

Abstract
A silanol-directed, Pd(II)-catalyzed C–H alkenylation of phenols is reported. This work features silanol, as a novel traceless directing group, and a directed o-C-H alkenylation of phenols. This new method allows for efficient synthesis of diverse alkenylated phenols, including an estrone derivative.
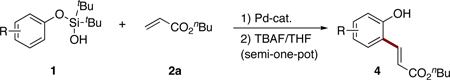
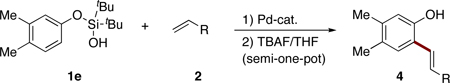
Ortho-alkenyl phenols are important building blocks for synthetic organic chemistry.1 Traditionally, these synthons can be assembled via a combined Claisen rearrangement of O-allylphenols to C-allylphenols followed by a transition metal-catalyzed double bond isomerization process (eq 1).2 This method is not general, as the Claisen rearrangement may produce a mixture of ortho- and para-allylphenols. Besides, the stereoselectivity of the isomerization step is ambiguous. Another common route to ortho-alkenyl phenols involves consecutive ortho-halogenation/Mizoroki-Heck cross-coupling reaction3 with alkenes (eq 2). The requisite of extra ortho-prefunctionalization step and concomitant over-bromination byproducts significantly limit wide applicaton of this approach.4 More directly, orhto-alkenylation reaction of phenols with terminal alkynes can be promoted by a Lewis acid, such as SnCl4.5 An obvious drawback of this method is an employment of stoichiometric amounts6 of a toxic tin reagent. Herein we wish to report a silanol group-directed Pd-catalyzed ortho C-H alkenylation of phenols to produce diverse ortho-alkenyl derivatives in good to high yields (eq 3).
 |
Transition metal-catalyzed directed C–H7 alkenylaton8 reactions have emerged as attractive alternative to the Mizoroki-Heck reaction. A directing group is usually introduced to control the regioselectivity as well as to enhance the reactivity of the reaction.9 We were intrigued by the possibility to develop a method that would employ an easily removable directing group at the phenol, which would allow for a general synthesis of alkenylated phenols.10,11 Recently, we reported a traceless/modifiable silicon-tethered directing group12 (PyDipSi) for ortho-acyloxylation and halogenation of arenes.13 Hence, we envisioned that employment of a temporary silicon-tethered directing group for phenols might be beneficial as it can efficiently be removed under mild conditions. In a recent report, Yu disclosed an elegant hydroxyl-directed ortho-C–H alkenylation of β-phenethylalcohols en route to alkenylaed arenes and/or benzopyrans (eq 4).14,15 Inspired by the
 |
(4) |
successful alcohol-directed C-H functionalization reactions14,15 and efficient silicon-tethered directing group employment in C-H functionalizations,13 we hypothesized that silanol may serve as an ideal easily removable directing group for C-H alkenylation of phenols.16
To test this hypothesis, silanol17 1a (1 equiv) was treated with butyl acrylate (2a, 2 equiv) under the conditions employing amino acid-derived ligand developed by Yu14 (10 mol% Pd(OAc)2, 20 mol% (+)Menthyl(O2C)-Leu-OH (L1), 1 equiv Li2CO3, 4 equiv AgOAc, in C6F6 at 100 °C). To our delight, the desired ortho-alkenylated product 3a was formed in 52% NMR yield (Table 1, entry 1). Solvent optimization indicated PhCF3 to be similarly efficient (entry 2), whereas employment of other solvents, such as toluene, dioxane, THF, t-AmylOH, and DMF gave poor yields. Finally, switching to DCE improved the yield of the reaction (78% NMR yield, entry 7).
Table 1.
Solvent Screening for Silanol-Directed Alkenylationa
 | |||
|---|---|---|---|
| entry | solvent (0.1 M) | conversion,%b | yield, %c |
| 1 | C6F6 | 77 | 52 |
| 2 | PhCF3 | 79 | 50 |
| 3 | PhMe | 43 | 24 |
| 4 | dioxane | 18 | <3 |
| 5 | THF | 4 | <3 |
| 6 | t-AmylOH | 26 | <3 |
| 7 | DCE | 90 | 78 |
| 8 | DMF | 55 | 0 |
1a/2a = 1 : 2, L1 = (+)Menthyl(O2C)-Leu-OH.
Consumption of starting material 1a measured by GC/MS.
1H NMR yield.
Next, the removal of the silanol directing group was examined. Expectedly, desilylation of 3a with TBAF proceeded uneventfully, producing the unprotected phenol 4a in 84% yield (eq 5) or in 66% yield over two steps. It deserves mentioning that better efficiency was achieved by carrying out two steps C–H alkenylation/desilylation in semi-one-pot fashion18 (Table 2, entry 1).
 |
(5) |
Table 2.
Phenol Scope for Silanol-Directed Alkenylation
 | |||||
|---|---|---|---|---|---|
| entry | substrate | product | yield, %a |
||
| 1 | 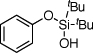 |
1a | 4a | 72 | |
| 2 | 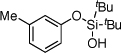 |
1b | 4b | 94 | |
| 3 | 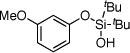 |
1c | 4c | 97 | |
| 4 | 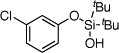 |
1d | 4d | 53b | |
| 5 | 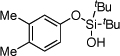 |
1e | 4e | 97 | |
| 6 | 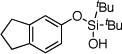 |
1f | 4f | 88b | |
| 7 | 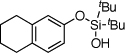 |
1g | 4g | 97 | |
| 8 | 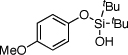 |
1h | 4h | 81 | |
| 9 | 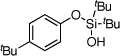 |
1i | 4i | 89 | |
| 10 | 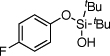 |
1j | 4j | 58b | |
| 11 | 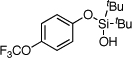 |
1k | 4k | 52b | |
Isolated yield.
The yield was measured by 1H NMR analysis using CH2Br2 as internal standard.
After developing the semi-one-pot procedure for the Pd-catalyzed silanol-directed C-H alkenylation/deprotection sequence, the scope of this new method was investigated. Table 2 summarizes olefinations of various phenol-derived silanols with butyl acrylate (2a) to produce the corresponding 2-hydroxy butyl cinnamates 4. It was found that diverse alkyl-, methoxy-, trifluoromethoxy-, chloro- and fluoro- substituents (entries 1–5, 8–11) were tolerated well under these reaction conditions. Moreover, 5-indanol and tetrahydro-2-naphthol reacted smoothly to afford the olefinated phenols in good to excellent yields (entries 6 and 7). Notably, meta-substituted substrates (entries 2–4) reacted regioselectively at the sterically less hindered C–H site. In general, electron-rich phenols gave better yields of the olefinated products compared to their electron-deficient counterparts. Remarkably, in contrast to most of the reported C-H alkenylation reactions,19 this Pd(II)-catalyzed olefination reaction is mono-selective. Most likely, the bulky tert-butyl groups at the silanol moiety prevent orientation of the silanol directing group toward the less hindered C-H site, thus effectively stopping the reaction at the monoalkenylation stage.
Next, we turned our attention to the scope of olefins. It was found that a wide range of electron-deficient alkenes could be successfully employed in this transformation (Table 3). Thus, vinylsulfonate 2b and vinylsulfone 2c readily reacted with silanol 1e to give the olefinated products in very good yields (entries 1, 2). Acrolein (2d) and alkyl vinyl ketones 2e and 2f are also capable reactants in this olefination reaction (entries 3–5). Moreover, styrene and its derivatives, smoothly reacted with 1e to give (E)-2-styrylphenols 4p-4s in reasonable yields (entries 6–9). 1,1-Disubstituted acrylate 2k reacted with 1e to give expected product 4u,20 along with its isomer 4v in 45% and 39% NMR yields, respectively.9b
Table 3.
Alkene Scope for Silanol-Directed Alkenylation
 | |||||
|---|---|---|---|---|---|
| entry | substrate | product | yield, %a |
||
| 1 | 2b | 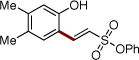 |
4l | 96 | |
| 2 | 2c | 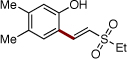 |
4m | 87b | |
| 3 | 2d | 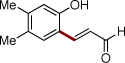 |
4n | 70b | |
| 4 | 2e | 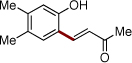 |
4o | 67b | |
| 5 | 2f | 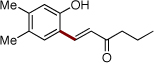 |
4p | 69b | |
| 6 | 2g | 4q | 64c,d | ||
| 7 | 2h | 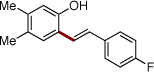 |
4r | 79 | |
| 8 | 2i | 4s | 83 | ||
| 9 | 2j | 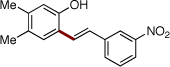 |
4t | 66 | |
| 10 | 2k | 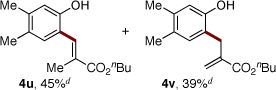 |
|||
Isolated yield.
Alkene 2 (4 equiv), Boc-Val-OH (20 mol %) as the ligand, 110 °C.
Styrene (4 equiv), 120 °C.
1H NMR yield.
Furthermore, the reaction of 1e with diethyl maleate (2l) under the standard reaction conditions produced alkenylated product 5, which upon desilylation/cyclization, led to the formation of lactone 6 in 58% yield (eq 6).20 It should be mentioned that this example represents the first synthesis of a benzofuranone from a simple phenol featuring a C–H activation strategy.
 |
(6) |
Finally, an application of this novel alkenylation methodology on the olefination of a more complex substrate estrone was tested. Thus, the corresponding silanol 7 underwent a smooth alkenylation/desilylation reaction sequence to produce the olefinated estrone 8
 |
(7) |
as a single regioisomer in 89% yield (eq 7).21 This example showcases the viability of employment of this method for a late-stage modification of complex phenol-containing bioactive molecules toward a diversity-oriented drug discovery.22
In summary, we have shown that the di-tert-butylsilanol can serve as a new and efficient directing group for the palladium-catalyzed ortho-alkenylation of phenols. Employment of this directing group is very convenient as it can easily be removed under mild conditions. A synthetic usefullness of this novel alkenylation method was further demonstrated in the efficient synthesis of benzofuranone and alkenylated estrone derivative.
Supplementary Material
ACKNOWLEDGMENT
We thank the National Institutes of Health (GM-64444) for financial support of this work.
Footnotes
Supporting Information. Detailed experimental procedures and charcterization data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Koehler K, Gordon S, Brandt P, Carlsson B, Backsbro-Saeidi A, Apelqvist T, Agback P, Grover GJ, Nelson W, Grynfarb M, Farnegardh M, Rehnmark S, Malm J. J. Med. Chem. 2006;49:6635. doi: 10.1021/jm060521i. [DOI] [PubMed] [Google Scholar]; (b) Gan FF, Chua YS, Scarmagnani S, Palaniappan P, Franks M, Poobalasingam T, Bradshaw TD, Westwell AD, Hagen T. Biochem. Biophys. Res. Commun. 2009. 387:741. doi: 10.1016/j.bbrc.2009.07.104. [DOI] [PubMed] [Google Scholar]; (c) Botyanszki J, Shi D-F, Roberts CD, Schmitz FU. 20070032488 U.S. Pat. Appl. Publ. US. ; (d) Finkelstein BL, Benner EA, Hendrixson MC, Kranis KT, Rauh JJ, Sethuraman MR, McCann SF. Biorg. Med. Chem. 2002;10:599. doi: 10.1016/s0968-0896(01)00326-1. [DOI] [PubMed] [Google Scholar]
- 2.For Claisen rearrangement of O-allylphenols to C-allylphenols, see: Martín Castro AM. Chem. Rev. 2004;104:2939. doi: 10.1021/cr020703u. For examples of C-allylphenols to C-vinylphenols, see: Gauthier D, Lindhardt AT, Olsen EPK, Overgaard J, Skrydstrup T. J. Am. Chem. Soc. 2010;132:7998. doi: 10.1021/ja9108424.
- 3.(a) Mizoroki T, Mori K, Ozaki A. Bull. Chem. Soc. Jpn. 1971;44:581. [Google Scholar]; (b) Heck RF, Nolley JP., Jr J. Org. Chem. 1972;37:2320. [Google Scholar]
- 4.For an example on regioselective formation of halophenols, see: de Rege FMG, Buchwald SL. Tetrahedron. 1995;51:4291.
- 5.(a) Yamaguchi M, Hayashi A, Hirama M. J. Am. Chem. Soc. 1995;117:1151. [Google Scholar]; (b) Kobayashi K, Yamaguchi M. Org. Lett. 2001;3:241. doi: 10.1021/ol006888d. [DOI] [PubMed] [Google Scholar]
- 6.The developed catalytic version of this process (see ref. 5b) is limited to vinylation reaction only.
- 7.For general reviews on transition metal-catalyzed C–H activation of arenes, see: Kakiuchi F, Chatani N. Adv. Synth. Catal. 2003;345:1077. Dick AR, Sanford MS. Tetrahedron. 2006;62:2439. Godula K, Sames D. Science. 2006;312:67. doi: 10.1126/science.1114731. Yu J-Q, Giri R, Chen X. Org. Biomol. Chem. 2006;4:4041. doi: 10.1039/b611094k. Alberico D, Scott ME, Lautens M. Chem. Rev. 2007;107:174. doi: 10.1021/cr0509760. Campeau L-C, Stuart DR, Fagnou K. Aldrichimica Acta. 2007;40:35. Ackermann L, Vicente R, Kapdi AR. Angew. Chem., Int. Ed. 2009;48:9792. doi: 10.1002/anie.200902996. Daugulis O, Do H-Q, Shabashov D. Acc. Chem. Res. 2009;42:1074. doi: 10.1021/ar9000058. McGlacken GP, Bateman LM. Chem. Soc. Rev. 2009;38:2447. doi: 10.1039/b805701j. Mkhalid IAI, Barnard JH, Marder TB, Murphy JM, Hartwig JF. Chem. Rev. 2010;110:890. doi: 10.1021/cr900206p. Ashenhurst JA. Chem. Soc. Rev. 2010;39:540. doi: 10.1039/b907809f. Satoh T, Miura M. Synthesis. 2010:3395. Sun C-L, Li B-J, Shi Z-J. Chem. Rev. 2011;111:1293. doi: 10.1021/cr100198w.
- 8.For reviews on transition metal-catalyzed C–H alkenylation of arenes, see: Oestreich M, editor. The Mizoroki-Heck Reaction. Chicester, U.K.: John Wiley and Sons; 2009. Beccalli EM, Broggini G, Martinelli M, Sottocornola S. Chem. Rev. 2007;107:5318. doi: 10.1021/cr068006f. Messaoudi S, Brion J-D, Alami M. Eur. J. Org. Chem. 2010:6495. Seregin IV, Gevorgyan V. Chem. Soc. Rev. 2007;36:1173. doi: 10.1039/b606984n. Chen X, Engle KM, Wang D-H, Yu J-Q. Angew. Chem., Int. Ed. 2009;48:5094. doi: 10.1002/anie.200806273. Colby DA, Bergman RG, Ellman JA. Chem. Rev. 2010;110:624. doi: 10.1021/cr900005n. Lyons TW, Sanford MS. Chem. Rev. 2010;110:1147. doi: 10.1021/cr900184e. Satoh T, Miura M. Chem. Eur. J. 2010;16:11212. doi: 10.1002/chem.201001363.
- 9.For examples of removable directing group assisted C–H activation, see: Ihara H, Suginome M. J. Am. Chem. Soc. 2009;131:7502. doi: 10.1021/ja902314v. García-Rubia A, Urones B, Gómez Arrayás R, Carretero JC. Chem. Eur. J. 2010;16:9676. doi: 10.1002/chem.201001126. García-Rubia A, Fernández-Ibáñez MÁ, Gómez Arrayás R, Carretero JC. Chem. Eur. J. 2011;17:3567. doi: 10.1002/chem.201003633. Dai H-X, Stepan AF, Plummer MS, Zhang Y-H, Yu J-Q. J. Am. Chem. Soc. 2011;133:7222. doi: 10.1021/ja201708f.
- 10.The employment of a phenoxyl group as a directing group for C–H activation is less common since it would generate a highly-strained, four-membered metallacycle, see: Vicente J, Abad J-A, Förtsch W, Jones PG, Fischer AK. Organometallics. 2001;20:2704.
- 11.For o-arylation of phenols employing phosphorous-containing additives, see: Bedford RB, Coles SJ, Hursthouse MB, Limmert ME. Angew. Chem., Int. Ed. 2003;42:112. doi: 10.1002/anie.200390037. Bedford RB, Limmert ME. J. Org. Chem. 2003;68:8669. doi: 10.1021/jo030157k. Oi S, Watanabe S-I, Fukita S, Inoue Y. Tetrahedron Lett. 2003;44:8665. For o-alkylation of phenols, see: Lewis LN, Smith JF. J. Am. Chem. Soc. 1986;108:2728. Dorta R, Tongi A. Chem. Commun. 2003;760 doi: 10.1039/b211672c. Carrión MC, Cole-Hamilton DJ. Chem. Commun. 2006:4527. doi: 10.1039/b610038d. Lewis JC, Wu J, Bergman RG, Ellman JA. Organometallics. 2005;24:5737. For pioneering work regarding o-deuteration of phenols, see: Lewis LN. Inorg. Chem. 1985;24:4433. For o-borylation of phenols employing a silicon-tethered directing group, see: Boebel TA, Hartwig JF. J. Am. Chem. Soc. 2008;130:7534. doi: 10.1021/ja8015878.
- 12.For early work on employment of removable silyl-directing group in Heck reaction, see: Itami K, Mitsudo K, Kamei T, Koike T, Nokami T, Yoshida J-i. J. Am. Chem. Soc. 2000;122:12013. For a review, see: Itami K, Yoshida J-i. Synlett. 2006:157.
- 13.(a) Chernyak N, Dudnik AS, Huang C, Gevorgyan V. J. Am. Chem. Soc. 2010;132:8270. doi: 10.1021/ja1033167. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dudnik AS, Chernyak N, Huang C, Gevorgyan V. Angew. Chem., Int. Ed. 2010;49:8729. doi: 10.1002/anie.201004426. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Huang C, Chernyak N, Dudnik AS, Gevorgyan V. Adv. Synth. Catal. 2011;353:1285. doi: 10.1002/adsc.201000975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lu Y, Wang D-H, Engle KM, Yu J-Q. J. Am. Chem. Soc. 2010;132:5916. doi: 10.1021/ja101909t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.For hydroxyl group as a directing group, see also: Terao Y, Wakui H, Satoh T, Miura M, Nomura M. J. Am. Chem. Soc. 2001;123:7725. doi: 10.1021/ja016914i. Satoh T, Kawamura Y, Miura M, Nomura M. Angew. Chem., Int. Ed. 1997;36:1740. Wang X, Lu Y, Dai H-X, Yu J-Q. J. Am. Chem. Soc. 2010;132:12203. doi: 10.1021/ja105366u. Lu Y, Leow D, Wang X, Engle KM, Yu J-Q. Chem. Sci. 2011;2:967.
- 16.To the best of our knowledge, there are no reports on employment of silanol as a directing group in C-H functionalization reactions.
- 17.Silanols 1 were prepared in a semi-one-pot reaction of phenols with di-t-butylchlorosilane, followed by bromination and hydrolysis. See Supporting Information for details. For published two-step procedure, see: Petit M, Chouraqui G, Aubert C, Malacria M. Org. Lett. 2003;5:2037. doi: 10.1021/ol034207j.
- 18.Upon completion of the first step, the mixture was filtered through a celite plug, concentrated and treated with TBAF solution in THF. See Supporting Information for details.
- 19.For rare examples on monoselective alkenylation, see: Shi B-F, Zhang Y-H, Lam JK, Wang D-H, Yu J-Q. J. Am. Chem. Soc. 2010;132:460. doi: 10.1021/ja909571z. Wang D-H, Engle KM, Shi B-F, Yu J-Q. Science. 2010;327:315. doi: 10.1126/science.1182512. For not completely selective mono vs bis-alkenylation, see: García-Rubia A, Arrayás RG, Carretero JC. Angew. Chem., Int. Ed. 2009;48:6511. doi: 10.1002/anie.200902802. Engle KM, Wang D-H, Yu J-Q. Angew. Chem., Int. Ed. 2010;49:6169. doi: 10.1002/anie.201002077. Patureau FW, Glorius F. J. Am. Chem. Soc. 2010;132:9982. doi: 10.1021/ja103834b. Mochida S, Hirano K, Satoh T, Miura M. J. Org. Chem. 2011;76:3024. doi: 10.1021/jo200509m. See also refs. 9b, d.
- 20.The stereochemistry was determined by NOE experiments.
- 21.(a) Edsall AB, Mohanakrishnan AK, Yang D, Fanwick PE, Hamel E, Hanson AD, Agoston GE, Cushman M. J. Med. Chem. 2004;47:5126. doi: 10.1021/jm049647a. [DOI] [PubMed] [Google Scholar]; (b) Ciana C-L, Phipps RJ, Brandt JR, Meyer F-M, Gaunt MJ. Angew. Chem., Int. Ed. 2011;50:458. doi: 10.1002/anie.201004703. [DOI] [PubMed] [Google Scholar]; (c) Zhou C-Y, Li J, Peddibhotla S, Romo D. Org. Lett. 2010;12:2104. doi: 10.1021/ol100587j. [DOI] [PubMed] [Google Scholar]
- 22.(a) Schreiber SL. Nature. 2009;457:153. doi: 10.1038/457153a. [DOI] [PubMed] [Google Scholar]; (b) Galloway WRJD, Spring DR. Nature. 2011;470:43. doi: 10.1038/470042a. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.