

Abstract
We have reported that heme-dependent activation of apo-neuronal nitric oxide synthase (apo-nNOS) to the active holo-enzyme dimer is dependent upon factors present in reticulocyte lysate and other cytosols. Here, we find that both Hsp70 and thioredoxin are components of the activation system. The apo-nNOS activating activity of reticulocyte lysate is retained in a pool of fractions containing Hsp70 that elute from DE52 prior to Hsp90. All of the activating activity and 20–30% of the Hsp70 elute in the flow-through fraction upon subsequent ATP-agarose chromatography. Apo-nNOS activation by this flow-through fraction is inhibited by pifithrin-μ, a small molecule inhibitor of Hsp70, suggesting that a non-ATP-binding form of Hsp70 is involved in heme-dependent apo-nNOS activation. Previous work has shown that apo-nNOS can be activated by thiol-disulfide exchange, and we show substantial activation with a small molecule dithiol modeled on the active motifs of thioredoxin and protein disulfide isomerase. Further fractionation of the ATP-agarose flow-through on Sephacryl S-300 separates free thioredoxin from apo-nNOS activating activity, Hsp70, and a small amount of thioredoxin, all of which are eluted throughout the macromolecular peak. Incubation of apo-nNOS with the macromolecular fraction in combination with either the thioredoxin-containing fraction or with purified recombinant human thioredoxin restores full heme-dependent activating activity. This supports a model in which Hsp70 binding to apo-nNOS stabilizes an open state of the heme/substrate binding cleft to facilitate thioredoxin access to the active site cysteine that coordinates with heme iron, permitting heme binding and dimerization to the active enzyme.
The chaperone protein Hsp901 regulates the function, turnover and trafficking of a wide variety of signaling proteins (reviewed in Ref. 1). Hsp90 regulates proteins by modulating ligand binding clefts to favor an open state of the cleft, increasing the efficiency of binding of ligands such as steroids or ATP (reviewed in Ref. 2). Two types of interaction with Hsp90 occur. Classic client proteins, such as the glucocorticoid receptor (GR), have metastable clefts that are recognized by the Hsp90/Hsp70-based chaperone machinery and form relatively stable complexes with Hsp90 that can be isolated from cell lysates and analyzed biochemically. These client protein•Hsp90 heterocomplexes are constantly undergoing cycles of assembly and disassembly in the cytoplasm and nucleoplasm, and client protein function and turnover are stringently regulated by Hsp90 (2). Some other signaling proteins that have more stable ligand binding clefts undergo much more dynamic cycling with Hsp90, forming heterocomplexes that rapidly disassemble such that no (or only trace amounts of) Hsp90 heterocomplexes can be isolated from cell lysates. The function and turnover of dynamically cycling proteins, such as the nitric oxide synthase (NOS) enzymes, are not as affected by Hsp90 inhibition as the classic client proteins (2).
The NOS enzymes, including endothelial NOS (eNOS), neuronal NOS (nNOS) and inducible NOS (iNOS), are enzymes whose activity is enhanced by Hsp90, as shown both by studies in intact cells (3–6) and by direct activation assays with purified proteins (3,6–11). NOS activity is Ca2+/calmodulin (CaM)-dependent, and several signaling pathways initiate nNOS and eNOS activity by raising intracellular Ca2+ concentration. Studies with purified proteins show that CaM and Hsp90 increase the binding of each other to both eNOS and nNOS (7,8,10,12). Binding of Hsp90 to NOS is different from its binding to the classic client proteins. The assembly of stable Hsp90 complexes with steroid receptors, for example, requires ATP, Hsp70, and p23, and both receptor-bound Hsp70 and Hsp90 must pass through at least one complete ATPase cycle (1). In contrast, both direct binding of purified Hsp90 to purified eNOS and nNOS and activation of their activities have been demonstrated in the absence of ATP, Hsp70, and p23 (3,10,12).
The NOS enzymes function as cytochrome P450-type hemoproteins to catalyze the formation of nitric oxide (NO) and citrulline from L-arginine, O2 and NADPH (reviewed in Ref. 13). These enzymes are active as homodimers, with each monomer binding tightly 1 eq each of FAD, FMN, tetrahydrobiopterin and heme. The prosthetic heme is the site of oxygen activation, which is required for the metabolism of L-arginine. Hsp90 binds to the oxygenase domain (14), which contains the heme/substrate binding cleft, and the binding of CaM to NOS enhances electron flux from flavin bound to the reductase domain to heme bound within the cleft (15).
For the past decade, our laboratory has studied the action of Hsp90 and Hsp70 on nNOS (reviewed in Refs. 16 and 17). Treatment of cells with Hsp90 inhibitors decreases nNOS catalytic activity, increases nNOS turnover, and inhibits activation of apo-nNOS by exogenous heme (4). nNOS degradation is via the ubiquitin-proteasome pathway (4,18–20), and both CHIP and parkin function as Hsp70-dependent E3 ligases for nNOS ubiquitination (19,20). Reaction of certain mechanism-based inactivators in the heme/substrate binding cleft causes opening of the cleft to yield a more unfolded state of the protein (2,17), triggering nNOS ubiquitination and proteasomal degradation (18,21). Hsp90 and Hsp70 have opposing actions on nNOS ubiquitination, with Hsp70 stimulating and Hsp90 inhibiting (11).
The effects of Hsp90 in stimulating nNOS enzymatic activity and stabilizing nNOS to ubiquitination and degradation both ensue from the same interaction of Hsp90 with the ligand (heme/substrate) binding cleft (11). Ligand binding clefts are hydrophobic clefts that are dynamic in that they shift to varying extents between closed and open states. When clefts open during this molecular breathing process, hydrophobic residues of the protein interior are exposed to solvent and continued opening may progress to nascent stages of unfolding. The extent to which the binding cleft is open determines ligand (substrate) access and protein function, but these clefts are inherent sites of conformational instability. Dynamic Hsp90 binding to nNOS stabilizes the open cleft, facilitating substrate access and enzyme activity while impeding further unfolding and Hsp70-dependent ubiquitination (reviewed in Ref. 17).
It has proven to be difficult to define a mechanistic basis for the observation that treatment of cells with an Hsp90 inhibitor, such as geldanamycin or radicicol, inhibits activation of apo-nNOS by heme. This inhibition of heme activation was noted in Sf9 cells which have very low levels of endogenous heme. Addition of exogenous heme to the culture medium causes an increase in nNOS activity in infected cells (4). The ferrous carbonyl complex of nNOS is not formed when Hsp90 is inhibited, indicating that functional heme insertion into heme-deficient apo-nNOS is not occurring in the inhibitor-treated Sf9 cells (22). However, studies of heme insertion into iNOS show that GAPDH plays a key role in handing off heme to apo-iNOS in a process that is itself regulated by NO (23). Thus, in cells the activation of apo-NOS by heme is more complicated than just Hsp90 stabilization of an open cleft to facilitate heme access.
Under cell-free conditions purified apo-nNOS can be completely activated with high concentrations of dithiothreitol (4.0 mM DTT) and heme in the presence of SOD and catalase to protect the heme (24). This would be consistent with formation of a disulfide bond(s) during apo-nNOS purification that prevents heme-dependent activation. We also found that Sf9 cytosol (25) or a DE52-retained fraction of rabbit reticulocyte lysate (4) promote functional heme insertion into nNOS, and they both convert the GR to a state where the hydrophobic ligand binding cleft is open to access by steroid. However, Hsp90 is required for GR activation but not for apo-nNOS activation by the lysate. We concluded that in vivo, where access by free heme is limited, Hsp90 is required for sustained opening of the heme binding cleft and nNOS activation. But, because a high concentration of DTT alone facilitates heme-dependent activation of purified apo-nNOS, it is clear that the endogenous apo-nNOS activating activity in reticulocyte lysate ultimately reflects a thiol-disulfide interchange.
Zgoda et al. (26) have shown that hemin-mediated reactivation of heme-stripped, microsomal CYP2B1 requires glutathione (GSH) as well as GRP94, the geldanamycin-sensitive homolog of Hsp90 in the endoplasmic reticulum. They suggest that GSH acts either directly or indirectly via a putative microsomal thiol reductase to regenerate the conserved active site cysteine-thiol in a reduced state that coordinates with heme iron, permitting heme binding and regeneration of enzyme activity (26). The NOS enzymes have a similar conserved, active site cysteine-thiol required for heme binding, and it is an intriguing notion that a similar chaperone-dependent thiol-disulfide interchange machinery may exist in cytosol for heme-dependent apo-nNOS activation.
In this paper we fractionate the DE52-retained fraction of reticulocyte lysate to identify the component(s) that facilitates heme activation of apo-nNOS. The activity is accounted for by the pool of fractions containing Hsp70 that elute from DE52 prior to Hsp90. When this pool is passed through a column of ATP-agarose, apo-nNOS activating activity is present in the flow-through material (ATPFT), which contains 20–30% of the Hsp70, as opposed to the ATP-agarose retained material (ATPRet), which contains the ATP-binding Hsp70 that is active in the multichaperone machinery that generates the steroid binding activity of the GR. Heme-dependent apo-nNOS activation by the ATPFT fraction is inhibited by the Hsp70 inhibitor pifithrin-μ. Apo-nNOS activation is also stimulated by the Hsp70 co-chaperone Hip (Hsp70 interacting protein), suggesting that an ADP-bound form of Hsp70 is part of the endogenous activating system. Further fractionation of the ATPFT fraction on Sephacryl S-300 reveals that the Hsp70 and a small amount of thioredoxin as well as the apo-nNOS activating activity are eluted throughout the high molecular mass material. A small molecular mass pool was collected that also contains thioredoxin, most likely in its monomeric free state. When the high molecular mass pool is combined with the small molecular mass pool containing free thioredoxin, the full heme-dependent apo-nNOS activating activity of the ATPFT material is reconstituted. Full activation is also achieved by combining the large molecular mass pool with purified human thioredoxin, suggesting that thioredoxin is the major endogenous component of the activating system promoting the reduced state of the active cysteine-thiol required for heme-binding. This leads to a model whereby binding of nNOS by Hsp70 stabilizes an open state of the heme/substrate binding cleft to facilitate thioredoxin-dependent thiol-disulfide interchange with a cysteine disulfide within the cleft.
EXPERIMENTAL PROCEDURES
Materials
Untreated rabbit reticulocyte lysate was from Green Hectares (Oregon, WI). (6R)-5,6,7,8-Tetrahydro-L-biopterin was purchased from Dr. Schirek’s Laboratory (Jona, Switzerland). [1,2,4,6,7-3H]Dexamethasone (85 Ci/mmol) was from GE Healthcare (Amersham Place, UK), and Sephacryl S-300 HR was from GE Healthcare (Uppsala, Sweden). Pifithrin-μ, ATP-agarose, and purified human recombinant N-terminal His-tagged thioredoxin were purchased from Sigma. Ni-NTA agarose was from Qiagen. (±)-trans-1,2-bis(mercaptoacetamido)cyclohexane (BMC) was from US Biological (Swampscott, MA). 125I-conjugated goat anti-mouse and anti-rabbit IgGs were obtained from Perkin Elmer Life Sciences (Boston, MA). The FiGR monoclonal IgG was provided by Dr. Jack Bodwell (Dartmouth Medical School, Lebanon, NH). The cDNA for rat nNOS was provided by Dr. Solomon Snyder (The Johns Hopkins Medical School, Baltimore, MD). The AC88 monoclonal IgG against Hsp90 and N27F3-4 anti-72/73-kDa Hsp70 monoclonal IgG (anti-Hsp70) were from Enzo Life Sciences International (Plymouth Meeting, PA). Human Hip cDNA was provided by Dr. David Smith (Mayo Clinic, Scottsdale, AZ). The rhodacyanine dye analogs MKT-077 and YM-1 were synthesized in the Gestwicki laboratory.
Expression and Purification of Apo-nNOS and Hip
Lysates were prepared from Sf9 cells infected with recombinant nNOS baculovirus as described previously (4). Lysates were centrifuged at 100,000 × g for 1 h, the supernatant fraction was loaded onto a 2′,5′-ADP-Sepharose column (2 ml), and the apo-nNOS was eluted with 10 mM 2′-AMP in high salt buffer (4). Eluted fractions were concentrated and loaded onto Sephacryl S-300 HR column equilibrated with 50 mM Tris-HCl, pH 7.4, containing 100 mM NaCl, 10% glycerol, 0.1 mM EDTA, 0.1 mM dithiothreitol. The apo-nNOS-containing fraction was concentrated with Amicon Ultra (MWCO, 100,000), and the concentrated sample was stored at −80 °C
Amino-terminally His-tagged Hip was expressed from plasmid pET-28a-Hip in Escherichia coli BL21 (DE3) and purified on Ni-NTA agarose. After extensive washing with PBS containing 25 mM imidazole, the bound protein was eluted with 100 mM imidazole in PBS, and desalted on PD-10 columns with buffer containing 10 mM Tris, pH 7.5, 1 mM DTT and 1 mM EDTA.
Activation of Apo-nNOS with Heme and nNOS Activity Assay
Purified apo-nNOS (5 μg) was preincubated for 1 h at 30 °C with 10 μl of heme buffer (10 mM Hepes, pH 7.4, 3 μM heme as a heme-bovine serum albumin complex (27), 100 mM KCl, 0.1 mM EGTA, 0.1 mM DTT, and 50 μM tetrahydrobiopterin (expressed as final concentrations)) and 15 μl of HKD0.1 buffer (10 mM Hepes, pH 7.4, 100 mM KCl, and 0.1 mM DTT) or 2 μl of the appropriate fraction of reticulocyte lysate adjusted to 15 μl with HKD0.1 buffer. Aliquots of 5 μl were removed for assay of nNOS activity. NO formation was assayed by the NO-mediated conversion of oxyhemoglobin to methemoglobin. nNOS-containing samples were added to an assay solution containing 200 μM CaCl2, 250 μM L-arginine, 100 μM tetrahydrobiopterin, 100 units/ml catalase, 5 μg/ml calmodulin, 25 μM oxyhemoglobin, and an NADPH-regenerating system consisting of 400 μM NADP+, 10 mM glucose-6-phosphate, and 1 unit/ml glucose-6-phosphate dehydrogenase (expressed as final concentrations) in a total volume of 180 μl of 50 mM potassium phosphate, pH 7.4. The mixture was incubated at 37 °C and the rate of oxidation of oxyhemoglobin was monitored by measuring the absorbance of λ401–411 nm with a microtiter plate reader. nNOS activity is expressed as nmol NO/min/mg ± S.E. for three or more experiments.
Immunoadsorption of GR
Receptors were immunoadsorbed from 50-μl aliquots of Sf9 cytosol (prepared from cells expressing mouse GR as previously described (25)) by rotation for 2 h at 4 °C with 18 μl of protein A-Sepharose precoupled to 9 μl of FiGR ascites suspended in 200 μl of TEG buffer (10 mM TES, pH 7.6, 50 mM NaCl, 4 mM EDTA, 10% glycerol). Prior to incubation with DE52-retained fractions of reticulocyte lysate, immunoadsorbed receptors were stripped of associated Hsp90 by incubating the immunopellet for an additional 2 h at 4 °C with 350 μl of 0.5 M NaCl in TEG buffer. The pellets were then washed once with 1 ml TEG buffer followed by a second wash with 1 ml of Hepes buffer (10 mM Hepes, pH 7.4).
GR•Hsp90 Heterocomplex Reconstitution
FiGR immunopellets containing GR stripped of chaperones were incubated with the appropriate fraction of reticulocyte lysate adjusted to 55 μl with HKD5.0 buffer (10 mM Hepes, pH 7.4, 100 mM KCl, 5 mM dithiothreitol) containing 20 mM sodium molybdate and 5 μl of an ATP-regenerating system (50 mM ATP, 100 units/ml creatine phosphokinase, 20 mM magnesium acetate, and 250 mM creatine phosphate in 10 mM Hepes, pH 7.4). The assay mixtures were incubated for 20 min at 30 °C with suspension of the pellets by shaking the tubes every 2 min. At the end of the incubation, the pellets were washed twice with 1 ml of ice-cold TEGM buffer (TEG with 20 mM sodium molybdate) and assayed for steroid binding capacity. For experiments where GR•Hsp90 reconstitution was carried out with purified Hsp90, Hop, Hsp40, p23 and the flow-through or retained fraction of reticulocyte lysate, proteins were purified as described by Kanelakis and Pratt (28).
Assay of Steroid Binding Capacity
Immunopellets to be assayed for steroid binding were incubated overnight at 4 °C in 50 μl of HEM buffer (10 mM Hepes, pH 7.4, 1 mM EDTA, 20 mM molybdate) plus 100 nM [3H]dexamethasone. Samples were then washed three times with 1 ml of TEGM buffer and counted by liquid scintillation spectrometry. The steroid binding is expressed as counts/min of [3H]dexamethasone bound/FiGR immunopellet prepared from 50 μl of cytosol ± S.E. for three experiments.
DE52, ATP-agarose, and Sephacryl S-300 Chromatography of Reticulocyte Lysate
Rabbit reticulocyte lysate (50 ml) was adsorbed to a 2.6 × 18-cm column of DE52 equilibrated with HE buffer (10 mM Hepes, 1 mM EDTA, pH 7.35); the column was washed with 250 ml of HE buffer with 20 mM sodium molybdate followed by 150 ml of HE buffer alone, and proteins were eluted with a 400 ml gradient of 0–0.5 M KCl. Hsp90 and Hsp70 were detected in the DE52-retained material by resolving an aliquot of each fraction by SDS-PAGE and Western blotting with appropriate antibodies, and fractions were combined in three pools designated A–C as described by Dittmar et al. (29). Pooled fractions were dialyzed against HKD0.1 buffer (10 mM Hepes, 100 mM KCl, 0.1 mM DTT) and concentrated to 2 ml.
To further fractionate the heme-dependent nNOS activating activity, the DE52 fraction A pool was applied to a 50 ml column of ATP-agarose, which was then washed with 150 ml of HEK buffer (HE containing 500 mM KCl). The flow-through fractions were combined, dialyzed against HKD0.1 buffer, concentrated by Amicon Ultra (MWCO, 10,000) to 3 ml, and designated ATPFT. The Hsp70 retained by ATP-agarose was eluted with 200 ml HEK buffer containing 5 mM ATP, dialyzed against HKD0.1 buffer, concentrated to 1 ml, and designated ATPRet.
To further fractionate the ATPFT, a 1 ml aliquot was concentrated to 350 μl and loaded onto a Sephacryl S-300 HR column (2.6 × 100 cm) equilibrated with HKD0.1 buffer. Fractions (2.5 ml) were collected at a flow rate of 0.5 ml/min and concentrated to a final volume of 150 μl. Hsp70, Hsp40, Hip and thioredoxin were detected by resolving an aliquot of each fraction by SDS-polyacrylamide gel electrophoresis and immunoblotting with appropriate antibodies. Fifteen μl of each fraction was assayed for apo-nNOS activating activity.
Immunoblotting
Aliquots of various preparations were boiled in SDS sample buffer with 10% μ-mercaptoethanol, and proteins were resolved on 7% SDS-polyacrylamide gels. Proteins were then transferred to Immobilon-P membranes and probed with the appropriate primary antibody. The immunoblots were then incubated a second time with 125I-conjugated goat anti-mouse or anti-rabbit IgG to visualize the immunoreactive bands.
RESULTS
nNOS Activating Activity Resides in the ATP-agarose Flow-through of Reticulocyte Lysate Fraction A
To begin identification of the apo-nNOS activating activity in the DE52-retained fraction of reticulocyte lysate, fractions were separated into three pools as described by Dittmar et al. (29) for reconstitution of GR steroid binding activating activity. Fraction pool A consists of those fractions eluting prior to Hsp90, and it contains the majority of the Hsp70, Hop, and Hsp40 components of the GR chaperone machinery (Figure 1A). Fraction pool B consists of fractions eluting at higher salt containing Hsp90, and fraction pool C consists of the fractions eluting at high salt, which contain p23, the highly anionic cochaperone of Hsp90 (Figure 1A). The GR and apo-nNOS activating activities of fractions A–C are shown in the top and bottom graphs in Figure 1B. As previously reported (29), all three fractions are required to reconstitute the GR activating activity of reticulocyte lysate (Figure 1B, top). In contrast, fraction A, which by itself has no GR activating activity, accounts fully for the apo-nNOS activating activity (Figure 1B, bottom). Moreover, in contrast to the reconstitution of GR steroid binding activity by reticulocyte lysate, the Hsp90-specific inhibitor radicicol does not inhibit the heme-dependent activation of apo-nNOS (Figure 1C).
Figure 1.

DE52 fractionation of reticulocyte lysate. Rabbit reticulocyte lysate was fractionated on a column of DE52 and fractions were combined into three pools designated A–C as described under Methods. (A) Fractions A, B, and C were immunoblotted for Hsp90, Hsp70, Hop, Hsp40, and p23. (B) GR (top) and apo-nNOS (bottom) activating activity of unfractionated reticulocyte lysate (RL) and the indicated combinations of fractions A, B, and C were assayed as described under Methods. (C) Apo-nNOS (solid bars) or GR (open bars) were activated with unfractionated reticulocyte lysate (RL) in the presence or absence of 10 μM radicicol (Rad). Data are mean ± SEM for three experiments.
In an attempt to determine if the Hsp70 component of reticulocyte lysate was necessary for apo-nNOS activating activity, fraction A was passed through a column of ATP-agarose. Adsorption to ATP-agarose and elution with ATP is normally the final step in our purification of Hsp70 for use in a purified 5-protein system – Hsp90, Hsp70, Hop, Hsp40, p23 – that yields efficient steroid receptor•Hsp90 heterocomplex assembly (28). The flow-through fraction that did not bind to ATP-agarose (ATPFT) and the ATP-agarose retained material from fraction A (ATPRet) were then tested for heme-dependent apo-nNOS activating activity. The flow-through fraction contained apo-nNOS activating activity, whereas the ATP-retained material did not (Figure 2A). Repassage of the ATPFT through a new ATP-agarose column did not remove activating activity or Hsp70 (Figure 2A). Thus, there is a non-ATP-binding form of Hsp70 that we have always discarded in our studies of GR activation with purified chaperone proteins. When the Hsp90-free GR is incubated with a purified 4-protein system without Hsp70, the non-ATP-binding Hsp70 in the ATPFT does not yield activation of GR steroid binding, whereas the ATP-binding Hsp70 in the ATPRet material does activate the receptor (Figure 2B).
Figure 2.
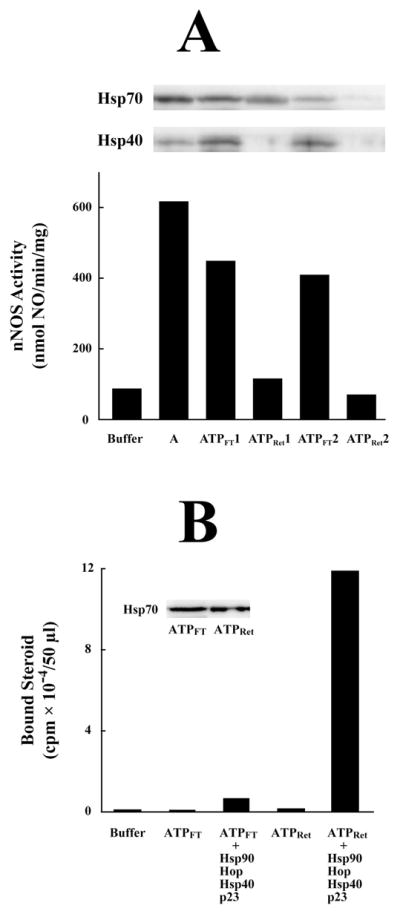
ATP-agarose fractionation of apo-nNOS activating activity in DE52 fraction A. (A) DE52 fraction pool A from reticulocyte lysate was applied to an ATP-agarose column, and the flow-through (ATPFT) and retained (ATPRet) fractions were prepared as described under Methods. The first ATP-agarose flow-through (ATPFT1) was applied to a fresh ATP-agarose column to prepare a second flow-through (ATPFT2) and retained (ATPRet2) preparation. The apo-nNOS activating activity of DE52 fraction A and the ATP-agarose flow-through and retained fractions was assayed. The Western blots show the Hsp70 and Hsp40 in aliquots of each preparation. (B) The Hsp70 in the ATPFT fraction does not activate the steroid binding activity of the GR in the presence of the other components of the chaperone machinery. Stripped GR was incubated with purified Hsp90, Hop, Hsp40 and p23 in the presence of ATPFT or ATPRet fractions as indicated. The amounts of ATPFT and ATPRet fractions were adjusted to contain similar amounts of Hsp/Hsc70 as indicated by the immunoblot in the inset.
Evidence that Hsp70 is Involved in Apo-nNOS Activation
To determine if Hsp70 in the ATPFT was required for heme-dependent apo-nNOS activation, purified apo-nNOS was incubated with ATPFT and various concentrations of pifithrin-μ, a small molecule inhibitor of Hsp70 (30). As shown in Figure 3A (closed squares), pifithrin-μ inhibited a major portion of the apo-nNOS activation by ATPFT in a concentration-dependent manner. Pifithrin-μ did not affect the activity of purified holo-nNOS under the same conditions (Figure 3A, open squares), showing that it is the activation of apo-nNOS and not the activity of the heme-bound nNOS dimer that is inhibited.
Figure 3.

Evidence that Hsp70 plays a role in apo-nNOS activation. (A) Pifithrin-μ inhibits a major portion of the apo-nNOS activating activity of the ATPFT fraction. For samples indicated by solid squares, the ATPFT fraction was preincubated for 30 min on ice with various concentrations of pifithrin-μ, the samples were then incubated with purified apo-nNOS for 1 h at 30 °C, and the apo-nNOS activating activity was assayed. The open squares show the effect of pifithrin-μ on the activity of purified holo-nNOS incubated with ATPFT. (B) The Hsp70 cochaperone Hip promotes apo-nNOS activation. Purified apo-nNOS was incubated for 1 h at 30 °C with various concentrations of Hip (open circles) or with the ATPFT fraction plus Hip (closed circles), and the apo-nNOS activating activity was assayed. The inset shows a western blot of Hip in the ATPFT and ATPRet fractions.
Hip is an Hsp70-binding protein that stabilizes the ADP-bound state of Hsp70 (31) and its overexpression enhances the Hsp70-dependent reactivation of heat-inactivated luciferase in cells (32). As shown in Figure 3B, incubation of purified apo-nNOS with purified Hip increases heme-dependent apo-nNOS activation (open circles). As reported previously (4), a substantial amount of insect Hsp70 copurifies with nNOS, providing nNOS-bound chaperone for Hip interaction. The increase in apo-nNOS activation by Hip is consistent with the notion that Hsp70 is involved in the heme insertion mechanism. The ATPFT fraction contains the reticulocyte lysate Hip (Figure 3B, inset), and addition of purified Hip to the ATPFT does not increase its apo-nNOS activating activity (Figure 3B, solid circles).
Apo-nNOS Activation by Thiol-Disulfide Interchange
Zgoda et al. (26) showed that reactivation of heme-stripped CYP2B1 reflected a GRP94-dependent thiol-disulfide interchange. Similarly, cysteine residues located within the ligand binding cleft of the GR can form disulfide bonds that must be reduced for the receptor to have steroid binding activity (reviewed in Ref. 33). The endogenous reducing factor is thioredoxin (34), and the GR must be bound by Hsp90 for thioredoxin-dependent activation of steroid binding activity. Because we have previously reported that complete activation of apo-nNOS is achieved with high concentrations of dithiothreitol (4.0 mM DTT) and heme (24), it is likely that the endogenous apo-nNOS activating activity in reticulocyte lysate ultimately reflects a thiol-disulfide interchange with an active site cysteine required for heme binding. In this work, we have kept the DTT concentration at 0.1 mM, a level of DTT that by itself does not promote heme-dependent apo-nNOS activation (24). In Figure 4 (solid circles), purified apo-nNOS was incubated with various amounts of the ATPFT fraction to determine a maximum nNOS activation. Using the same preparation of purified apo-nNOS, we incubated with various concentrations of (±)-trans-1,2-bis(mercaptoacetamido)cyclohexane (BMC). BMC is a small molecule dithiol modeled on the active-site motifs of thioredoxin and protein disulfide isomerase that has been used as a reducing agent for disulfides (35,36). As shown in Figure 4 (open circles), BMC activated apo-nNOS to ~400 nmol NO/min/mg nNOS, showing that under our conditions at least half of the activation can be achieved with a small thiol-disulfide exchange reagent alone. Thus, at this point, we had the notion that the activating activity in the ATPFT fraction reflected the combined action of a non-ATP-binding form of Hsp70 and a disulfide reducing activity. One possible cellular reductant is GSH, which can be found in mM concentrations in some cells. Reduced GSH was ineffective in providing the reducing activity needed for apo-nNOS activation (closed squares).
Figure 4.

Activation of apo-nNOS with BMC. Full activation of apo-nNOS by ATPFT, partial activation by BMC, and no activation by GSH. Purified apo-nNOS was incubated for 1 h at 30 °C with various concentrations of the ATPFT fraction (closed circles) or BMC (open circles), or GSH (closed squares), and nNOS activity was assayed. BMC is maximally effective in the presence of glutathione (1 mM GSH; 0.2 mM GSSG) (36), and in the curve defined by open circles, glutathione was present at these concentrations in all samples. The effect of different concentrations of reduced GSH is also shown in the curve defined by closed squares.
Distribution of Apo-nNOS Activating Activity and Hsp70 on Fractionation of ATPFT Through Sephacryl S-300
We wanted to determine if the apo-nNOS activating activity distributed on molecular sieve chromatography as an Hsp70-based machinery of a discrete size in the same way that the Hsp90•Hop•Hsp70 machinery in reticulocyte lysate that activates GR steroid binding distributes as a discrete chaperone complex (37). Thus, the ATPFT fraction was applied to a column of Sephacryl S-300 and the eluted fractions were assayed for apo-nNOS activating activity and Hsp70. As shown in Figure 5, the majority of the apo-nNOS activating activity and the majority of the Hsp70 eluted between fractions 22 and 32, but both activating activity and Hsp70 are also present in fractions eluting between the void volume and the 440,000 dalton molecular mass standard. As both activating activity and Hsp70 seem to distribute throughout the bulk protein peak, the Hsp70 involved in apo-nNOS activation appears to be derived from Hsp70 that is associated with exposed hydrophobic regions of multiple proteins in the ATPFT.
Figure 5.

Sephacryl S-300 chromatography of ATPFT. The ATPFT fraction was chromatographed on Sephacryl S-300, all fractions were assayed for total protein (dashed line) and fractions 12–40 were assayed for apo-nNOS activating activity (solid bars). The arrows indicate the peak elution of molecular mass markers expressed as kDa. Samples of each fraction from the void volume through the smallest marker peak were immunoblotted for Hsp70, Hsp40, Hip and thioredoxin. The inset shows the relative distribution of Hsp70 (solid line) and thioredoxin (dotted line) in fractions 12–38. Fractions defined by arrows A and B shown at bottom were combined and contracted to form the Seph A and Seph B samples assayed in Figure 6.
In considering the possibility that thioredoxin is responsible for the thiol-disulfide interchange activity in the ATPFT fraction, we immunoblotted each Sephacryl S-300 fraction for this disulfide reducing agent. The great majority of the thioredoxin elutes as the free protein at the smallest molecular mass marker (Figure 5). However, immunoblotting revealed small amounts of thioredoxin in all of the higher molecular mass fractions as well (Figure 5), and the relative distribution of thioredoxin in fractions 12–38 was similar to the relative distribution of Hsp70 (Figure 5, inset). Although the chaperone and the reducing factor do not distribute as an Hsp70-based heterocomplex with a discrete size like the Hsp90/Hsp70-based chaperone machinery, they both appear to be present in multiple, high molecular mass protein-bound forms.
Evidence that Thioredoxin is Involved in Apo-nNOS Activation
The Sephacryl S-300 fractions indicated by the arrows in Figure 5 were pooled to yield a macromolecular fraction (Seph A) containing Hsp70 and some thioredoxin and a small molecule fraction (Seph B) containing the large peak of free thioredoxin. As shown in Figure 6A, Seph A or Seph B alone supported some heme-dependent activation of nNOS and the combination of Seph A and Seph B yielded as much activation as the original ATPFT fraction from which they were prepared. That thioredoxin is the active component of the Seph B fraction is suggested by the data of Figure 6B. In this case, apo-nNOS was incubated with Seph A and purified recombinant human thioredoxin (open circles) to yield the level of activation achieved with the original ATPFT fraction.
Figure 6.

Activation of apo-nNOS with Sephacryl S-300 fractions A and B and with Seph A plus thioredoxin. (A) Activation of apo-nNOS with Seph A and Seph B. nNOS activity was assayed after incubating purified apo-nNOS for various times at 30 °C with an amount (5 μl) of Seph A (■) or Seph B (□) that yielded maximum activation and with both Seph A and Seph B (○). Incubation with buffer alone (●) and ATPFT (●) were also carried out. (B) Activation of apo-nNOS with Seph A and thioredoxin. Apo-nNOS was incubated for 1 h at 30 °C with various concentrations of purified recombinant human thioredoxin (●) or with Seph A plus thioredoxin (○) and nNOS activity was assayed.
Apo-nNOS Activation by Purified Activating Reagents
Figure 7 shows heme-dependent activation of apo-nNOS by a purified activating system. In this case, apo-nNOS was incubated with purified Hsp70 in the presence of Hsp40 to promote ATPase activity and conversion to the ADP-bound conformation. In this system, we have substituted the Hsp70 cochaperone Hip with one of two rhodacyanine dye analogs, MKT-077 or YM-1. These compounds bind to Hsp70 family chaperones (38), and, as part of ongoing studies by the Gestwicki lab in the pharmacological targeting of Hsp70 (39), they have been shown to act like Hip in stabilizing Hsp70 association with unfolded protein. Either purified thioredoxin or BMC was added to boost thiol-disulfide exchange. The combination of Hsp70/40, MKT-077 and either thioredoxin or BMC yields the level of activation achieved with the ATPFT fraction. Although this activation has not been achieved by reconstituting the activating system of reticulocyte lysate with purified lysate proteins, the data of Figure 7 are consistent with the notion that Hsp70 and thioredoxin are critical components of the native heme-dependent apo-nNOS activating system.
Figure 7.

Activation of apo-nNOS with purified activating reagents. Purified apo-nNOS was incubated for 1 h at 30 °C with the additions noted under each column and nNOS activity was assayed. YM-1 (25 μM), MKT-077 (25 μM), thioredoxin (Trx, 25 μM), and BMC (3 mM) were present at their maximally effective concentrations.
DISCUSSION
We have previously shown that heme-dependent apo-nNOS conversion to the active nNOS homodimer is promoted by reticulocyte lysate or Sf9 cytosol (4,25) and that activation can be achieved with a high concentration of DTT in the absence of lysate or cytosol (24). Here, we have presented data consistent with the proposal that the abundant ubiquitous chaperone Hsc/Hsp70 and the ubiquitous thiol-disulfide exchange protein thioredoxin are two components of the apo-nNOS activating system in reticulocyte lysate. The identification of each of these components is subject to caveats that are inherent to the activating system.
Because apo-nNOS exists in a cytosolic heterocomplex containing Hsp70 and a substantial amount of the chaperone copurifies with the enzyme (4), some Hsp70 is present under all activating conditions. Heme-dependent activation of apo-nNOS involves a non-ATP-binding form of Hsp70 (Figure 2A) that is bound to multiple proteins in the lysate (Figure 5). This likely reflects ADP-bound Hsp70 that is associated with exposed hydrophobic regions of proteins in the ATPFT fraction. It is the ADP-bound conformation of the chaperone that interacts with Hip and has the highest affinity for hydrophobic peptides. In the apo-nNOS activation assay, the ADP-bound Hsp70 may exchange and interact with hydrophobic residues that are exposed to solvent in the open state of the heme/substrate binding cleft of apo-nNOS. This could serve to dynamically stabilize the open cleft conformation facilitating the access of thioredoxin to regenerate the conserved active site cysteine thiol of nNOS that coordinates with heme iron. Chaperone stabilization of the open state of the cleft may also facilitate heme entry into the cleft in addition to promoting heme retention through binding to the active site cysteine thiol.
Another possibility is that the non-ATP-binding Hsp70 in the ATPFT fraction is a modified form of the chaperone. For example, Hsp70 is subject to acetylation and deacetylation, and it is hyperacetylated in cells treated with certain histone deacetylase inhibitors (40). A similar acetylation of Hsp90 markedly reduces its ATP binding affinity (41,42) and disrupts the chaperone’s function (43–45). Specifically, acetylation of Hsp90 converts the chaperone from stable cycling into complex with the glucocorticoid receptor to dynamic cycling (42). We have been unable to detect acetylation of the Hsp70 in the ATPFT fraction by immunoblotting with anti-acetylated lysine antibodies. However, we have been unable to immunoadsorb more than a small fraction of the ATPFT Hsp70 using several different anti-Hsp/Hsc70 antibodies. This inability to immunoadsorb and concentrate the Hsp70 in the ATPFT fraction may be due to its binding to multiple lysate proteins (Figure 5), and it compromises our ability to detect any possible acetylation.
An additional caveat is that some reducing agent in the form of both DTT and thioredoxin is present in all of the lysate fractions with apo-nNOS activating activity. Although the 0.1 mM DTT present in our buffers is not by itself sufficient to activate apo-nNOS (24), it is possible that nNOS-bound Hsp70 and the non-ATP-binding Hsp70 in the various lysate fractions facilitates access of DTT to the active site cysteine of apo-nNOS. Similarly, the small amount of thioredoxin that is present in the Seph A fraction (Figure 5) may contribute to the apo-nNOS activating activity seen with Seph A alone (Figure 6).
Studies with specific inhibitors of the Hsp90 family proteins strongly suggest that cytosolic Hsp90 or GRP94, its homolog in the endoplasmic reticulum, play some role, respectively, in apo-nNOS activation (4,22,25) and in heme-dependent activation of heme-free CYP2B1 prepared by reaction with the heme targeting suicide substrate allylisopropylacetamide (26). In the case of apo-nNOS activation, treatment of intact Sf9 cells with geldanamycin or radicicol inhibited the enzyme’s activation by exogenous heme (22), and cytosol prepared from radicicol-treated Sf9 cells had reduced activity when incubated with purified apo-nNOS (25). Purified Hsp90 increases the enzymatic activity of purified holo-nNOS (10–12), and the decreased apo-nNOS activating activity of cytosol prepared from radicicol-treated cells may reflect this effect of Hsp90 on the active holo dimer. It is clear from Figure 1 that the heme-dependent apo-nNOS activating activity of reticulocyte lysate is retained in Fraction A, which contains Hsp70 but no Hsp90. It is also clear that inhibition of Hsp90 has no effect on the heme-dependent apo-nNOS activating activity of reticulocyte lysate. Thus, Hsp90 is not required for the reticulocyte lysate to promote heme entry into or retention in the heme/substrate binding cleft of apo-nNOS.
Activation of holo-nNOS activity by Hsp90 does not require ATP (10–12) and, thus, is a passive chaperoning effect that does not require Hsp90 to pass through its ATPase cycle, a cycle that is required for activation of GR steroid binding activity (1). Similarly, promotion of heme-dependent apo-nNOS activation by either reticulocyte lysate fractions or purified Hsp70 and thioredoxin does not require ATP. This suggests that the Hsp70 effect on apo-nNOS activation is also a passive chaperoning effect. Again, this stands in contrast with GR activation where Hsp70 first binds to the GR and passes through at least one ATPase cycle to prime the receptor for subsequent interaction with Hsp90 (1). Thus, with the GR, Hsp90 and Hsp70 work together as part of a multiprotein chaperone machinery to open and stabilize the ligand binding cleft, forming a stable GR•Hsp90 complex and facilitating steroid entry and high affinity steroid binding (1). In contrast, the effects of Hsp70 and Hsp90 on nNOS are independent of each other and do not require a chaperone machinery.
A scheme of our proposal for purified apo-nNOS activation is presented in Figure 8. We envision that the hydrophobic heme/substrate binding cleft of apo-nNOS opens and closes during a normal molecular breathing cycle. And, as hydrophobic moieties of the cleft interior are exposed, ADP-bound Hsp70 binds to the open state of the cleft, facilitating thioredoxin access to the cleft interior to maintain the active site cysteine in the reduced state. Maintaining the open cleft state facilitates heme entry and the cysteine thiol coordinates heme iron to retain heme. This interpretation complements the study of hemin-mediated activation of CYP2B1 by the Correia laboratory (26), and together these studies may suggest a general model for chaperone-assisted maintenance of active site cysteine thiols by cellular reductants, such as glutathione and thioredoxin.
Figure 8.

Activation of purified apo-nNOS by Hsp70 and thioredoxin. During apo-nNOS purification a disulfide is formed within the heme/substrate binding cleft favoring a closed state of the cleft. Hsp70 binding traps an open state of the cleft to facilitate thioredoxin mediated generation of thiols, with subsequent heme entry and ligation.
Acknowledgments
We thank Drs. Solomon Snyder, David Smith, David Toft and Jack Bodwell for providing plasmids and antibodies used in this work. We thank Dr. Craig Harris for assistance in some of the assays.
Footnotes
This work was supported by National Institutes of Health grants GM077430 and DA022354 (to Y.O.) and NS059060 (to J.E.G.) and National Science Foundation grant MCB-0844512 (to J.E.G.).
Abbreviations: Hsp, heat shock protein; GR, glucocorticoid receptor; NOS, nitric oxide synthase; CaM, Ca2+/calmodulin; Hop, Hsp organizer protein; Hip, Hsp70 interacting protein; CHIP, C-terminus of Hsp70-interacting protein; GSH, glutathione; BMC, (±)-trans-1,2-bis(mercaptoacetamido)cyclohexane; Trx, thioredoxin.
References
- 1.Pratt WB, Toft DO. Regulation of signaling protein function and trafficking by the hsp90/hsp70-based chaperone machinery. Exp Biol Med. 2003;222:111–133. doi: 10.1177/153537020322800201. [DOI] [PubMed] [Google Scholar]
- 2.Pratt WB, Morishima Y, Osawa Y. The Hsp90 chaperone machinery regulates signaling by modulating ligand binding clefts. J Biol Chem. 2007;283:22885–22889. doi: 10.1074/jbc.R800023200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Garcia-Cardena G, Fan R, Shah V, Sorrentino R, Cirino G, Papapetropoulos A, Sessa WC. Dynamic activation of endothelial nitric oxide synthase by Hsp90. Nature. 1998;392:821–824. doi: 10.1038/33934. [DOI] [PubMed] [Google Scholar]
- 4.Bender AT, Silverstein AM, Demady DR, Kanelakis KC, Noguchi S, Pratt WB, Osawa Y. Neuronal nitric oxide synthase is regulated by the hsp90-based chaperone system in vivo. J Biol Chem. 1999;274:1472–1478. doi: 10.1074/jbc.274.3.1472. [DOI] [PubMed] [Google Scholar]
- 5.Brouet A, Sonveaux P, Dessy C, Ballingand JL, Feron O. Hsp90 ensures the transition from the early Ca2+-dependent to the late phosphorylation-dependent activation of the endothelial nitric-oxide synthase in vascular endothelial growth factor-exposed endothelial cells. J Biol Chem. 2001;276:32663–32669. doi: 10.1074/jbc.M101371200. [DOI] [PubMed] [Google Scholar]
- 6.Yoshida M, Xia Y. Heat shock protein 90 as an endogenous protein enhancer of inducible nitric oxide synthase. J Biol Chem. 2003;278:36953–36958. doi: 10.1074/jbc.M305214200. [DOI] [PubMed] [Google Scholar]
- 7.Gratton JP, Fontana J, O’Conner DS, Garcia-Cardena G, McCabe TJ, Sessa WC. Reconstitution of an endothelial nitric-oxide synthase (eNOS), hsp90, and caveolin-1 complex in vitro, evidence that hsp90 facilitates calmodulin stimulated displacement of eNOS from caveolin-1. J Biol Chem. 2000;275:22268–22272. doi: 10.1074/jbc.M001644200. [DOI] [PubMed] [Google Scholar]
- 8.Takahashi S, Mendelsohn ME. Calmodulin-dependent and -independent activation of endothelial nitric-oxide synthase by heat shock protein 90. J Biol Chem. 2003;278:9339–9344. doi: 10.1074/jbc.M212651200. [DOI] [PubMed] [Google Scholar]
- 9.Takahashi S, Mendelsohn ME. Synergistic activation of endothelial nitric-oxide synthase (eNOS) by HSP90 and Akt. J Biol Chem. 2003;278:30821–30827. doi: 10.1074/jbc.M304471200. [DOI] [PubMed] [Google Scholar]
- 10.Song Y, Zweir JL, Xia Y. Heat shock protein 90 augments neuronal nitric oxide synthase by enhancing Ca2+/calmodulin binding. Biochem J. 2001;355:357–360. doi: 10.1042/0264-6021:3550357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peng HM, Morishima Y, Clapp KM, Lau M, Pratt WB, Osawa Y. Dynamic cycling with Hsp90 stabilizes neuronal nitric oxide synthase through calmodulin-dependent inhibition of ubiquitination. Biochemistry. 2009;48:8483–8490. doi: 10.1021/bi901058g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Song Y, Zweier JL, Xia Y. Determination of the enhancing action of Hsp90 on neuronal nitric oxide synthase by EPR spectroscopy. Am J Physiol. 2001;281:C1819–C1824. doi: 10.1152/ajpcell.2001.281.6.C1819. [DOI] [PubMed] [Google Scholar]
- 13.Marletta MA. Nitric oxide synthase structure and mechanism. J Biol Chem. 1993;268:12231–12234. [PubMed] [Google Scholar]
- 14.Fontana J, Fulton D, Chen Y, Fairchild TA, McCabe TJ, Fujita N, Tsuruo T, Sessa WC. Molecular mapping studies reveal that the M domain of hsp90 serves as a molecular scaffold to regulate Akt-dependent phosphorylation of endothelial nitric oxide synthase and NO release. Circ Res. 2002;90:866–873. doi: 10.1161/01.res.0000016837.26733.be. [DOI] [PubMed] [Google Scholar]
- 15.Abu-Soud HM, Stuehr DJ. Nitric oxide synthases reveal a role for calmodulin in controlling electron transfer. Proc Natl Acad Sci USA. 1993;90:10769–10772. doi: 10.1073/pnas.90.22.10769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Osawa Y, Lowe ER, Everett AC, Dunbar AY, Billecke SS. Proteolytic degradation of nitric oxide synthase: effect of inhibition and role of hsp90-based chaperones. J Pharmacol Exptl Ther. 2003;304:493–497. doi: 10.1124/jpet.102.035055. [DOI] [PubMed] [Google Scholar]
- 17.Pratt WB, Morishima Y, Peng HM, Osawa Y. Proposal for a role of the Hsp90/Hsp70-based chaperone machinery in making triage decisions when proteins undergo oxidative and toxic damage. Exp Biol Med. 2010;235:278–289. doi: 10.1258/ebm.2009.009250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Noguchi S, Jianmongkol S, Bender AT, Kamada Y, Demady DR, Osawa Y. Guanabenz-mediated inactivation and enhanced proteolytic degradation of neuronal nitric oxide synthase. J Biol Chem. 2000;275:2376–2380. doi: 10.1074/jbc.275.4.2376. [DOI] [PubMed] [Google Scholar]
- 19.Peng HM, Morishima Y, Jenkins GJ, Dunbar AY, Lau M, Patterson C, Pratt WB, Osawa Y. Ubiquitination of neuronal nitric-oxide synthase by CHIP, a chaperone-dependent E3 ligase. J Biol Chem. 2004;279:52970–52977. doi: 10.1074/jbc.M406926200. [DOI] [PubMed] [Google Scholar]
- 20.Morishima Y, Wang AM, Yu Z, Pratt WB, Osawa Y, Lieberman AP. CHIP deletion reveals functional redundancy of E3 ligases in promoting degradation of both signaling proteins and expanded glutamine proteins. Hum Mol Genet. 2008;17:3942–3952. doi: 10.1093/hmg/ddn296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bender AT, Demady DR, Osawa Y. Ubiquitination of neuronal nitric oxide synthase in vitro and in vivo. J Biol Chem. 2000;275:17407–17411. doi: 10.1074/jbc.M000155200. [DOI] [PubMed] [Google Scholar]
- 22.Billecke SS, Bender AT, Kanelakis KC, Murphy PJM, Lowe ER, Kamada Y, Pratt WB, Osawa Y. Hsp90 is required for heme binding and activation of apo-neuronal nitric-oxide synthase. J Biol Chem. 2002;277:20504–20509. doi: 10.1074/jbc.M201940200. [DOI] [PubMed] [Google Scholar]
- 23.Chakravarti R, Aulak KS, Fox PL, Stuehr DJ. GAPDH regulates cellular heme insertion into inducible nitric oxide synthase. Proc Natl Acad Sci USA. 2010;107:18004–18009. doi: 10.1073/pnas.1008133107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bender AT, Nakatsuka M, Osawa Y. Heme insertion, assembly, and activation of apo-neuronal nitric-oxide synthase in vitro. J Biol Chem. 2000;275:26018–26023. doi: 10.1074/jbc.275.34.26018. [DOI] [PubMed] [Google Scholar]
- 25.Billecke SS, Draganov DI, Morishima Y, Murphy PJM, Dunbar AY, Pratt WB, Osawa Y. The role of Hsp90 in heme-dependent activation of apo-neuronal nitric oxide synthase. J Biol Chem. 2004;279:30252–30258. doi: 10.1074/jbc.M403864200. [DOI] [PubMed] [Google Scholar]
- 26.Zgoda VG, Arison B, Mkrtchian S, Ingelman-Sundberg, Correia MA. Hemin-mediated restoration of allylisopropylacetamide-inactivated CYP2B1: a role for glutathione and GRP94 in the heme-protein assembly. Arch Biochem Biophys. 2002;408:58–68. doi: 10.1016/s0003-9861(02)00489-7. [DOI] [PubMed] [Google Scholar]
- 27.Richards MK, Marletta MA. Characterization of neuronal nitric oxide synthase and a C415H mutant purified from a baculovirus overexpression system. Biochemistry. 1994;33:14723–14732. doi: 10.1021/bi00253a010. [DOI] [PubMed] [Google Scholar]
- 28.Kanelakis KC, Pratt WB. Regulation of glucocorticoid receptor ligand binding activity by the hsp90/hsp70-based chaperone machinery. Meth Enzymol. 2003;364:159–173. doi: 10.1016/s0076-6879(03)64010-3. [DOI] [PubMed] [Google Scholar]
- 29.Dittmar KD, Hutchison KA, Owens-Grillo JK, Pratt WB. Reconstitution of the steroid receptor•hsp90 heterocomplex assembly system of rabbit reticulocyte. J Biol Chem. 1996;271:12833–12839. doi: 10.1074/jbc.271.22.12833. [DOI] [PubMed] [Google Scholar]
- 30.Leu JI, Pimkina J, Frank A, Murphy ME, George DL. A small molecule inhibitor of inducible heat shock protein 70. Mol Cell. 2009;36:15–27. doi: 10.1016/j.molcel.2009.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hohfeld J, Minami Y, Hartl FU. Hip, a novel cochaperone involved in the eukaryotic Hsc70/Hsp40 reaction cycle. Cell. 1995;83:589–598. doi: 10.1016/0092-8674(95)90099-3. [DOI] [PubMed] [Google Scholar]
- 32.Nollen EAA, Kabakov AE, Brunsting JF, Kanon B, Hohfeld J, Kampinga HH. Modulation of in vivo HSP70 chaperone activity by Hip and BAG-1. J Biol Chem. 2001;276:4677–4682. doi: 10.1074/jbc.M009745200. [DOI] [PubMed] [Google Scholar]
- 33.Simons SS, Pratt WB. Glucocorticoid receptor thiols and steroid binding activity. Meth Enzymol. 1995;251:406–422. doi: 10.1016/0076-6879(95)51144-x. [DOI] [PubMed] [Google Scholar]
- 34.Grippo JF, Holmgren A, Pratt WB. Proof that the endogenous, heat-stable glucocorticoid receptor activating factor is thioredoxin. J Biol Chem. 1985;260:93–97. [PubMed] [Google Scholar]
- 35.Lamoureux GV, Whitesides GM. Synthesis of dithiols as reducing agents for disulfides in neutral aqueous solution and comparison of reduction potentials. J Org Chem. 1993;58:633–641. [Google Scholar]
- 36.Woycechowsky KJ, Wittrup KD, Raines RT. A small-molecule catalyst of protein folding in vitro and in vivo. Chem Biol. 1999;6:871–879. doi: 10.1016/s1074-5521(00)80006-x. [DOI] [PubMed] [Google Scholar]
- 37.Murphy PJM, Kanelakis KC, Galigniana MD, Morishima Y, Pratt WB. Stoichiometry, abundance and functional significance of the hsp90/hsp70-based multiprotein machinery in reticulocyte lysate. J Biol Chem. 2001;276:30092–30098. doi: 10.1074/jbc.M103773200. [DOI] [PubMed] [Google Scholar]
- 38.Wadhwa R, Sugihara T, Yoshida A, Nomura H, Reddel RR, Simpson R, Maruta H, Kaul SC. Selective toxicity of MKT-077 to cancer cells is mediated by its binding to the Hsp70 family protein mot-2 and reactivation of p53 function. Cancer Res. 2000;60:6818–6821. [PubMed] [Google Scholar]
- 39.Patury S, Miyata Y, Gestwicki JE. Pharmacological targeting of the Hsp70 chaperone. Curr Top Med Chem. 2009;9:1337–1351. doi: 10.2174/156802609789895674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang Y, Wang SY, Zhang XH, Zhao M, Hou CM, Xu YJ, Du ZY, Yu XD. FK228 inhibits Hsp90 chaperone function in K562 cells via hyperacetylation of Hsp70. Biochem Biophys Res Commun. 2007;356:998–1003. doi: 10.1016/j.bbrc.2007.03.076. [DOI] [PubMed] [Google Scholar]
- 41.Yu X, Guo ZS, Marcu MG, Neckers L, Nguyen DM, Chen GA, Schrump DS. Modulation of p53, ErbB1, ErbB2, and Raf-1 expression in lung cancer cells by depsipeptide FR901228. J Natl Cancer Inst. 2002;94:504–513. doi: 10.1093/jnci/94.7.504. [DOI] [PubMed] [Google Scholar]
- 42.Murphy PJM, Morishima Y, Kovacs JJ, Yao TP, Pratt WB. Regulation of the dynamics of hsp90 action on the glucocorticoid receptor by acetylation/deacetylation of the chaperone. J Biol Chem. 2005;280:33792–33799. doi: 10.1074/jbc.M506997200. [DOI] [PubMed] [Google Scholar]
- 43.Bali P, Pranpat M, Bradner J, Balasis M, Fiskus W, Guo F, Rocha K, Kumaraswamy S, Boyapalle S, Atadja P, Seto E, Bhalla K. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90. J Biol Chem. 2005;280:26729–26734. doi: 10.1074/jbc.C500186200. [DOI] [PubMed] [Google Scholar]
- 44.Rao R, Fiskus W, Yang Y, Lee P, Joshi R, Fernandez P, Mandawat A, Atadja P, Bradner JE, Bhalla K. HDAC6 inhibition enhances 17-AAG-mediated abrogation of Hsp90 chaperone function in human leukemia cells. Blood. 2008;112:1886–1893. doi: 10.1182/blood-2008-03-143644. [DOI] [PubMed] [Google Scholar]
- 45.Kovacs JJ, Murphy PJM, Gaillard S, Zhao X, Wu JT, Nicchitta CV, Yoshida M, Toft DO, Pratt WB, Yao TP. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol Cell. 2005;18:601–607. doi: 10.1016/j.molcel.2005.04.021. [DOI] [PubMed] [Google Scholar]