left untreated. Cells were then incubated with various levels of arsenic (as sodium arsenite) for an additional 24 h and LC50 values were from cytolethality curves (B). LC50 values were correlated with V-PROLI/NO concentrations (C) or nitrite production (D) using Pearson's r correlation. Results are presented as the mean ± SEM of four separate determinations. *, significantly different (p < 0.05) from untreated control.
3.3. Correlations between V-PROLI/NO concentrations, nitrite production, Cyp1a1 transcript levels and arsenic cytotoxicity
Cells were treated with various levels of V-PROLI/NO for 24 h followed by arsenic (as sodium arsenite) treatment for an additional 24 h, where upon Cyp1a1 transcript levels, nitrite production and LC50 values for arsenic were measured. Cyp1a1 transcript levels were increased by the highest concentration of V-PROLI/NO treatment alone (200 μM). Arsenic treatment alone also increased Cyp1a1 transcript level about 50% (Figure 4A). Pretreatment with V-PROLI/NO facilitated further concentration-related increases in Cyp1a1 transcript to a maximum of about 400%. In addition, Cyp1a1 transcript levels were clearly correlated with V-PROLI/NO treatment concentration (Figure 4B) and nitrite production (as an indication of NO production; Figure 4C). A strong correlation was observed between Cyp1a1 transcript levels and increasing LC50 for arsenic (Figure 4D). These strong correlations suggest that V-PROLI/NO is metabolized to release NO by Cyp1a1 in TRL 1215 cells which then protects against arsenic. Further, V-PROLI/NO is inducing Cyp1a1 expression in these cells. However, Cyp2e1 transcript levels did not show any increases after treatment with V-PROLI/NO or arsenic alone or sequentially (data not shown). Thus, it appears V-PROLI/NO is metabolized by and, at least in part, induces Cyp1a1. Indeed, when Cyp1a1 activity was blocked by α-naphthoflavone the protective effects of V-PROLI/NO against arsenic toxicity were abolished (Figure 5).
Fig. 4.
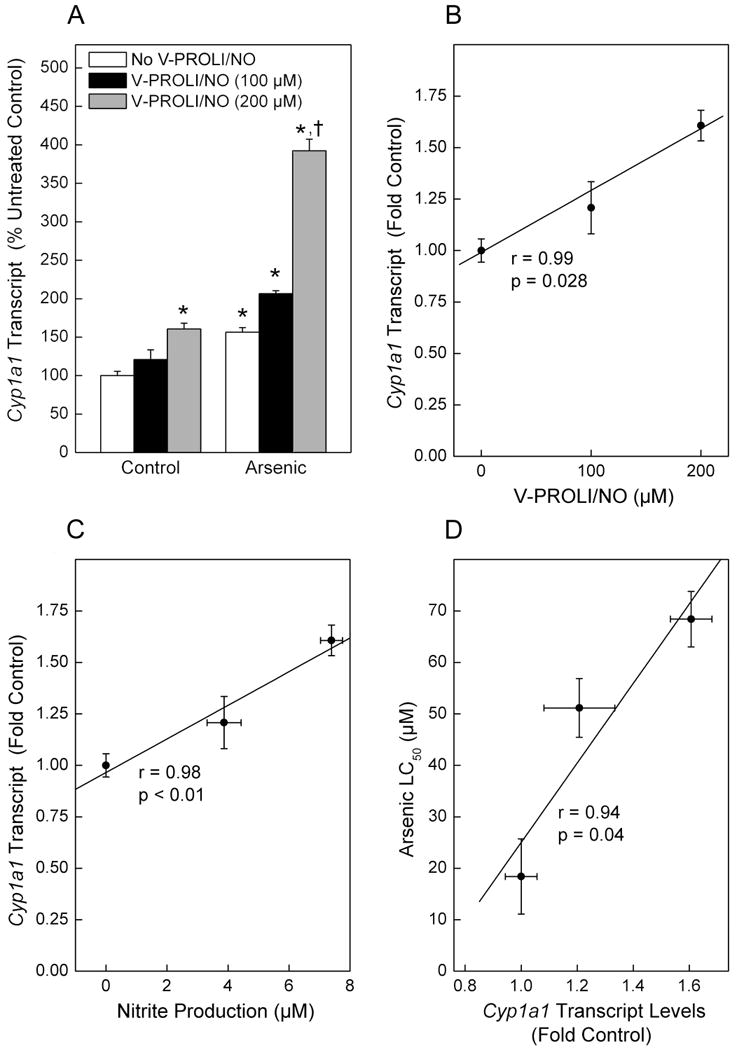
Effect of V-PROLI/NO and arsenic on Cyp1a1 transcript levels and correlations between V-PROLI/NO concentrations, nitrite production, Cyp1a1 transcript levels and arsenic cytotoxicity.
Cells were first treated with various levels of V-PROLI/NO for 24 h followed by arsenic treatment (70 μM as sodium arsenite; 24 h). Cyp1a1 transcript levels were then determined by real-time PCR (A). Nitrite produced after V-PROLI/NO treatment as an indication of NO production was measured. Cyp1a1 transcript levels were then correlated with V-PROLI/NO concentration (B) or nitrite production (C) or arsenic LC50 (D) using Pearson's r correlation. Data are expressed as a mean percent of untreated control (no V-PROLI/NO or arsenic) ± SEM of three separate determinations. *, significantly different (p < 0.05) from untreated (no arsenic and no V-PROLI/NO) control. †, significantly different (p < 0.05) from the arsenic-alone (no V-PROLI/NO) control. Cyp1a1 transcript levels were significantly correlated with V-PROLI/NO concentration, NO production or LC50.
Fig. 5.
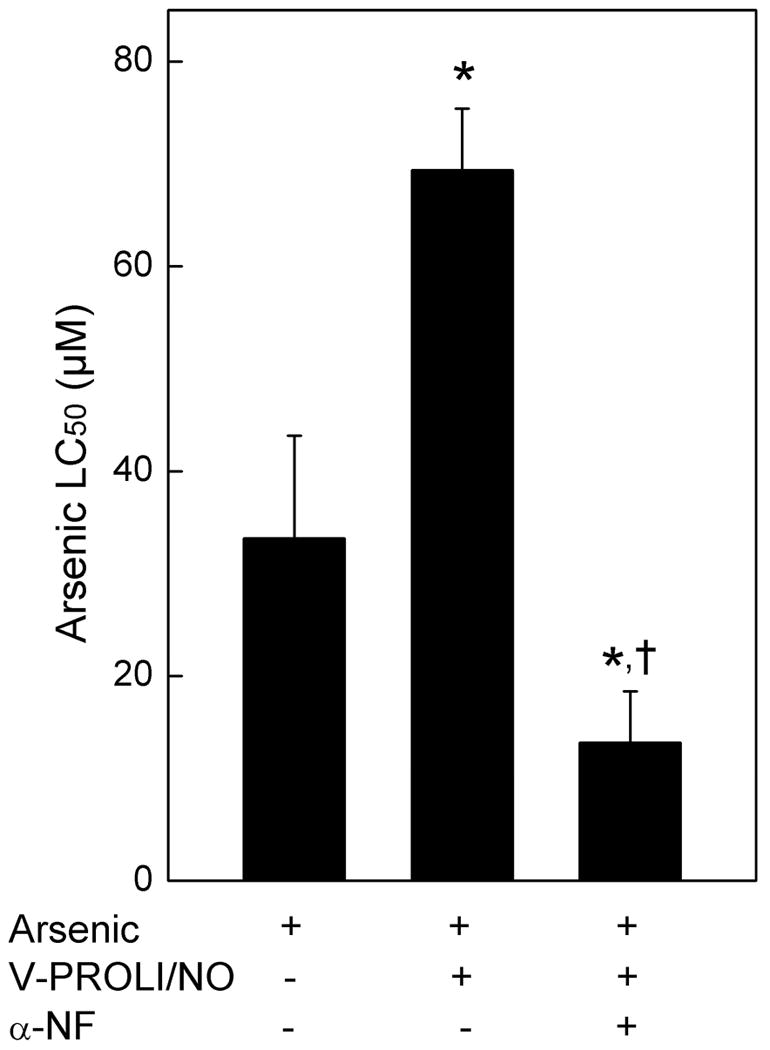
Effect of α-naphthoflavone (α-NF), a Cyp1a1 inhibitor, on protective effects of V-PROLI/NO against arsenic toxicity.
Cells were pretreated with α-NF (25μM; 1 h) then exposed to V-PROLI/NO (200 μM; 24h) or left untreated. Cells were then incubated with various levels of arsenic (as sodium arsenite) for an additional 24 h and LC50 values were calculated from cytolethality curves. Results are presented as the mean ± SEM of four separate determinations. *, significantly different (p < 0.05) from arsenic treatment. †, significantly different (p < 0.05) from V-PROLI/NO pretreatment and arsenic treatment.
3.4. Effect of V-PROLI/NO on Gst-π and HO-1 transcript levels and MT protein levels
Cells were treated with various levels of V-PROLI/NO for 24 h followed by arsenic (as sodium arsenite) treatment for an additional 24 h, after which Gst-π and HO-1 transcript levels and MT protein levels were measured (Figure 6). Arsenic-related metabolic stress response genes, Gst-π and HO-1 transcript levels were both markedly increased by V-PROLI/NO pretreatment in a fashion that was related to the level of arsenic exposure (Figure 6A and 6B). Arsenic alone modestly increased MT, while pretreatment with V-PROLI/NO facilitated additional increases in MT levels in an arsenic concentration-dependent manner after subsequent arsenic exposure (Figure 6C).
Fig. 6.
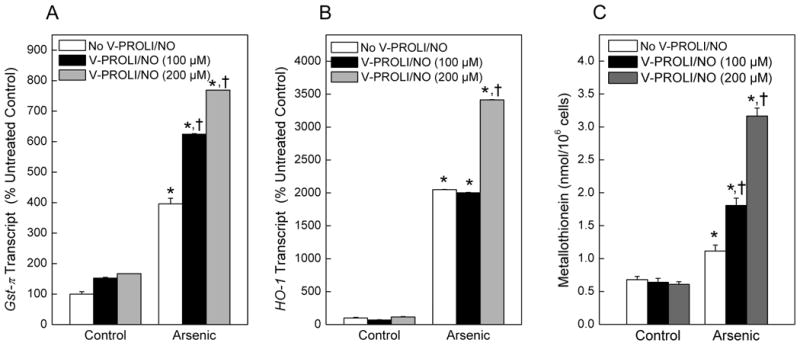
Effect of V-PROLI/NO pretreatment on Gst-π or HO-1 transcript levels and MT levels.
Cells were first treated with various levels of V-PROLI/NO for 24 h followed by arsenic treatment (70 μM as sodium arsenite; 24 h). Gst-π (A) and HO-1 (B) transcript levels were then determined by real-time PCR. MT levels were measured by the cadmium-hemoglobin assay (C). Data are expressed as a percent of control (no V-PROLI/NO or arsenic) ± SEM of three separate determinations. *, significantly different (p < 0.05) from untreated (no arsenic and no V-PROLI/NO) control. †, significantly different (p < 0.05) from the arsenic-alone control (no V-PROLI/NO).
3.5. Effect of V-PROLI/NO pretreatment on arsenic-induced apoptosis and apoptotic signaling
Cells were pretreated with V-PROLI/NO for 24 h then arsenic-induced apoptosis at the cellular level was measured (Figure 7A). V-PROLI/NO was able to block arsenic-induced apoptosis in a fashion related to V-PROLI/NO pretreatment concentration. The levels of phosphorylated JNK1/2 or total JNK1/2, key factors in apoptosis, were also determined (Figure 7B). V-PROLI/NO at 200 μM diminished the activation of JNK1/2 by phosphorylation induced by arsenic. This is likely an important factor in reducing apoptotic cell death. The membranes used to define phosphorylated forms were stripped and then reprobed with regular antibody against JNK1/2 which acts as loading control. As shown in Figure 7B, middle, no major differences were observed. Thus, the decrease in phosphorylated-JNK is probably a decrease in phosphorylation of the native protein in V-PROLI/NO treated cells rather than a reduction in total cellular JNK protein. The levels of both phosphorylated ERK1/2 and p38 did not show any significant differences after treatment with V-PROLI/NO or arsenic alone or sequentially (data not shown).
Fig. 7.
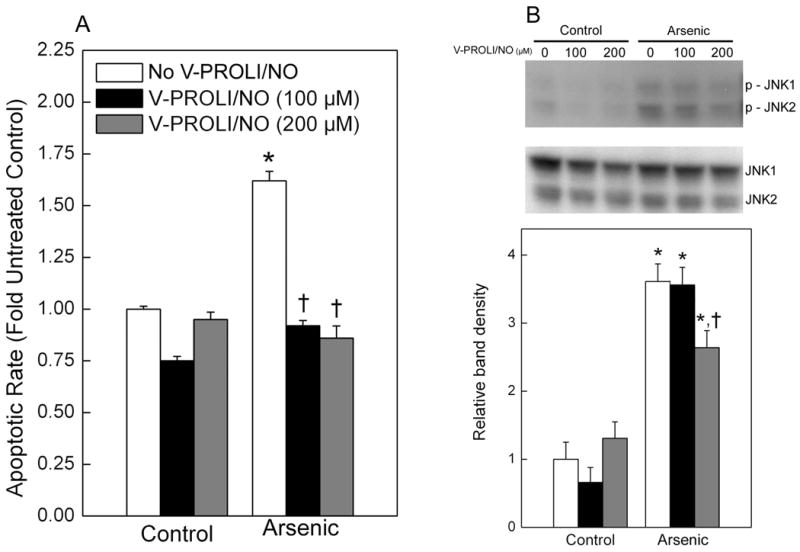
Effect of V-PROLI/NO pretreatment on arsenic-induced apoptosis and level of phosphorylated JNK1/2 induced by arsenic.
Cells were pretreated with V-PROLI/NO (24 h) then arsenic (70 μM as sodium arsenite; 24 h) and apoptosis was measured by ELISA (A). The levels of phosphorylated JNK1/2 were determined by Western blot analysis (B, Top). After development, the membranes were stripped and reprobed with regular antibodies against JNK1/2 (B, Middle). Blots represent a typical result of four independent experiments. The phosphorylated JNK1/2 protein immunoblots were analyzed by scanning densitometry (B, Bottom) and values were then standardized to untreated control as 1. Data are expressed as a percent of untreated control (no V-PROLI/NO or arsenic) ± SEM of four separate determinations. *, significantly different (p < 0.05) from untreated (no arsenic and no V-PROLI/NO) control. †, significantly different (p < 0.05) from the arsenic-alone control (no V-PROLI/NO).
V-PROLI/NO protects against arsenic toxicity in rat liver cells, reducing cytolethality, apoptosis and dysregulation of MAPKs, through generation of NO formed after metabolism by liver cell enzymes, possibly including Cyp1a1. CYP450 required for NO production from V-PROLI/NO treatment in the rat and human appears to differ. Rat and human liver cells act upon V-PROLI/NO via a CYP450-related mechanism to produce NO and subsequently reduce arsenic toxicity.
4. Discussion
Recently, arsenic has been re-introduced into cancer chemotherapy, showing stunning efficacy in the treatment of certain leukemias [1,5]. However, inorganic arsenic can have profound toxic effects even following short-term use. For instance, potentially fatal hepatic toxicity has been reported in a subset of patients receiving arsenic chemotherapy [5]. Individual variation in susceptibility to arsenic-induced toxicity clearly exists, possibly related to polymorphisms, arsenic methylation capacity [1] or perhaps ability to express MT [21]. Thus, development of adjuvant pharmacological agents that effectively and specifically limit arsenical hepatotoxicity could potentially increase arsenical chemotherapeutic efficacy by blocking or reducing toxic side effects. In this regard, the V-PROLI/NO was clearly able to mitigate arsenic toxicity at the cellular level in rat liver cells in the present study. Many NO donor prodrugs have been designed with pharmacological potential [9]. Indeed, our prior work indicates that O2-vinyl 1-(pyrrolidin-1-yl)diazen-1-ium-1,2-diolate (V-PYRRO/NO), a liver-selective NO-releasing agent metabolized by human CYP450 [26], mitigates the toxicity of various compounds in liver cells both in vivo and in vitro, including arsenic [27,28]. V-PROLI/NO is a relatively new NO donor prodrug whose CYP450-induced NO-donating metabolite PROLI/NO has vasodilatory and antithrombotic effects [29-31]. The present study showed that V-PROLI/NO pretreatment reduced arsenic-induced toxicity in rat liver cells in vitro, which would seem to either eliminate a direct vasodilator effect or perhaps augment such an effect in vivo. The results also indicate that V-PROLI/NO releases NO apparently, at least in part, via metabolism by Cyp1a1 in rat liver cells. Interestingly, metal exposure alters Cyp1a1 enzyme activity [11]. Cyp1a1 also plays an important role in detoxification of environmental carcinogens and cancer prevention [12]. We have previously shown V-PROLI/NO can prevent arsenic toxicity in human liver HepG2 cells in vitro where NO is apparently released through a CYP2E1-mediated metabolic mechanism [13]. Thus, it seems that there are species differences in precise CYP450 requirements for NO production from V-PROLI/NO in the rat and human. However, in both cases a CYP450 helps to prevent arsenic cytotoxicity by metabolizing the liver-selective NO donor V-PROLI/NO. Species variation in CYP450 could be due to a number of reasons and this species divergence may be helpful for extrapolating animal data to humans. Nonetheless, these data confirm the direct utility of V-PROLI/NO at the level of the liver cell to reduce the toxicity of an effective cancer chemotherapeutic with potentially limiting hepatoxicity.
V-PROLI/NO-induced tolerance to arsenic was clearly related to NO release, likely by the metabolic action of the liver cells on the NO prodrug. In the present work, V-PROLI/NO protected against the adverse effects of arsenic directly within TRL 1215 cells, including cytolethality likely due to reduced apoptosis from JNK pathway down-regulation. Thus, at least in vitro, V-PROLI/NO acts to reduce arsenic toxicity in a key cell site for limiting toxic side effects of arsenicals. In this regard, heme plays a vital role in regulating the formation of NO via NO synthases and is important in mediating oxidative metabolism of xenobiotics and drugs by CYP450 [32]. Heme oxygenase (HO) catalyzes degradation of heme [32]. HO-1 is well-known marker of arsenic-induced oxidative stress [33] and is typically thought to be involved with adaptation to the toxic metalloid. The expression of HO-1 was clearly enhanced by treatment of cells with V-PROLI/NO prior to arsenic in the present study. Similarly, GST-π is a member of GST super family of enzymes that catalyzes cellular reduced glutathione (GSH)-dependent detoxification of substrates [23]. GSH is involved in cellular protection against reactive oxygen species (ROS). Previously, we found that MRP1/ABCC1 transports inorganic arsenic as a tri-GSH conjugate, and GSH S-transferase P1-1 (GSTP1-1) may have a synergistic role in this process [34]. Thus, arsenic efflux by GSTP1-1 and ABCC1 reduce arsenic toxicity. The present study showed that although V-PROLI/NO alone had minimal impact on Gst-π and HO-1, it markedly facilitated arsenic-induced expression of these same adaptive genes when cells were subsequently exposed to the metalloid. This would provide cellular protective response of V-PROLI/NO against arsenic toxicity since arsenic can induce ROS when biomethylated [24] and these ROS are implicated in various pathologies including carcinogenesis, genotoxicity and cytoxicity [24,35].
The JNK signal transduction pathway when activated is linked to apoptosis [14,16]. Arsenic activates JNK in various cells and this helps lead to arsenic-induced apoptosis [15,36]. Cells that undergo arsenic-induced malignant transformation are often highly resistant to arsenic-induced apoptosis through perturbation of JNK1/2 activity [16]. V-PROLI/NO also protects human liver cells from arsenic-induced toxicity and apoptosis and this protection is apparently through generation of NO and the concurrent blockade of arsenic-activation of the apoptosis-related JNK pathway [13]. The present results indicate that V-PROLI/NO also generates NO within rat liver cells and down regulates levels of phosphorylated JNK1/2 induced by arsenic, an event probably linked to reduced arsenic-induced apoptosis. These observations are in accord with previous data indicating that NO negatively regulates c-Jun N-terminal kinase/stress-activated protein kinase [37] and, in particular, impacts JNK signaling [38]. Thus, the release of NO from V-PROLI/NO is related to a reduction in arsenic-induced apoptosis via perturbed signaling events.
Arsenic has been shown to induce MT both in vitro and in vivo [18,39), possibly through binding to MT [20]. MT–I/II double knock-out (MT-null) mice are more sensitive than wild-type mice to chronic arsenic-induced hepatotoxicity [40]. Humans that poorly express MT appear more sensitive to arsenic-induced skin lesions than persons with higher MT levels [21]. In this regard, although arsenic alone in the present study increased MT levels directly in rat liver cells, V-PROLI/NO pretreatment facilitated additional increases in MT with subsequent arsenic exposure. Thus, the protective effect of V-PROLI/NO might be, in part, a result of facilitation of MT expression which, in turn, allows greater sequestration of arsenic.
In conclusions, the present work shows that exposure of rat liver cells to the NO-releasing prodrug, V-PROLI/NO, protects against the adverse effects of arsenic including cytolethality, apoptosis and JNK pathway activation, apparently by generation of NO possibly via Cyp1a1. V-PROLI/NO pretreatment also enhanced arsenic adaptation as seen with increases in MT, Gst-π and HO-1. Since hepatotoxicity is a limiting side effect of arsenical chemotherapy, the potential use of V-PROLI/NO as an adjuvant in chemotherapy should be explored in vivo.
Acknowledgments
The authors thank Drs. Erik Tokar and Yang Sun for critical review of this manuscript. This research was supported in part by the National Toxicology Program, National Institute of Environmental Health Sciences (NIEHS) and by the Intramural Research Program of the National Institutes of Health (NIH), National Cancer Institute, Center for Cancer Research. Additional support came from contract HHSN261200800001E with SAIC Frederick, Inc. This article may be the work product of an employee or a group of employees of the NIEHS, NIH, however, the statements contained herein do not necessarily represent the statements, opinions or conclusions of the NIEHS, NIH or the United States Government. The content of this publication does not necessarily reflect the views or the policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Abbreviations
- Cyp1a1
cytochrome P450 1a1
- CYP2E1
cytochrome P450 2E1
- CYP450
cytochrome P450
- ERKs
extracellular signal-regulated kinases
- GSH
cellular glutathione
- GST
Glutathione S-transferase
- GSTP1-1
GSH S-transferase P1-1
- HO
heme oxygenase
- HO-1
heme oxygenase-1
- JNKs/SAPKs
c-Jun NH2-terminal kinases/stress-activated protein kinases
- MAPKs
Mitogen-activated protein kinases
- MT
Metallothionein
- NO
nitric oxide
- ROS
reactive oxygen species
- V-PROLI/NO
O2-vinyl 1-[2-(carboxylato)pyrrolidin-1-yl]diazen-1-ium-1,2-diolate
- V-PYRRO/NO
O2-vinyl 1-(pyrrolidin-1-yl)diazen-1-ium-1,2-diolate
Footnotes
Conflict of interest statement: None.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hede K. Chinese folk treatment reveals power of arsenic to treat cancer, new studies under way. J Natl Cancer Inst. 2007;99:667–668. doi: 10.1093/jnci/djk191. [DOI] [PubMed] [Google Scholar]
- 2.McNeely SC, Belshoff AC, Taylor BF, Fan TW, McCabe MJ, Jr, Pinhas AR, States JC. Sensitivity to sodium arsenite in human melanoma cells depends upon susceptibility to arsenite-induced mitotic arrest. Toxicol Appl Pharmacol. 2008;229:252–261. doi: 10.1016/j.taap.2008.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Shen ZX, Chen GQ, Ni JH, Li XS, Xiong SM, Qiu QY, Zhu J, Tang W, Sun GL, Yang KQ, Chen Y, Zhou L, Fang ZW, Wang YT, Ma J, Zhang P, Zhang TD, Chen SJ, Chen Z, Wang ZY. Use of arsenic trioxide (As2O3) in the treatment of acute promyelocytic leukemia (APL). II. Clinical efficacy and pharmacokinetics in relapsed patients. Blood. 1997;89:3354–3360. [PubMed] [Google Scholar]
- 4.Wang ZY, Chen Z. Acute promyelocytic leukemia: from highly fatal to highly curable. Blood. 2008;111:2505–2515. doi: 10.1182/blood-2007-07-102798. [DOI] [PubMed] [Google Scholar]
- 5.Mathews V, Desire S, George B, Lakshmi KM, Rao JG, Viswabandya A, Bajel A, Srivastava VM, Srivastava A, Chandy M. Hepatotoxicity profile of single agent arsenic trioxide in the treatment of newly diagnosed acute promyelocytic leukemia, its impact on clinical outcome and the effect of genetic polymorphisms on the incidence of hepatotoxicity. Leukemia. 2006;20:881–883. doi: 10.1038/sj.leu.2404165. [DOI] [PubMed] [Google Scholar]
- 6.Liu J, Waalkes MP. Liver is a target of arsenic carcinogenesis. Toxicol Sci. 2008;105:24–32. doi: 10.1093/toxsci/kfn120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kozoni V, Rosenberg T, Rigas B. Development of novel agents based on nitric oxide for the control of colon cancer. Acta Pharmacol Sin. 2007;28:1429–1433. doi: 10.1111/j.1745-7254.2007.00696.x. [DOI] [PubMed] [Google Scholar]
- 8.Rigas B. Novel agents for cancer prevention based on nitric oxide. Biochem Soc Trans. 2007;35:1364–1368. doi: 10.1042/BST0351364. [DOI] [PubMed] [Google Scholar]
- 9.Keefer LK. Progress toward clinical application of the nitric oxide-releasing diazeniumdiolates. Annu Rev Pharmacol Toxicol. 2003;43:585–607. doi: 10.1146/annurev.pharmtox.43.100901.135831. [DOI] [PubMed] [Google Scholar]
- 10.Chakrapani H, Showalter BM, Kong L, Keefer LK, Saavedra JE. V-PROLI/NO, a prodrug of the nitric oxide donor, PROLI/NO. Org Lett. 2007;9:3409–3412. doi: 10.1021/ol701419a. [DOI] [PubMed] [Google Scholar]
- 11.Anwar-Mohamed A, Elbekai RH, El-Kadi AO. Regulation of CYP1A1 by heavy metals and consequences for drug metabolism. Expert Opin Drug Metab Toxicol. 2009;5:501–521. doi: 10.1517/17425250902918302. [DOI] [PubMed] [Google Scholar]
- 12.Androutsopoulos VP, Tsatsakis AM, Spandidos DA. Cytochrome P450 CYP1A1: wider roles in cancer progression and prevention. BMC Cancer. 2009;9:187–204. doi: 10.1186/1471-2407-9-187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Qu W, Liu J, Dill AL, Saavedra JE, Keefer LK, Waalkes MP. V-PROLI/NO, a nitric oxide donor prodrug, protects liver cells from arsenic-induced toxicity. Cancer Sci. 2009;100:382–388. doi: 10.1111/j.1349-7006.2008.01050.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME. Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 1995;270:1326–1331. doi: 10.1126/science.270.5240.1326. [DOI] [PubMed] [Google Scholar]
- 15.Samet JM, Graves LM, Quay J, Dailey LA, Devlin RB, Ghio AJ, Wu W, Bromberg PA, Reed W. Activation of MAPKs in human bronchial epithelial cells exposed to metals. Am J Physiol. 1998;275:L551–558. doi: 10.1152/ajplung.1998.275.3.L551. [DOI] [PubMed] [Google Scholar]
- 16.Qu W, Bortner CD, Sakurai T, Hobson MJ, Waalkes MP. Acquisition of apoptotic resistance in arsenic-induced malignant transformation: role of the JNK signal transduction pathway. Carcinogenesis. 2002;23:151–159. doi: 10.1093/carcin/23.1.151. [DOI] [PubMed] [Google Scholar]
- 17.Klaassen CD, Liu J, Choudhuri S. Metallothionein: An intracellular protein to protect against cadmium toxicity. Ann Rev Pharmacol Toxicol. 1999;39:267–294. doi: 10.1146/annurev.pharmtox.39.1.267. [DOI] [PubMed] [Google Scholar]
- 18.Kreppel H, Bauman JW, Liu J, McKim JM, Jr, Klaassen CD. Induction of metallothionein by arsenicals in mice. Fundam Appl Toxicol. 1993;20:184–189. doi: 10.1006/faat.1993.1025. [DOI] [PubMed] [Google Scholar]
- 19.Jiang G, Gong Z, Li XF, Cullen WR, Le XC. Interaction of trivalent arsenicals with metallothionein. Chem Res Toxicol. 2003;16:873–880. doi: 10.1021/tx034053g. [DOI] [PubMed] [Google Scholar]
- 20.Ngu TT, Stillman MJ. Arsenic binding to human metallothionein. J Am Chem Soc. 2006;128:12473–12483. doi: 10.1021/ja062914c. [DOI] [PubMed] [Google Scholar]
- 21.Liu J, Cheng ML, Yang Q, Shan K, Shen J, Zhou Y, Zhang X, Dill A, Waalkes MP. Blood metallothionein transcript as a biomarker for metal sensitivity: Low blood metallothionein transcripts in arsenicosis patients from Guizhou, China, Environ. Health Perspect. 2007;115:1101–1106. doi: 10.1289/ehp.10035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Katakai K, Liu J, Nakajima K, Keefer LK, Waalkes MP. Nitric oxide induces metallothionein (MT) gene expression apparently by displacing zinc bound to MT. Toxicol Lett. 2001;119:103–108. doi: 10.1016/s0378-4274(00)00301-5. [DOI] [PubMed] [Google Scholar]
- 23.Eaton DL, Bammler TK. Concise review of the glutathione S-transferases and their significance to toxicology. Toxicol Sci. 1999;49:156–164. doi: 10.1093/toxsci/49.2.156. [DOI] [PubMed] [Google Scholar]
- 24.Kojima C, Ramirez DC, Tokar EJ, Himeno S, Drobná Z, Stýblo M, Mason RP, Waalkes MP. Requirement of arsenic biomethylation for oxidative DNA damage. J Natl Cancer Inst. 2009;101:1670–1681. doi: 10.1093/jnci/djp414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eaton DL, Toal BF. Evaluation of the Cd/hemoglobin affinity assay for the rapid determination of metallothionein in biological tissues. Toxicol Appl Pharmacol. 1982;66:134–142. doi: 10.1016/0041-008x(82)90068-0. [DOI] [PubMed] [Google Scholar]
- 26.Inami K, Nims RW, Srinivasan A, Michael L, Citro ML, Saavedra JE, Arthur I, Cederbaum AI, Keefer LK. Metabolism of a liver-selective nitric oxide-releasing agent, V-PYRRO/NO, by human microsomal cytochromes P450. Nitric Oxide. 2006;14:309–315. doi: 10.1016/j.niox.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 27.Saavedra JE, Billiar TR, Williams DL, Kim YM, Watkins SC, Keefer LK. Targeting nitric oxide (NO) delivery in vivo. Design of a liver-selective NO donor prodrug that blocks tumor necrosis factor-alpha-induced apoptosis and toxicity in the liver. J Med Chem. 1997;40:1947–1954. doi: 10.1021/jm9701031. [DOI] [PubMed] [Google Scholar]
- 28.Qu W, Liu J, Fuquay R, Saavedra JE, Keefer LK, Waalkes MP. The nitric oxide prodrug, V-PYRRO/NO, mitigates arsenic-induced liver cell toxicity and apoptosis. Cancer Lett. 2007;256:238–45. doi: 10.1016/j.canlet.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Waterhouse DJ, Saavedra JE, Davies KM, Citro ML, Xu X, Powell DA, Grimes GJ, Potti GK, Keefer LK. Injectable formulation of disodium 1-[2-(carboxylato)pyrrolidin-1-yl]diazen-1-ium-1,2-diolate (PROLI/NO), an ultrafast nitric oxide donor prodrug. J Pharm Sci. 2006;95:108–115. doi: 10.1002/jps.20486. [DOI] [PubMed] [Google Scholar]
- 30.Saavedra JE, Southan GJ, Davies KM, Lundell A, Markou C, Hanson SR, Adrie C, Hurford WE, Zapol WM, Keefer LK. Localizing antithrombotic and vasodilatory activity with a novel, ultrafast nitric oxide donor. J Med Chem. 1996;39:4361–4365. doi: 10.1021/jm960616s. [DOI] [PubMed] [Google Scholar]
- 31.Adrie C, Hirani WM, Holzmann A, Keefer LK, Zapol WM, Hurford WE. Selective pulmonary vasodilation by intravenous infusion of an ultrashort half-life nucleophile/nitric oxide adduct. Anesthesiol. 1998;88:190–195. doi: 10.1097/00000542-199801000-00027. [DOI] [PubMed] [Google Scholar]
- 32.Zhu Y, Silverman RB. Revisiting heme mechanisms. A perspective on the mechanisms of nitric oxide synthase (NOS), Heme oxygenase (HO), and cytochrome P450s (CYP450s) Biochem. 2008;47:2231–2243. doi: 10.1021/bi7023817. [DOI] [PubMed] [Google Scholar]
- 33.Meng D, Wang X, Chang Q, Hitron A, Zhang Z, Xu M, Chen G, Luo J, Jiang B, Fang J, Shi X. Arsenic promotes angiogenesis in vitro via a heme oxygenase-1-dependent mechanism. Toxicol Appl Pharmacol. 2010;244:291–299. doi: 10.1016/j.taap.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 34.Leslie EM, Haimeur A, Waalkes MP. Arsenic transport by the human multidrug resistance protein 1 (MRP1/ABCC1). Evidence that a tri-glutathione conjugate is required. J Biol Chem. 2004;279:32700–32708. doi: 10.1074/jbc.M404912200. [DOI] [PubMed] [Google Scholar]
- 35.Qu W, Kasprzak KS, Kadiiska M, Liu J, Chen H, Maciag A, Mason RP, Waalkes MP. Mechanisms of arsenic-induced cross-tolerance to nickel cytotoxicity, genotoxicity, and apoptosis in rat liver epithelial cells. Toxicol Sci. 2001;63:189–195. doi: 10.1093/toxsci/63.2.189. [DOI] [PubMed] [Google Scholar]
- 36.Huang C, Ma WY, Li J, Dong Z. Arsenic induces apoptosis through a c-Jun NH2-terminal kinase-dependent, p53-independent pathway. Cancer Res. 1999;59:3053–3058. [PubMed] [Google Scholar]
- 37.Park HS, Huh SH, Kim MS, Lee SH, Choi EJ. Nitric oxide negatively regulates c-Jun N-terminal kinase/stress-activated protein kinase by means of S-nitrosylation. Proc Natl Acad Sci USA. 2000;97:14382–14387. doi: 10.1073/pnas.97.26.14382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lo YY, Wong JM, Cruz TF. Reactive oxygen species mediate cytokine activation of c-Jun NH2-terminal kinases. J Biol Chem. 1996;271:15703–15707. doi: 10.1074/jbc.271.26.15703. [DOI] [PubMed] [Google Scholar]
- 39.Falnoga I, Stibilj E, Tušek-Žnidarič M, Šlejkovec Z, Mazej D, Jaćimović R, Ščančar J. Effect of arsenic trioxide on metallothionein and its conversion to different arsenic metabolites in hen liver. Biol Trace Elem Res. 2001;78:241–254. doi: 10.1385/bter:78:1-3:241. [DOI] [PubMed] [Google Scholar]
- 40.Liu J, Liu Y, Goyer RA, Achanzar W, Waalkes MP. Metallothionein-I/II null mice are more sensitive than wild-type mice to the hepatotoxic and nephrotoxic effects of chronic oral or injected inorganic arsenicals. Toxicol Sci. 2000;55:460–467. doi: 10.1093/toxsci/55.2.460. [DOI] [PubMed] [Google Scholar]