

Abstract
Protein function is intimately coupled to protein localization. Although some proteins are restricted to a specific location or subcellular compartment, many proteins are present as a freely diffusing population in free exchange with a sub-population that is tightly associated with a particular subcellular domain or structure. In situ subcellular fractionation allows the visualization of protein compartmentalization and can also reveal protein sub-populations that localize to specific structures. For example, removal of soluble cytoplasmic proteins and loosely held nuclear proteins can reveal the stable association of some transcription factors with chromatin. Subsequent digestion of DNA can in some cases reveal association with the network of proteins and RNAs that is collectively termed the nuclear scaffold or nuclear matrix.
Here we describe the steps required during the in situ fractionation of adherent and non-adherent mammalian cells on microscope coverslips. Protein visualization can be achieved using specific antibodies or fluorescent fusion proteins and fluorescence microscopy. Antibodies and/or fluorescent dyes that act as markers for specific compartments or structures allow protein localization to be mapped in detail. In situ fractionation can also be combined with western blotting to compare the amounts of protein present in each fraction. This simple biochemical approach can reveal associations that would otherwise remain undetected.
Protocol
I. Preparation for fractionation
This section describes the preparation of poly-L-Lysine coated microscope coverslips and the attachment of cells prior to fractionation. If required the cells can be transiently transfected with protein expression vectors either before or after attachment.
A. Preparation of poly-L-Lysine coated coverslips
Prepare a solution of 1mg/ml poly-L-Lysine in distilled water.
Coat clean coverslips with poly-L-Lysine by incubating them in the solution for at least 1 hour on a rocking platform at 22°C.
Wash the coated coverslips with sterile distilled water twice and follow with a single wash with 96% ethanol.
Air dry the coated coverslips on a piece of filter paper and keep them in a dry container for future use (once dry they can be stacked).
B. Attaching cells to coverslips
Non-adherent cells (here we use K562 cells)
Place a poly-L-Lysine coated coverslip in a well in a 6-well plate with the coated surface facing up.
Seed K562 cells at a density of 7x105 cells/well in Dulbecco's Modified Eagles Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), and PS (penicillin 100units/ml, streptomycin 100mg/ml). In the case of K562 cells transient transfection can be performed using electroporation (0.4cm electroporation cuvettes with 1x107 cells in 200μl of media at 250V / 975μF) before seeding the cells.
Incubate the cells for 24 hours at 37°C in 5% CO2.
Pour off the medium and wash the cells twice with ice cold phosphate buffered saline (PBS).
Follow with the subcellular fractionation protocol.
Adherent cells (here we use Cos-7 cells)
Place an uncoated coverslip in a well in a 6-well plate.
Wash twice in PBS prewarmed at 37°C.
Detach the cells by digesting with trypsin (0.03% EDTA, 0.25% trypsin) in PBS at 37°C for 2 to 5 minutes. It is important not to over-digest the cells and to stop the digestion as soon as predominantly individual floating cells are present.
Stop the digestion by seeding the cells at a density of 3x105 cells/well in a 6-well plate in DMEM supplemented with 10% FBS and PSG (penicillin 100 units/ml, streptomycin 100ug/ml and L-glutamine 292ug/ml), on normal coverslips. Adherent cells generally attach well onto normal microscope coverslips, however, poly-L-Lysine coated coverslips can be used if required.
Incubate the cells for 24 hours at 37°C and 5% CO2. In the case of Cos-7 cells transient transfection can be performed using Fugene 6 (Roche) if required and the cells left to recover for a further 24 hours at 37°C and 5% CO2.
Pour off the medium and wash the cells twice with ice cold PBS.
Follow with the subcellular fractionation protocol.
II. Subcellular fractionation
In situ fraction is performed according to the schematic shown in Figure 1 and is based on a method described previously5 with modifications described previously7 and in the detailed protocol below. It is recommended to transfer the coverslips to a fresh 6-well plate following each fractionation step removing the coverslip before removing the liquid. Although only four coverslips are required for imaging, additional coverslips are required to produce whole cell extract and nuclear matrix fractions for western blotting if this is to be performed in parallel.
Prepare and filter cytoskeleton buffer (CSK): 10mM PIPES, 300mM Sucrose, 100mM NaCl, 3mM MgCl2, 1mM EGTA. CSK buffer should be freshly prepared on the day of fractionation.
If required the cells from one coverslip can be used to prepare whole cell extract for western blotting by gently placing 200ul TES buffer (1% SDS, 2mM EDTA, 20mM Tris-HCl pH 7.4) directly onto the coverslip and incubating for 1 or 2 minutes at 22°C. The whole cell extract can then be removed and the proteins precipitated by adding 250ul of 1M (NH4)2SO4 and incubating on ice until the other western samples are ready.
Wash the remaining coverslips with 1ml ice cold PBS twice at 22°C by tilting the 6 well-plate two or three times.
Remove the coverslip representing whole cells and transfer to a fresh 6-well plate. Follow with the cell fixation and immuno-fluorescence protocol.
Gently remove the PBS from the remaining wells.
Extract the cytoplasmic and loosely held nuclear proteins by gently placing 200ul CSK buffer + 0.1% (V/V) Triton X-100 directly onto each remaining coverslip and incubating on ice for 1 minute.
Remove the cytoplasmic and loosely held nuclear extract and precipitate as described in step 2.
Wash the coverslips with 1ml ice cold PBS twice at 22°C by tilting the 6 well-plate.
Remove the coverslip representing tightly held nuclear material and follow with the cell fixation and immuno-fluorescence protocol.
Gently remove the PBS from the remaining wells then extract the tightly held proteins by gently placing 200ul CSK buffer + 0.5% (V/V) Triton X-100 directly onto each remaining coverslip and incubating on ice for 20 minutes.
Remove the tightly held extract and precipitate as described in step 2.
Wash the coverslips with 1ml ice cold PBS twice at 22°C by tilting the 6 well-plate.
Remove the coverslip representing chromatin fraction and follow with the cell fixation and immuno-fluorescence protocol.
Gently remove the PBS from the remaining wells then place 200ul CSK buffer + 100ug/ml of DNase I onto the remaining coverslips and incubate for 30 minutes at 37°C.
Remove the chromatin extract and precipitate as described in step 2.
Wash the coverslips with ice cold PBS twice at 22°C by tilting the 6 well-plate.
Remove the coverslip representing the nuclear matrix fraction and follow with the cell fixation and immuno-fluorescence protocol.
If required the matrix proteins can be removed from an additional matrix coverslip for western blotting using 200ul TES buffer as described in step 2.
Samples for western blot should be centrifuged at 13000 rpm in a bench top microcentrifuge at 4°C and the pellets resuspended in 40ul SDS loading buffer (62.5mM Tris-HCl pH 6.8, 2% SDS (w/v), 50 mM DTT, 10% glycerol, 0.01% bromophenol blue(w/v)) before analysis by SDS-PAGE.
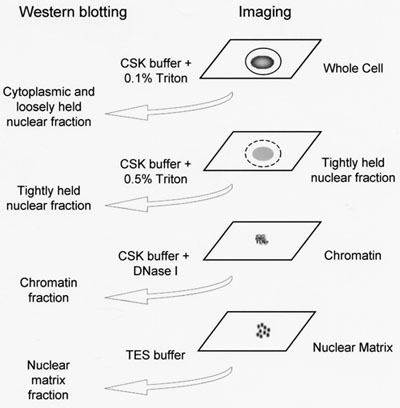 Figure 1. A schematic representation of in situ subcellular fractionation. Cytoskeleton buffer (CSK) consists of 10mM PIPES, 300mM Sucrose, 100mM NaCl, 3mM MgCl2, 1mM EGTA. TES buffer consists of 1% SDS, 2mM EDTA, 20mM Tris-HCl pH 7.4.
Figure 1. A schematic representation of in situ subcellular fractionation. Cytoskeleton buffer (CSK) consists of 10mM PIPES, 300mM Sucrose, 100mM NaCl, 3mM MgCl2, 1mM EGTA. TES buffer consists of 1% SDS, 2mM EDTA, 20mM Tris-HCl pH 7.4.
III. Cell fixation and immuno-fluorescence
Gently add 1ml of 4% formaldehyde/PBS to each coverslip and incubate at 4°C for 30 minutes.
Wash each coverslip gently 2-3 times with 1ml of PBS at 22°C.
Add 400ul of 0.1% Triton X-100/PBS to each coverslip and incubate at 22°C for 10 minutes.
Wash each coverslip gently 2-3 times with 1ml of PBS at 22°C.
Add 400ul of 3% Bovine Serum Albumin (BSA)/PBS and incubate at 22°C for 20 minutes to reduce potential background staining.
Wash each coverslip gently 2-3 times with 1ml of PBS at 22°C.
Place 100ul of the primary antibody in PBS directly onto the coverslip and incubate at 22°C for 1 hour. Primary and secondary antibodies should be diluted in PBS to the required concentration. This may need to be optimized for each antibody used.
Wash each coverslip gently 2-3 times with 1ml of PBS at 22°C.
Place 200ul of secondary antibody in PBS and incubate at 22°C away from light for 1 hour.
Wash each coverslip gently 2-3 times with 1ml of PBS at 22°C.
Mount each coverslip (cells down) onto a glass microscope slide using Vectashield mounting medium containing DAPI (4',6-diamidino-2-phenylindole hydrochloride).
Wipe excess mounting medium from around the coverslip using a paper towel and incubate the slide at 22°C for at least 30 minutes away from light.
Fix the coverslip to the glass slide using clear acrylic nail polish.
Keep the slides away from light before visualization by fluorescence microscopy.
IV. Representative results
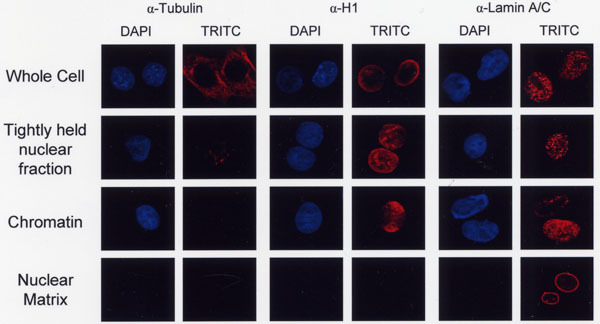 Figure 2. Subcellular fractionation of non-transfected COS-7 cells. Fractionated Cos-7 cells were fixed with formaldehyde and immunostained using α-Tubulin, α-Histone H1 or α-Lamin A/C antibodies conjugated to TRITC followed by treatment with mounting medium containing DAPI. Images were acquired with 10x ocular lens and 63x objective lens using a Leica confocal microscope. Cell nuclei were visualized using a DAPI filter set while in the same field cells stained with marker antibodies were visualized using a TRITC filter set.
Figure 2. Subcellular fractionation of non-transfected COS-7 cells. Fractionated Cos-7 cells were fixed with formaldehyde and immunostained using α-Tubulin, α-Histone H1 or α-Lamin A/C antibodies conjugated to TRITC followed by treatment with mounting medium containing DAPI. Images were acquired with 10x ocular lens and 63x objective lens using a Leica confocal microscope. Cell nuclei were visualized using a DAPI filter set while in the same field cells stained with marker antibodies were visualized using a TRITC filter set.
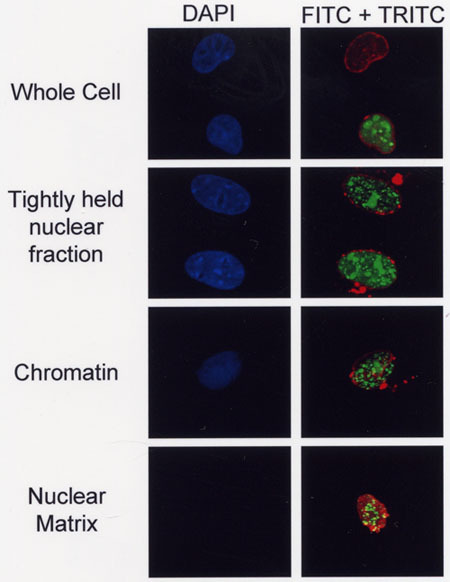 Figure 3. A GFP-6E2 fusion protein is associated with the nuclear matrix. Subcellular fractionation of Cos-7 cells expressing a fusion protein consisting of green fluorescent protein (GFP) fused to the human papillomavirus (HPV) type 6 E2 protein (GFP-6E2). Cell nuclei were visualized via DAPI staining while localization of GFP-6E2 in the transfected cells was directly visualized via GFP fluorescence. Lamin A/C was visualized using α-Lamin A/C antibodies conjugated to TRITC and a TRITC filter set. The merge images reveal that some of the GFP-6E2 protein is associated with the nuclear matrix (lower right panel). Images were obtained as described in Figure 2.
Figure 3. A GFP-6E2 fusion protein is associated with the nuclear matrix. Subcellular fractionation of Cos-7 cells expressing a fusion protein consisting of green fluorescent protein (GFP) fused to the human papillomavirus (HPV) type 6 E2 protein (GFP-6E2). Cell nuclei were visualized via DAPI staining while localization of GFP-6E2 in the transfected cells was directly visualized via GFP fluorescence. Lamin A/C was visualized using α-Lamin A/C antibodies conjugated to TRITC and a TRITC filter set. The merge images reveal that some of the GFP-6E2 protein is associated with the nuclear matrix (lower right panel). Images were obtained as described in Figure 2.
Discussion
Common problems and suggestions:
All or most of the cells are lost during washing. During washing steps liquids must be added slowly to the side of the 6-well plate avoiding the coverslip. Similarly liquids should be removed by carefully tilting the plate and slowly pipetting off excess fluid. Cell adhesion can be increased using poly-L-Lysine coated coverslips.
Genomic DNA is not completely digested. For some cell types the DNAse I digestion step may need to be extended and/or the DNAse I concentration increased in order to achieve complete removal of the genomic DNA.
Fixation and fractionation artifacts. It is important to note that some proteins can relocalize during fixation or fractionation 6. Proteins released from the nucleus following disruption of the nuclear membrane for example can bind to sites that would otherwise be located in well separated compartments. These effects can be minimized by comparing results obtained using different fixation methods for example using paraformaldehyde in place of formaldehyde. Live cell imaging can also be helpful for whole cells at least 3.
This technique is well suited to the detection of GFP fusion proteins. However, it is important to confirm that the fusion protein behaves in a similar manner to the untagged protein. It is also important to confirm by titration that the proteins are expressed at comparable levels since protein over-expression can alter dramatically sub-cellular localization.
Applications of the technique:
This underutilized technique allows protein localization to be determined with reference to well-characterized markers of different subcellular domains or structures and has applications across many areas of cell biology. For example, many transcription factors display a complex distribution with localization to the cytoplasm, loosely held nucleoplasm, chromatin and/or nuclear matrix 1,2,4. Localization in each of these domains can influence protein function as well as protein turnover and post-translational modifications.
Protein co-localization to specific domains such as the nuclear matrix can provide insights into protein function. Protein re-localization in response to specific signals and/or the expression of partner proteins can also shed light on protein function 1,4.
Disclosures
No conflicts of interest declared.
Acknowledgments
Anyaporn Sawasdichai and Nazefah Abdul Hamid are grateful to the Royal Thai Government and the Government of Malaysia respectively for Ph.D. Scholarships. This work was funded by a Wellcome Trust project grant awarded to PSJ and KG. We are also grateful to the University of Bristol Wolfson Bioimaging Facility.
References
- Donaldson MM, Boner W, Morgan IM. TopBP1 regulates human papillomavirus type 16 E2 interaction with chromatin. J Virol. 2007;81:4338–4342. doi: 10.1128/JVI.02353-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javed A, Guo B, Hiebert S, Choi JY, Green J, Zhao SC, Osborne MA, Stifani S, Stein JL, Lian JB, van Wijnen AJ, Stein GS. Groucho/TLE/R-esp proteins associate with the nuclear matrix and repress RUNX (CBF(alpha)/AML/PEBP2(alpha)) dependent activation of tissue-specific gene transcription. J Cell Sci. 2000;113:2221–2231. doi: 10.1242/jcs.113.12.2221. [DOI] [PubMed] [Google Scholar]
- Kowalczyk AM, Roeder GE, Green K, Stephens DJ, Gaston K. Measuring the induction or inhibition of apoptosis by HPV proteins. Methods Mol. Med. 2005;119:419–432. doi: 10.1385/1-59259-982-6:419. [DOI] [PubMed] [Google Scholar]
- McLarren KW, Theriault FM, Stifani S. Association with the nuclear matrix and interaction with Groucho and RUNX proteins regulate the transcription repression activity of the basic helix loop helix factor Hes1. J Biol. Chem. 2001;276:1578–1584. doi: 10.1074/jbc.M007629200. [DOI] [PubMed] [Google Scholar]
- McNeil S, Guo B, Stein JL, Lian JB, Bushmeyer S, Seto E, Atchison ML, Penman S, van Wijnen AJ, Stein GS. Targeting of the YY1 transcription factor to the nucleolus and the nuclear matrix in situ: the C-terminus is a principal determinant for nuclear trafficking. J Cell Biochem. 1998;68:500–510. [PubMed] [Google Scholar]
- Melan MA, Sluder G. Redistribution and differential extraction of soluble proteins in permeabilized cultured cells. Implications for immunofluorescence microscopy. J Cell Sci. 1992;101:731–743. doi: 10.1242/jcs.101.4.731. [DOI] [PubMed] [Google Scholar]
- Zou N, Lin BY, Duan F, Lee KY, Jin G, Guan R, Yao G, Lefkowitz EJ, Broker TR, Chow LT. The hinge of the human papillomavirus type 11 E2 protein contains major determinants for nuclear localization and nuclear matrix association. J. Virol. 2000;74:3761–3770. doi: 10.1128/jvi.74.8.3761-3770.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]