

Abstract
Anti-neutrophil cytoplasm autoantibody (ANCA)-associated diseases are autoimmune conditions characterized by necrotizing inflammation of small blood vessels. The immunogenesis and etiology of these conditions are unknown, but our knowledge of the immunopathogenesis has increased considerably in recent years. In this review, we discuss the animal models currently used to investigate the mechanisms of vascular injury and to test novel therapies. We outline their advantages and limitations and propose potential directions for future research.
Introduction
Anti-neutrophil cytoplasm autoantibody (ANCA)-associated systemic small vessel vasculitis (SVV) (comprising Wegener granulomatosis and microscopic polyangiitis) is a group of related autoimmune disorders characterized by inflammatory necrosis of small blood vessels [1]. This results in dysfunction of supplied organs, and the principal clinical consequences are renal failure and lung hemorrhage. If the condition is untreated, the mortality at 1 year is almost 90% [2]. Despite modern treatment protocols, there are still mortality rates of 15% and 36% at 1 and 5 years, respectively [3], significantly higher than other autoimmune diseases and certain malignancies. The immunosuppressive therapies used carry a heavy burden of adverse events; one recent study found that death in the first year is three times more likely to be due to an adverse event than to the vasculitis itself [4]. In those patients not recovering renal function, renal replacement therapy carries an additional average annual cost of €31,000 to €40,000 ($42,240 to $54,500) per patient.
ANCAs are directed against enzymes stored in the azurophilic granules of neutrophils and the lysosomes of monocytes [5]. Several antigenic targets for ANCAs have been identified, but ANCAs directed against myeloper-oxidase (MPO) [6] and proteinase 3 (Pr3) [7,8] are most common. However, vasculitic lesions contain only scant immune deposits ('pauci-immune') and do not contain ANCAs. Therefore, it has been argued that these anti-bodies are unrelated to the actual vasculitic injury and that they are epi-phenomena and not part of the disease pathogenesis. The last two to three decades have witnessed the gradual emergence of an empirically supported paradigm that seeks to explain how these antibodies, which are so tightly associated with clinical disease, could exert a pathogenic effect by direct action on neutrophils.
ANCA-SVV pathogenesis
Evidence for a pathogenic role for ANCAs comes from numerous in vitro observations that support the contention that ANCA-mediated effector mechanisms contribute to endothelial injury (reviewed in [9]). The concept that has emerged from these observations is that ANCAs and proinflammatory stimuli (most likely of infectious origin) synergize to cause a destructive inflammatory process. The primary event in this process is that ANCA-mediated activation of neutrophils causes the generation of reactive oxygen species, release of proteases, and cytokine production. Full-blown ANCA-mediated neutro-phil activation requires priming with minor proinflammatory stimuli that induce translocation of the ANCA antigens to the cell surface, facilitating interaction with ANCAs [10]. Moreover, a recent study suggests that neutrophils from ANCA-SVV patients also have increased transcription of the ANCA antigens because of epigenetic modifications associated with gene silencing and thus increased autoantigen availability [11]. Following engagement of the F(ab')2 portion of ANCAs with ANCA antigens on the cell surface, and Fc receptor-mediated interactions, neutrophil activation is triggered [12]. Importantly, ANCAs also increase neutrophil adherence to endothelial monolayers, and co-incubation of ANCA-activated neutrophils and endothelial cells results in endothelial cell lysis [13]. There is a large body of in vitro experimental evidence to support this paradigm. However, to study the interplay between ANCAs, neutrophils, and infectious stimuli in the complex multicellular three-dimensional environment of renal and other tissues patrolled by elements of the innate and active immune system, animal models are required. Here, we will review the animal models of ANCA SVV that have been developed and address their advantages and limitations. In addition, we will discuss how these models have contributed to dissecting the pathogenic mechanisms involved in ANCA-mediated vasculitis and how they have provided us with a test bed for novel therapies.
Insights from animal models into the pathogenesis of pauci-immune SVV in the presence of ANCAs: pathogenicity of anti-MPO antibodies
Development of animal models of MPO-ANCA-mediated vasculitis has been an essential step in proving the direct pathogenic potential of anti-MPO antibodies in vivo. During the 1990 s, several rodent models were developed in an effort to model the effect of anti-MPO antibodies [14]. These involved inducing autoimmunity with mercuric chloride (a polyclonal B-cell stimulator) [15,16], planting of ANCA antigens within the kidney by direct infusion into the renal artery [17], or focusing the immune response to MPO on the kidney by administration of subnephritogenic doses of anti-glomerular basement membrane (anti-GBM) antibody [18]. These models provided evidence for the ability of anti-MPO antibodies to exacerbate renal injury. However, the development of crescentic nephritis was reliant upon the presence of immune complexes in the kidney, or the anti-MPO response was part of a broad, nonspecific, antibody profile. Therefore, these approaches, though informative, did not accurately model the pathology of anti-MPO-associated SVV in humans.
In 2002, Xiao and colleagues [19] demonstrated that systemic administration of purified murine anti-MPO IgG, obtained from murine MPO-immunized Mpo-/- mice, into recipient C57Bl/6 mice causes vasculitis. In this model, systemic injection of anti- MPO IgG resulted in urinary abnormalities (hematuria, leukocyturia, and albuminuria), early glomerular neutrophil accumulation, and focal necrotising crescentic glomerulo-nephritis in all recipient animals (Figure 1). In a subset of animals, vasculitic lesions were also observed in lungs, spleen, and ears. Shortly after this murine model was developed, Little and colleagues [20] developed a rat model of systemic anti-MPO-associated vasculitis ('experimental autoimmune vasculitis', or EAV) that was characterized by generation of an immune response to exogenously administered human MPO in adjuvant, followed over a period of 4 to 8 weeks by progressive pauci-immune cresentic glomerulonephritis and lung hemorrhage (Figure 2). The initial immune response in this model is to the foreign human MPO protein, but the anti-MPO antibodies that develop cross-react with rat MPO.
Figure 1.

Overview of the mouse model of anti-myeloperoxidase (anti-MPO) IgG-induced glomerulonephritis. CFA, complete Freund's adjuvant.
Figure 2.
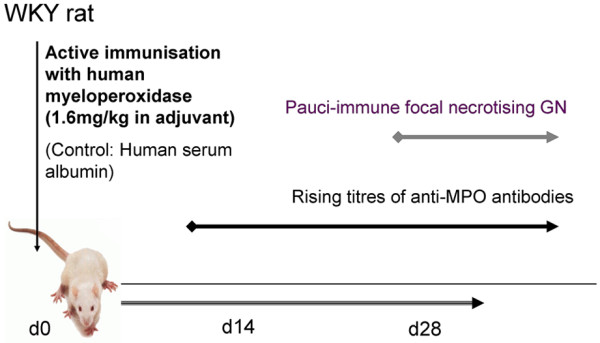
Overview of the experimental autoimmune vasculitis rat model of anti-myeloperoxidase (anti-MPO)-associated systemic vasculitis. d, day; GN, glomerulonephritis; WKY, Wistar Kyoto.
The histopathological findings in both of these models resemble, to a large extent, those in human ANCA SVV. For example, in agreement with the pauci-immune nature of the glomerular capillary lesions observed in human ANCA-associated glomerulonephritis, only a limited amount of immunoglobulins and complement factors is detected in the glomeruli of mice and rats with anti-MPO-mediated glomerulonephritis.
Both the mouse and rat models of MPO-ANCA SVV convincingly demonstrate the pathogenic potential of MPO-ANCAs but clearly also have their limitations. Essentially, both cannot be regarded as genuine auto-immune models, as they rely on active immunization strategies that depend upon the use of adjuvants for disease induction. Indeed, in the murine model, the MPO-deficient mouse has never been exposed to any MPO molecule before, so the MPO molecule is effectively a xeno-antigen and there is no requirement for breaking tolerance. As a consequence, high-affinity antibodies recognizing diverse epitopes are induced.
Second, the renal phenotype of both models is mild, thereby limiting the ability to use them to adequately test novel therapies. Without the use of additional inflammatory stimuli, such as lipopolysaccharide (LPS), crescent fraction is of the order of 5% to 10% and excretory renal function is preserved. This contrasts sharply with the relentless loss of kidney function observed in human ANCA-associated vasculitis, in which crescents often affect 100% of glomeruli. Therefore, an agent that successfully treats rodent vasculitis may not be effective in treating the established human condition. One important step in making the model's vasculitis severity more representative of human disease was published recently in abstract form by Xiao and colleagues [21], who made use of a different strain of mouse (129S6) that is known to be more sensitive to induction of glomerulo-nephritis than the C57/Bl6 strain used in the original experiments. Using the same approach of passive transfer of anti-murine MPO antibodies, the authors showed that the 129S6 strain develops crescents in 50% to 60% of glomeruli, reflecting the human situation more closely. These findings have not yet been replicated.
Finally, the passive transfer model developed by Xiao and colleagues [19] is induced by a single injection of anti-MPO IgG. Therefore, the model is useful for studying the induction of acute vascular injury by anti-MPO antibodies but is less suited for studying the chronic phase of the disease because of the lack of sustained autoantibody production. To address this issue, Schreiber and colleagues [22] employed a bone marrow (BM) transplantation approach to develop a model in which the effects of longer-term exposure to anti-MPO responses can be investigated. In these experiments, murine MPO-immunized Mpo-/- mice were irradiated and transplanted with BM from either MPO-deficient mice or wild-type mice. In the recipient mice, anti-MPO antibody production was preserved but only engraftment of MPO+ BM cells resulted in crescentic glomerulonephritis, thereby demonstrating that MPO+ BM cells are essential for the development of anti-MPO-mediated glomerulo-nephritis. However, in this model, the disease is again relatively mild, and the contribution of radiation-induced tissue injury and MPO+ T cells is unclear.
Insights from animal models into ANCA-SVV pathogenesis: mechanisms of anti-MPO-induced acute vascular injury
The MPO-ANCA vasculitis rodent models have proven to be very valuable for in vivo studies of effector mechanisms involved in the acute vascular inflammatory phase and for the evaluation of experimental therapies (summarized in Table 1, which is adapted from [23]). In the mouse model, neutrophils are the main effector cells, as neutrophil depletion completely prevented vasculitis induction upon injection of anti-MPO IgG [24]. Further-more, co-administration of LPS and anti-MPO IgG was found to severely aggravate glomerulonephritis development [25] in a Toll-like receptor 4 (TLR4)-dependent manner [26]. These observations support the contention that, following infection, proinflammatory stimuli and MPO-ANCAs synergize to cause full-blown vasculitis. To zoom in on the very early events in the interaction of neutrophils with the endothelium, intravital microscopy analysis of the mouse cremasteric microvasculature has been employed [27]. This study showed that, in the presence of a local inflammatory stimulus, anti-MPO IgG reduced neutrophil rolling while promoting adhesion and transendothelial migration of leukocytes. These MPO-ANCA-mediated neutrophil-endothelium interactions were found to depend upon β2 integrins and Fcγ receptors.
Table 1.
Summary of findings obtained using MPO-ANCA vasculitis animal models
| Result | Model | Reference | |
|---|---|---|---|
| Effector mechanisms | |||
| Neutrophil | Neutrophil depletion abrogates crescentic glomerulonephritis. | Mouse | [24] |
| T cells | CD4+ effector T cells contribute to anti-MPO-mediated crescentic glomeruloneprhitis. | Mouse (anti-GBM) | [55] |
| Th17 cells promote anti-MPO-mediated crescentic glomerulonephritis. | Mouse (anti-GBM) | [56] | |
| Proinflammatory stimuli | Lipopolysaccharide aggravates crescentic glomerulonephritis in a TLR4-dependent manner. | Mouse | [25] |
| Pertussis toxin/Mycobacterium tuberculosis aggravates crescentic glomerulonephritis. | Rat | [59] | |
| IgG glycosylation | IgG glycan hydrolysis attenuates crescentic glomeruloneprhitis. | Mouse | [38] |
| Leukocyte-endothelial interactions | Anti-MPO IgG increases leukocyte adhesion and migration in cremasteric venules. | Rat | [20] |
| Anti-MPO IgG increases leukocyte adhesion in glomerular capillaries. | Mouse | [27] | |
| Genetic susceptibility | Rat and mouse strains differ in susceptibility to anti-MPO-mediated crescentic glomerulonephritis. | Rat | [59] |
| Mouse | [21] | ||
| Targets for treatment | |||
| Complement pathway | Disruption of alternative complement pathway abrogates crescentic glomerulonephritis. | Mouse | [29] |
| Genetic ablation of C5aR attenuates crescentic glomerulonephritis. | Mouse (BM) | [30] | |
| PI3Kγ signalling | Genetic ablation of PI3Kγ attenuates crescentic glomerulonephritis. | Mouse (BM) | [33] |
| Experimental therapies | |||
| Anti-TNFα treatment | Anti-TNFα pretreatment attenuates crescentic glomerulonephritis. | Rat | [32] |
| Mouse | [25] | ||
| Anti-C5 treatment | Anti-C5 pretreatment abrogates and treatment attenuates crescentic glomerulonephritis. | Mouse | [31] |
| PI3Kγ inhibitor treatment | Treatment with a PI3Kγ inhibitor attenuates crescentic glomerulonephritis. | Mouse (BM) | [33] |
| P38 MAPK inhibitor treatment | Treatment with a P38 MAPK inhibitor attenuates crescentic glomerulonephritis. | Mouse | [36] |
anti-GBM refers to the mouse model in which low-dose anti-glomerular basement membrane administration triggers disease in myeloperoxidase-immunized mice. BM refers to the bone marrow transplantation myeloperoxidase-anti-neutrophil cytoplasm autoantibody (MPO-ANCA) mouse model. C5aR, C5a receptor; MAPK, mitogen-activated protein kinase; PI3Kγ, phosphatidylinositol 3 kinase-gamma; Th17, T helper 17; TLR4, Toll-like receptor 4; TNFα, tumor necrosis factor-alpha. Adapted from [23].
The ability of anti-MPO antibodies to increase leukocyte adhesion to and transmigration through endothelium is also supported by mesenteric intravital microscopy experiments in the EAV rat model [20]. In addition, workers from Monash University in Australia have used renal intravital microscopy to visualize an acute increase in leukocyte adhesion within a more clinically relevant organ, the kidney, following infusion of anti-MPO antibodies [28]. It is conventionally thought that leukocytes do not roll or adhere in glomerular capillaries, but this group provided evidence to support a nonclassical, α4 integrin-mediated mechanism of neutrophil capture in the glomeruli. Intravital microscopy cannot yet observe events in glomeruli in the normal mouse, and these experiments have used a hydronephrotic kidney model that is likely to markedly alter the glomerular responses, thereby making it difficult to interpret.
Insights from animal models in ANCA-SVV pathogenesis: an unexpected role for complement
ANCA SVV is a pauci-immune condition. One does not observe deposition of complement components at vasculitic sites, and levels of complement in the blood remain unperturbed unlike, for example, in systemic lupus erythematosus. In addition, the paradigm describing the pathogenesis pathway mentioned above does not include a role for complement. Therefore, the finding that mice depleted from circulating C3 by cobra venom factor as well as mice deficient in the common complement pathway component C5, its receptor C5aR, or the alter-native pathway component factor B were completely protected against anti-MPO-induced glomerulonephritis was unexpected [29,30]. Moreover, in this model, administration of a C5-inhibiting antibody markedly attenuated glomerulonephritis development even when treatment was started after disease induction [31]. The exact mechanism by which anti-MPO antibodies require complement for their action remains to be worked out, but these in vivo experiments have illuminated a novel therapeutic target.
Insights from animal models in ANCA-SVV pathogenesis: testing of novel therapies
The rodent models of anti-MPO-mediated glomerulonephritis described above have proven to be useful tools for testing experimental therapies. For example, therapeutic interventions aimed at blocking the proinflammatory effects of tumor necrosis factor-alpha (TNFα) have been evaluated in both the MPO-ANCA mouse model [25] and the EAV rat model [32]. In both, anti-TNFα treatment was beneficial and ameliorated disease severity, although this strategy appears to be more effective in rats. More recently, interventions have focused on the signalling pathways involved in ANCA-mediated neutrophil activation. Employing in vitro assays and the BM transplantation anti-MPO mouse model, Schreiber and colleagues [33] demonstrated a pivotal role for phoshatidylinositol 3 kinase-gamma (PI3Kγ) in MPO-ANCA-mediated neutrophil activation and glomerulonephritis development. In these studies, transplantation of BM from PI3Kγ-deficient mice into irradiated MPO-immunized MPO-/- mice prevented glomerulonephritis. Similar effects were observed in mice transplanted with wild-type BM upon oral treatment with a PI3Kγ-specific inhibitor (AS605240), suggesting that inhibition of PI3Kγ might be a therapeutic option in ANCA-SVV patients.
Another signalling pathway implicated in ANCA-mediated neutrophil activation is the P38 mitogen-activated protein kinase (MAPK) pathway. In vitro, inhibition of P38 MAPK abrogates ANCA-induced neutrophil activation, and there is evidence that the P38 MAPK pathway is activated in glomerular lesions of ANCA-SVV patients [34,35]. Using the anti-MPO IgG/LPS model, van der Veen and colleagues [36] tested the effects of an orally administered P38 MAPK inhibitor on glomerulonephritis development. In this study, P38 MAPK inhibition was found to ameliorate disease severity, although the effects were rather moderate, reducing glomerular crescent formation by approximately 30%. These data suggest that, besides p38 MAPK activity, other signalling pathways, such as the PI3Kγ (see above) and SYK [37] pathways, are activated in MPO-ANCA-mediated inflammation and are perhaps more important in the disease process.
An alternative strategy to block the pathogenic effects of MPO-ANCAs was described recently by van Timmeren and colleagues [38], who focused on the autoantibodies themselves. In this study, the bacterial enzyme endo-glycosidase S (EndoS) was used to specifically hydrolyze the conserved asparagine-linked glycans on IgG heavy chains, abolishing Fc receptor-mediated activation of leukocytes and complement [39]. In vitro, EndoS treatment of ANCA IgG markedly attenuated ANCA-mediated neutrophil activation without affecting antigen binding capacity whereas injection of EndoS-pretreated anti-MPO IgG in mice prevented glomerulonephritis development. Moreover, systemic EndoS administration early after disease induction rescued mice from disease progression. Overall, these data suggest that modulation of IgG glycosylation by EndoS is a promising strategy to interfere with the early ANCA-mediated inflammatory processes [38].
Finally, as mentioned above, therapeutic approaches aimed at inhibiting complement activation may prove to be fruitful. However, the applicability of results from these and other novel therapies to human disease is hampered by the relatively mild phenotype in both models, both of which have renal disease that is much less severe than the kidney-threatening disease seen in human SVV.
Insights from animal models in ANCA-SVV pathogenesis: investigating anti-Pr3 antibody-associated vasculitis
Strategies to develop Pr3-ANCA vasculitis models, in contrast to MPO-ANCA vasculitis models, have been unsuccessful so far. Using an approach similar to that of the murine anti-MPO antibody model, Pfister and colleagues [40] obtained anti-Pr3 antibodies from Pr3/elastase double-knockout mice by immunizing with murine recombinant Pr3. The anti-Pr3 antibodies were passively transferred to wild-type recipient mice and found to aggravate subcutaneous panniculitis induced by intradermal injection of TNFα. However, the presence of circulating anti-Pr3 antibodies, in contrast to that of anti-MPO antibodies, did not lead to vasculitic lesions in the lungs or kidneys. Along the same line, van der Geld and colleagues [41] showed that immunization of mice and rats with chimeric human-mouse Pr3 elicited an antibody response to mouse Pr3 and rat granulocytes. Again, how-ever, no signs of vasculitis development were observed in these animals. More recently, Primo and colleagues [42] attempted to use splenocyte transfer from recombinant Pr3-immunized mice to immunodeficient NOD/SCID (nonobese diabetic/severe combined immunodeficiency disease) mice that lack a functioning endogenous immune system. This splenocyte transfer approach was also adopted by Xiao and colleagues [19] in the anti-MPO model but was abandoned because of the development of numerous immune deposits in the vasculitic lesions, thereby rendering this model nonrepresentative of human vasculitis. Anti-Pr3 antibodies were detectable at high levels in recipients of splenocytes from Pr3-immunized mice, all of which developed necrotising glomerulonephritis. These experiments support a possible pathogenic effect of anti-Pr3 antibodies but cannot be considered definitive, because of the issue of immune complex deposition following splenocyte transfer. therefore, as convincing animal models are still lacking, our knowledge of the pathogenesis of vasculitis induced by anti-Pr3 antibodies remains limited.
ANCA SVV: outstanding issues
Clinical and experimental studies in the field of ANCA SVV have contributed much to our current knowledge of disease pathogenesis, but many questions remain. First, despite several attempts and approaches (vide supra), no convincing animal model for Pr3-ANCA SVV has yet been established. This raises the fundamental question of whether the pathogenesis of MPO-ANCA vasculitis and that of Pr3-ANCA vasculitis are the same. It has been recognized that Pr3-and MPO-ANCA SVV patients differ to some extent in their clinical presentation and histopathological characteristics of the vasculitic lesions [43]. Compared with patients with MPO-ANCAs, patients with Pr3-ANCAs more frequently present with extrarenal manifestations and respiratory tract granulomas and tend to have a higher rate of disease relapses. Moreover, systemic injection of high-affinity anti-Pr3 antibodies, unlike that of anti-MPO antibodies, in mice does not cause vasculitis. The underlying mechanisms accounting for these differences are not easily explained but may be due to differences in the ability of MPO-ANCAs and Pr3-ANCAs to interact with their target antigens, to activate their target cells, or to evoke cellular immune responses [43]. the discrepancy in pathogenic potential between anti-Pr3 and anti-MPO antibodies in animal models may also be caused by differences in physicochemical properties of the antigens themselves. For example, the iso-electric points of murine and human Pr3 (approximately 7) are far less than that of MPO (greater than 10) [40], and this theoretically could result in differential interactions of the antigens with negatively charged cell structures. Thus, clinical and experimental findings suggest that the pathogenesis of MPO-ANCA SVV and that of Pr3-ANCA SVV may not be the same, although adequate animal models need to be established to confirm this.
Second, why ANCA SVV primarily targets small-to medium-sized blood vessels and affects susceptible organs such as the kidneys and lungs is unclear. However, it is likely that the target of these diseases, the endothelium, actively participates in the induction and progression of vasculitis. Through expression of adhesion molecules and generation of cytokines and chemokines, activated endothelial cells are important players in driving the inflammatory response. It is also well appreciated that endothelial cells from different vascular beds are quite heterogeneous in their response to inflammatory stimuli, and this is most likely due to organ-and function-specific adaptations [44]. With this in mind, it will be interesting to compare the phenotype of endothelial cells in vascular beds that are affected in ANCA SVV with those that are resistant. One approach could be to analyze gene and protein expression profiles of affected vascular beds in human tissues and experimental models of ANCA SVV [45]. In the end, such analyses may reveal new vascular bed-specific targets for treatment. Third, the reason that ANCAs develop in the first place remains unknown, although a number of theories on ANCA immunogenesis have been proposed [46],
In a concept analogous to the idiotype network championed by Shoenfeld [47], Pendergraft and colleagues [48] described the presence of antibodies to a peptide encoded by (a portion of) the antisense DNA to the neutrophil granule constituent, Pr3. In this 'theory of autoantigen complementarity', the Pr3-ANCAs are part of an idiotypic network, and the authors postulated that some antigens expressed on infectious agents (for example, Staphylo-coccus aureus) may act as the complementary antigen. Several units around the world are in the process of replicating this finding of antibodies to complementary Pr3 in patients with crescentic glomerulonephritis.
A second theory invokes molecular mimicry between exogenous proteins and the ANCA antigens. This theory assumes that an initial immune response is evoked against pathogen-derived peptides that are highly homologous to peptide sequences within the ANCA antigens, resulting in a cross-reactive immune response against the ANCA self-antigens. A recent study by Kain and colleagues [49] suggests that such a mechanism may operate in ANCA SVV. In that study, the authors observed that circulating autoantibodies against lysosomal-associated membrane protein 2 (LAMP-2), a heavily glycosylated type 1 membrane protein involved in cellular adhesion and homeostasis, are highly prevalent in patients with active focal necrotising crescentic glomerulonephritis, most of which were also seropositive for either MPO- or Pr3-ANCAs. Subsequent experiments revealed potential pathogenic effects of anti-LAMP-2 antibodies. In vitro, anti-LAMP-2 antibodies caused neutrophil and endothelial cell activation, and injection of polyclonal rabbit anti-LAMP-2 antibodies induced a mild form of pauci-immune crescentic glomerulonephritis in rats. Of particular interest is the observation that a major epitope recognized by anti-LAMP-2 antibodies has strong homology with FimH-1, an adhesin of common Gram-negative bacteria. Upon immunization with FimH-1, rats developed antibodies directed against FimH-1 that cross-reacted with LAMP-2 and caused crescentic nephritis. Overall, these intriguing data suggest that infections with Gram-negative bacteria may provoke an autoimmune response to LAMP-2 that in turn induces vasculitis. However, since bacterial Gram-negative infections are common and ANCA SVV is rare, other factors must also be involved in disease induction [50]. Clearly, more studies are needed to extend these observations, and confirmation in other patient cohorts is eagerly awaited.
Animal models for ANCA SVV: what is next?
Despite their limitations, the current animal models of ANCA vasculitis are likely to be useful for further elucidation of mechanisms and factors involved in disease pathogenesis and for identifying targets for treatment. In this respect, the murine model is likely to be useful in identifying therapeutic targets at the point of acute vascular injury, whereas the EAV rat model may be more useful in identifying therapies that can be administered over a more prolonged period of time to disrupt the MPO-specific immune response. The most pressing current need is the development of a reliable model of anti-Pr3-associated vasculitis. In addition, we believe that the following issues will be important to address over the coming 5 to 10 years:
The crucial role of neutrophils in ANCA-SVV pathogenesis is well established but other effector cells are likely to contribute as well. Besides neutrophils, ANCAs can activate monocytes in vitro to produce oxygen radicals [51], proinflammatory cytokines, and chemokines [52]. In addition, macrophages are important cellular components of the inflammatory infiltrate in vasculitic lesions and contribute to glomerular crescent formation [53]. Thus, it is of interest to explore the role of monocytes/macrophages in disease progression in the ANCA vasculitis models. To this end, strategies to deplete monocytes/macrophages or modify their functions could be applied in these models.
A rather unexplored area in the SVV models is the role of T cells in disease pathogenesis. In the original mouse model developed by Xiao and colleagues [19], adoptive transfer of splenocytes from mMPO-immunized MPO-deficient mice into mice that lack mature B and T cells (RAG1-/- mice) caused severe glomerulonephritis. In this model, adoptive transfer of pure B cells also induces disease manifestations whereas transfer of pure CD4+ T cells does not [54]. These results indicate that, in this model, MPO-specific CD4+ T cells are not required for disease induction but do not rule out a role for these cells in the maintenance and propagation of the immune response. A study by Ruth and colleagues [55] indeed suggests that MPO-ANCA- and MPO-specific CD4+ T cells may work together in a unique way. These authors demonstrated that immunization of C57Bl6 mice with human MPO in adjuvant induces a humoral (MPO-ANCA) as well as a cellular (MPO-specific CD4+ T-cell reactivity) immune response. In these mice, an additional challenge with a subnephritogenic dose of heterologous anti-GBM anti-bodies caused glomerular MPO deposition and triggered the development of severe crescentic glomerulonephritis. Interestingly, similar experiments performed in B cell-deficient mice still resulted in crescentic glomerulonephritis despite the absence of MPO-ANCAs in these mice. On the basis of these results, the authors postulated that the effector phase of MPO-ANCA-associated glomerulonephritis is a two-step process requiring (a) MPO ANCA-mediated glomerular neutrophil recruitment and release of MPO and (b) CD4+ T-cell effector responses to induce crescentic glomerulonephritis [55]. The importance of CD4+ effector T cells in anti-MPO glomerulonephritis may be confirmed through studies involving transfer of MPO-specific T cells from immunized Mpo-/- mice into wild-type recipients with or without anti-MPO IgG. Using a similar experimental setup, Gan and colleagues [56] recently investigated the role of T helper 17 (Th 17) cells in autoimmune anti-MPO glomerulonephritis. Th17 cells are a recently identified Th subset characterized by the production of effector cytokines such as interleukin (IL)-17A, IL-17F, IL-21, and IL-22. IL-17A is of particular interest because it has a wide range of proinflammatory properties promoting neutrophil and monocyte recruitment and stimulation of release of proinflammatory cytokines such as TNF and IL-1 by macrophages. Interestingly, increased serum levels of IL-17 and IL-23 in conjunction with increased percentages of circulating Th 17 cells have been detected in human ANCA SVV [57]. In their studies, Gan and colleagues [56] showed that immunization of C57Bl6 mice with murine MPO resulted in MPO-specific dermal delayed- type hypersensitivity and systemic IL-17A production. Upon injection of low-dose anti-GBM antibodies, these mice developed glomerulonephritis. In contrast, IL-17A-deficient mice were almost completely protected from disease induction, and this was due in part to reduced glomerular neutrophil recruitment. These results identify IL-17A as an important effector cytokine in the pathogenesis of MPO-ANCA glomerulonephritis and suggest that targeting IL-17A may be a therapeutic option. It should be noted, however, that the models employed by Ruth and colleagues and Gan and colleagues are different from the model originally described by Xiao and colleagues [19] because a subnephritogenic dose of heterologous anti-GBM antibodies is used to trigger disease manifestations. Thus, effectively, these are models involving immune complex deposition in addition to anti-MPO autoantibodies.
Besides studies into the role of effector cells, further elucidation of the pathogenic mechanism of the ANCA autoantibodies themselves is of interest. In the mouse model, the induced polyclonal anti-MPO antibodies are pathogenic, but it is unclear whether disease induction is dependent upon specific antibody isotypes or antigen epitopes. These issues could be addressed using monoclonal antibodies generated from murine MPO-immunized Mpo-/- mice combined with heavy-chain switch variants of these monoclonal antibodies [23].
The ANCA vasculitis animal models are also likely to contribute to the elucidation of genetic risk factors for disease development. Until now, genetic studies in human ANCA SVV have focused on candidate genes and have been hampered by small sample sizes [58]. Although large multicenter genome-wide studies in ANCA SVV have been initiated and are currently under way, the animal models may also offer opportunities. In the rat EAV model, Wistar Kyoto (WKY) rats have been shown to be highly susceptible to vasculitis development upon immunization with human MPO in complete Freund's adjuvant. In contrast, three other rat strains tested - Lewis, Wistar Furth, and Brown Norway - were found to be resistant to vasculitis development, although similar levels of anti-human MPO antibodies were detected [59]. Interestingly, since WKY and Lewis rats share the same major histocompatibility complex (MHC) 2 haplotype, these observations indicate that susceptibility to vasculitis development in this model is dependent on non-MHC-linked genes [59]. Similarly, preliminary studies in the anti-MPO IgG transfer mouse model have shown that 129S6 mice are much more susceptible to anti-MPO IgG-mediated glomerulonephritis induction than the originally used C57Bl6 mice [21]. Since both strains are of the H2b MHC haplotype, this again indicates that non-MHC genes are involved. Collectively, these observations in rats and mice now pave the way for more detailed genetic studies that will aid in the identification of genetic risk factors for human ANCA SVV.
Finally, the rodent models of ANCA SVV may continue to be used for the discovery and testing of new targets for treatment. Possible target candidates include mediators of signalling pathways other than PI3K and P38 MAPK that have been shown to be involved in ANCA-mediated activation of neutrophils in vitro. Also, new targets may be identified by analyzing vascular bed-specific gene and protein expression patterns or via genome-wide gene expression analysis of affected tissues. Since the alternative pathway of complement appears to be pivotal in anti-MPO-mediated glomerulonephritis in mice, a potential therapy could involve inhibition of components critical for this pathway, including factor B and properdin. Such therapies have been evaluated recently in other inflammatory models and could be attractive targets for ANCA vasculitis as well [60,61].
Conclusions
Boosted by the development of various animal models for MPO-ANCA SVV, our knowledge of the unique pathogenic mechanisms involved in ANCA-mediated vasculitis has increased tremendously and this will open new avenues for therapeutic strategies. At the same time, many questions concerning the pathogenesis and immunogenesis of ANCA SVV remain. The current MPO-ANCA models will continue to be helpful in providing answers to these questions, although further 'fine tuning' of the animal models is necessary. The development of a convincing in vivo model for Pr3-ANCA SVV is eagerly awaited.
Abbreviations
ANCA: anti-neutrophil cytoplasm autoantibody; BM: bone marrow; EAV: experimental autoimmune vasculitis; EndoS: endoglycosidase S; GBM: glomerular basement membrane; IL: interleukin; LAMP-2: lysosomal-associated membrane protein 2; LPS: lipopolysaccharide; MAPK: mitogen-activated protein kinase; MHC: major histocompatibility complex; MPO: myeloperoxidase; PI3K: phoshatidylinositol 3 kinase; Pr3: proteinase 3; SVV: small vessel vasculitis; Th17: T helper 17; TNF: tumor necrosis factor; WKY: Wistar Kyoto.
Competing interests
The authors declare that they have no competing interests.
Note
Autoimmune Basis of Rheumatic Diseases
This article is part of a review series on Vasculitis, edited by Cees Kallenberg, which can be found online at http://arthritis-research.com/series/vasculitis
This series forms part of a special collection of reviews covering major autoimmune rheumatic diseases, available at: http://arthritis-research.com/series/abrd
Contributor Information
Peter Heeringa, Email: p.heeringa@med.umcg.nl.
Mark A Little, Email: m.little@medsch.ucl.ac.uk.
Acknowledgements
PH is supported by a grant from the Dutch Organization of Scientific Research (NWO VIDI grant 917.66.341). MAL is supported by the Higher Education Funding Council of England. The authors thank Betty S van der Veen for preparing Table 1.
References
- Berden AE, Ferrario F, Hagen EC, Jayne DR, Jennette JC, Joh K, Neumann I, Noël LH, Pusey CD, Waldherr R, Bruijn JA, Bajema IM. Histopathologic classification of ANCA-associated glomerulonephritis. J Am Soc Nephrol. 2010;21:1628–1636. doi: 10.1681/ASN.2010050477. [DOI] [PubMed] [Google Scholar]
- Fauci AS, Haynes BF, Katz P, Wolff SM. Wegener's granulomatosis: prospective clinical and therapeutic experience with 85 patients for 21 years. Ann Intern Med. 1983;98:76–85. doi: 10.7326/0003-4819-98-1-76. [DOI] [PubMed] [Google Scholar]
- Little MA, Nazar L, Farrington K. Outcome in glomerulonephritis due to systemic small vessel vasculitis: effect of functional status and non-vasculitic co-morbidity. Nephrol Dial Transplant. 2004;19:356–364. doi: 10.1093/ndt/gfg551. [DOI] [PubMed] [Google Scholar]
- Little MA, Nightingale P, Verburgh CA, Hauser T, De Groot K, Savage C, Jayne D, Harper L. Early mortality in systemic vasculitis: relative contribution of adverse events and active vasculitis. Ann Rheum Dis. 2010;69:1036–1043. doi: 10.1136/ard.2009.109389. [DOI] [PubMed] [Google Scholar]
- Anti neutrophil cytoplasm antibodies. Proceedings of the 1st international workshop on ANCA. Copenhagen, 1988. APMIS Suppl. 1989;6:1–49. [PubMed] [Google Scholar]
- Falk RJ, Jennette JC. Anti-neutrophil cytoplasmic autoantibodies with specificity for myeloperoxidase in patients with systemic vasculitis and idiopathic necrotizing and crescentic glomerulonephritis. N Engl J Med. 1988;318:1651–1657. doi: 10.1056/NEJM198806233182504. [DOI] [PubMed] [Google Scholar]
- Jenne DE, Tschopp J, Ludemann J, Utecht B, Gross WL. Wegener's autoantigen decoded. Nature. 1990;346:520. doi: 10.1038/346520a0. [DOI] [PubMed] [Google Scholar]
- Jennette JC, Hoidal JR, Falk RJ. Specificity of anti-neutrophil cytoplasmic autoantibodies for proteinase 3. Blood. 1990;75:2263–2264. [PubMed] [Google Scholar]
- Jennette JC, Xiao H, Falk RJ. Pathogenesis of vascular inflammation by anti-neutrophil cytoplasmic antibodies. J Am Soc Nephrol. 2006;17:1235–1242. doi: 10.1681/ASN.2005101048. [DOI] [PubMed] [Google Scholar]
- Falk RJ, Terrell RS, Charles LA, Jennette JC. Anti-neutrophil cytoplasmic autoantibodies induce neutrophils to degranulate and produce oxygen radicals in vitro. Proc Natl Acad Sci USA. 1990;87:4115–4119. doi: 10.1073/pnas.87.11.4115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciavatta DJ, Yang J, Preston GA, Badhwar AK, Xiao H, Hewins P, Nester CM, Pendergraft WF, Magnuson TR, Jennette JC, Falk RJ. Epigenetic basis for aberrant upregulation of autoantigen genes in humans with ANCA vasculitis. J Clin Invest. 2010;120:3209–3219. doi: 10.1172/JCI40034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder AH, Heeringa P, Brouwer E, Limburg PC, Kallenberg CG. Activation of granulocytes by anti-neutrophil cytoplasmic antibodies (ANCA): a Fc gamma RII-dependent process. Clin Exp Immunol. 1994;98:270–278. doi: 10.1111/j.1365-2249.1994.tb06137.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage CO, Pottinger BE, Gaskin G, Pusey CD, Pearson JD. Autoantibodies developing to myeloperoxidase and proteinase 3 in systemic vasculitis stimulate neutrophil cytotoxicity toward cultured endothelial cells. Am J Pathol. 1992;141:335–342. [PMC free article] [PubMed] [Google Scholar]
- Heeringa P, Brouwer E, Tervaert JW, Weening JJ, Kallenberg CG. Animal models of anti-neutrophil cytoplasmic antibody associated vasculitis. Kidney Int. 1998;53:253–263. doi: 10.1046/j.1523-1755.1998.00743.x. [DOI] [PubMed] [Google Scholar]
- Esnault VL, Mathieson PW, Thiru S, Oliveira DB, Martin-Lockwood C. Autoantibodies to myeloperoxidase in brown Norway rats treated with mercuric chloride. Lab Invest. 1992;67:114–120. [PubMed] [Google Scholar]
- Mathieson PW, Thiru S, Oliveira DB. Mercuric chloride-treated brown Norway rats develop widespread tissue injury including necrotizing vasculitis. Lab Invest. 1992;67:121–129. [PubMed] [Google Scholar]
- Brouwer E, Huitema MG, Klok PA, de Weerd H, Tervaert JW, Weening JJ, Kallenberg CG. Antimyeloperoxidase-associated proliferative glomerulonephritis: an animal model. J Exp Med. 1993;177:905–914. doi: 10.1084/jem.177.4.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heeringa P, Brouwer E, Klok PA, Huitema MG, Van Den Born J, Weening JJ, Kallenberg CG. Autoantibodies to myeloperoxidase aggravate mild anti-glomerular-basement-membrane-mediated glomerular injury in the rat. Am J Pathol. 1996;149:1695–1706. [PMC free article] [PubMed] [Google Scholar]
- Xiao H, Heeringa P, Hu P, Liu Z, Zhao M, Aratani Y, Maeda N, Falk RJ, Jennette JC. Antineutrophil cytoplasmic autoantibodies specific for myeloperoxidase cause glomerulonephritis and vasculitis in mice. J Clin Invest. 2002;110:955–963. doi: 10.1172/JCI15918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little MA, Smyth CL, Yadav R, Ambrose L, Cook HT, Nourshargh S, Pusey CD. Antineutrophil cytoplasm antibodies directed against myeloperoxidase augment leukocyte-microvascular interactions in vivo. Blood. 2005;106:2050–2058. doi: 10.1182/blood-2005-03-0921. [DOI] [PubMed] [Google Scholar]
- Xiao H, Ciavatta DJ, Zeng Y, Johnson LA, De Villena FP, Falk R, Jennette JC. Genetic control of the severity of experimental anti-MPO necrotizing and crescentic glomerulonephritis. APMIS. 2009;117(suppl 127):90 (abstract). [Google Scholar]
- Schreiber A, Xiao H, Falk RJ, Jennette JC. Bone marrow-derived cells are sufficient and necessary targets to mediate glomerulonephritis and vasculitis induced by anti-myeloperoxidase antibodies. J Am Soc Nephrol. 2006;17:3355–3364. doi: 10.1681/ASN.2006070718. [DOI] [PubMed] [Google Scholar]
- van der Veen BS, Heeringa P. ANCA-small vessel vasculitides: what have we (not yet) learned from animal models? APMIS Suppl. 2009. pp. 21–26. [DOI] [PubMed]
- Xiao H, Heeringa P, Liu Z, Huugen D, Hu P, Maeda N, Falk RJ, Jennette JC. The role of neutrophils in the induction of glomerulonephritis by anti-myeloperoxidase antibodies. Am J Pathol. 2005;167:39–45. doi: 10.1016/S0002-9440(10)62951-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huugen D, Xiao H, van Esch A, Falk RJ, Peutz-Kootstra CJ, Buurman WA, Tervaert JW, Jennette JC, Heeringa P. Aggravation of anti-myeloperoxidase antibody-induced glomerulonephritis by bacterial lipopolysaccharide: role of tumor necrosis factor-alpha. Am J Pathol. 2005;167:47–58. doi: 10.1016/S0002-9440(10)62952-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers SA, van der Veen BS, O'Sullivan KM, Gan PY, Ooi JD, Heeringa P, Satchell SC, Mathieson PW, Saleem MA, Visvanathan K, Holdsworth SR, Kitching AR. Intrinsic renal cell and leukocyte-derived TLR4 aggravate experimental anti-MPO glomerulonephritis. Kidney Int. 2010;78:1263–1274. doi: 10.1038/ki.2010.327. [DOI] [PubMed] [Google Scholar]
- Nolan SL, Kalia N, Nash GB, Kamel D, Heeringa P, Savage CO. Mechanisms of ANCA-mediated leukocyte-endothelial cell interactions in vivo. J Am Soc Nephrol. 2008;19:973–984. doi: 10.1681/ASN.2007111166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuligowski MP, Kwan RY, Lo C, Wong C, James WG, Bourges D, Ooi JD, Abeynaike LD, Hall P, Kitching AR, Hickey MJ. Antimyeloperoxidase antibodies rapidly induce alpha-4-integrin-dependent glomerular neutrophil adhesion. Blood. 2009;113:6485–6494. doi: 10.1182/blood-2008-12-192617. [DOI] [PubMed] [Google Scholar]
- Xiao H, Schreiber A, Heeringa P, Falk RJ, Jennette JC. Alternative complement pathway in the pathogenesis of disease mediated by anti-neutrophil cytoplasmic autoantibodies. Am J Pathol. 2007;170:52–64. doi: 10.2353/ajpath.2007.060573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber A, Xiao H, Jennette JC, Schneider W, Luft FC, Kettritz R. C5a receptor mediates neutrophil activation and ANCA-induced glomerulonephritis. J Am Soc Nephrol. 2009;20:289–298. doi: 10.1681/ASN.2008050497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huugen D, van Esch A, Xiao H, Peutz-Kootstra CJ, Buurman WA, Tervaert JW, Jennette JC, Heeringa P. Inhibition of complement factor C5 protects against anti-myeloperoxidase antibody-mediated glomerulonephritis in mice. Kidney Int. 2007;71:646–654. doi: 10.1038/sj.ki.5002103. [DOI] [PubMed] [Google Scholar]
- Little MA, Bhangal G, Smyth CL, Nakada MT, Cook HT, Nourshargh S, Pusey CD. Therapeutic effect of anti-TNF-alpha antibodies in an experimental model of anti-neutrophil cytoplasm antibody-associated systemic vasculitis. J Am Soc Nephrol. 2006;17:160–169. doi: 10.1681/ASN.2005060616. [DOI] [PubMed] [Google Scholar]
- Schreiber A, Rolle S, Peripelittchenko L, Rademann J, Schneider W, Luft FC, Kettritz R. Phosphoinositol 3-kinase-gamma mediates antineutrophil cytoplasmic autoantibody-induced glomerulonephritis. Kidney Int. 2010;77:118–128. doi: 10.1038/ki.2009.420. [DOI] [PubMed] [Google Scholar]
- Kettritz R, Schreiber A, Luft FC, Haller H. Role of mitogen-activated protein kinases in activation of human neutrophils by antineutrophil cytoplasmic antibodies. J Am Soc Nephrol. 2001;12:37–46. doi: 10.1681/ASN.V12137. [DOI] [PubMed] [Google Scholar]
- Polzer K, Soleiman A, Baum W, Axmann R, Distler J, Redlich K, Kilian A, Kronke G, Schett G, Zwerina J. Selective p38MAPK isoform expression and activation in antineutrophil cytoplasmatic antibody-associated crescentic glomerulonephritis: role of p38MAPKalpha. Ann Rheum Dis. 2008;67:602–608. doi: 10.1136/ard.2007.077263. [DOI] [PubMed] [Google Scholar]
- van der Veen BS, Chen M, Müller R, van Timmeren MM, Petersen AH, Lee PA, Satchell SC, Mathieson PW, Saleem MA, Stegeman CA, Zwerina J, Molema G, Heeringa P. Effects of p38MAPK inhibition on ANCA pathogenicity in vitro and in vivo. Ann Rheum Dis. 2011;70:356–365. doi: 10.1136/ard.2010.129106. [DOI] [PubMed] [Google Scholar]
- Hewins P, Williams JM, Wakelam MJ, Savage CO. Activation of Syk in neutrophils by antineutrophil cytoplasm antibodies occurs via Fcgamma receptors and CD18. J Am Soc Nephrol. 2004;15:796–808. doi: 10.1097/01.ASN.0000113241.98702.77. [DOI] [PubMed] [Google Scholar]
- van Timmeren MM, van der Veen BS, Stegeman CA, Petersen AH, Hellmark T, Collin M, Heeringa P. IgG glycan hydrolysis attenuates ANCA-mediated glomerulonephritis. J Am Soc Nephrol. 2010;21:1103–1114. doi: 10.1681/ASN.2009090984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collin M, Shannon O, Bjorck L. IgG glycan hydrolysis by a bacterial enzyme as a therapy against autoimmune conditions. Proc Natl Acad Sci USA. 2008;105:4265–4270. doi: 10.1073/pnas.0711271105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfister H, Ollert M, Frohlich LF, Quintanilla-Martinez L, Colby TV, Specks U, Jenne DE. Antineutrophil cytoplasmic autoantibodies against the murine homolog of proteinase 3 (Wegener autoantigen) are pathogenic in vivo. Blood. 2004;104:1411–1418. doi: 10.1182/blood-2004-01-0267. [DOI] [PubMed] [Google Scholar]
- van der Geld YM, Hellmark T, Selga D, Heeringa P, Huitema MG, Limburg PC, Kallenberg CG. Rats and mice immunised with chimeric human/mouse proteinase 3 produce autoantibodies to mouse Pr3 and rat granulocytes. Ann Rheum Dis. 2007;66:1679–1682. doi: 10.1136/ard.2006.064626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Primo VC, Marusic S, Franklin CC, Goldmann WH, Achaval CG, Smith RN, Arnaout MA, Nikolic B. Anti-PR3 immune responses induce segmental and necrotizing glomerulonephritis. Clin Exp Immunol. 2010;159:327–337. doi: 10.1111/j.1365-2249.2009.04072.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franssen CF, Stegeman CA, Kallenberg CG, Gans RO, De Jong PE, Hoorntje SJ, Tervaert JW. Antiproteinase 3- and antimyeloperoxidase-associated vasculitis. Kidney Int. 2000;57:2195–2206. doi: 10.1046/j.1523-1755.2000.00080.x. [DOI] [PubMed] [Google Scholar]
- Molema G. Heterogeneity in endothelial responsiveness to cytokines, molecular causes, and pharmacological consequences. Semin Thromb Hemost. 2010;36:246–264. doi: 10.1055/s-0030-1253448. [DOI] [PubMed] [Google Scholar]
- van der Veen BS, Petersen AH, Belperio JA, Satchell SC, Mathieson PW, Molema G, Heeringa P. Spatiotemporal expression of chemokines and chemokine receptors in experimental anti-myeloperoxidase antibody-mediated glomerulonephritis. Clin Exp Immunol. 2009;158:143–153. doi: 10.1111/j.1365-2249.2009.03993.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lind van Wijngaarden RA, van Rijn L, Hagen EC, Watts RA, Gregorini G, Tervaert JW, Mahr AD, Niles JL, de Heer E, Bruijn JA, Bajema IM. Hypotheses on the etiology of antineutrophil cytoplasmic autoantibody associated vasculitis: the cause is hidden, but the result is known. Clin J Am Soc Nephrol. 2008;3:237–252. doi: 10.2215/CJN.03550807. [DOI] [PubMed] [Google Scholar]
- Shoenfeld Y. Idiotypic induction of autoimmunity: a new aspect of the idiotypic network. FASEB J. 1994;8:1296–1301. doi: 10.1096/fasebj.8.15.8001742. [DOI] [PubMed] [Google Scholar]
- Pendergraft WF III, Preston GA, Shah RR, Tropsha A, Carter CW Jr, Jennette JC, Falk RJ. Autoimmunity is triggered by cPR-3(105-201), a protein complementary to human autoantigen proteinase-3. Nat Med. 2004;10:72–79. doi: 10.1038/nm968. [DOI] [PubMed] [Google Scholar]
- Kain R, Exner M, Brandes R, Ziebermayr R, Cunningham D, Alderson CA, Davidovits A, Raab I, Jahn R, Ashour O, Spitzauer S, Sunder-Plassmann G, Fukuda M, Klemm P, Rees AJ, Kerjaschki D. Molecular mimicry in pauciimmune focal necrotizing glomerulonephritis. Nat Med. 2008;14:1088–1096. doi: 10.1038/nm.1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kallenberg CG, Stegeman CA, Heeringa P. Autoantibodies vex the vasculature. Nat Med. 2008;14:1018–1019. doi: 10.1038/nm1008-1018. [DOI] [PubMed] [Google Scholar]
- Weidner S, Neupert W, Goppelt-Struebe M, Rupprecht HD. Antineutrophil cytoplasmic antibodies induce human monocytes to produce oxygen radicals in vitro. Arthritis Rheum. 2001;44:1698–1706. doi: 10.1002/1529-0131(200107)44:7<1698::AID-ART294>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- Ralston DR, Marsh CB, Lowe MP, Wewers MD. Antineutrophil cytoplasmic antibodies induce monocyte IL-8 release. Role of surface proteinase-3, alpha1-antitrypsin, and Fcgamma receptors. J Clin Invest. 1997;100:1416–1424. doi: 10.1172/JCI119662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidner S, Carl M, Riess R, Rupprecht HD. Histologic analysis of renal leukocyte infiltration in antineutrophil cytoplasmic antibody-associated vasculitis: importance of monocyte and neutrophil infiltration in tissue damage. Arthritis Rheum. 2004;50:3651–3657. doi: 10.1002/art.20607. [DOI] [PubMed] [Google Scholar]
- Xiao H, Heeringa P, Hu PQ, Liu Z, Falk RJ, Jennette JC. Neutrophils but not T lymphocytes are important in the induction of necrotizing and crescentic glomerulonephritis by antineutrophil cytoplasmic autoantibodies specific for myeloperoxidase (MPO-ANCA) in mice. J Am Soc Nephrol. 2003;14:634A–634A (abstract). [Google Scholar]
- Ruth AJ, Kitching AR, Kwan RY, Odobasic D, Ooi JD, Timoshanko JR, Hickey MJ, Holdsworth SR. Anti-neutrophil cytoplasmic antibodies and effector CD4+ cells play nonredundant roles in anti-myeloperoxidase crescentic glomerulonephritis. J Am Soc Nephrol. 2006;17:1940–1949. doi: 10.1681/ASN.2006020108. [DOI] [PubMed] [Google Scholar]
- Gan PY, Steinmetz OM, Tan DS, O'Sullivan KM, Ooi JD, Iwakura Y, Kitching AR, Holdsworth SR. Th17 cells promote autoimmune anti-myeloperoxidase glomerulonephritis. J Am Soc Nephrol. 2010;21:925–931. doi: 10.1681/ASN.2009070763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogueira E, Hamour S, Sawant D, Henderson S, Mansfield N, Chavele KM, Pusey CD, Salama AD. Serum IL-17 and IL-23 levels and autoantigen-specific Th17 cells are elevated in patients with ANCA-associated vasculitis. Nephrol Dial Transplant. 2010;25:2209–2217. doi: 10.1093/ndt/gfp783. [DOI] [PubMed] [Google Scholar]
- Holle JU, Wieczorek S, Gross WL. Genetic association studies in ANCA-associated vasculitides: what we have learnt so far and what needs to be done in the future. Clin Exp Rheumatol. 2010;28(1 Suppl 57):5–7. [PubMed] [Google Scholar]
- Little MA, Smyth L, Salama AD, Mukherjee S, Smith J, Haskard D, Nourshargh S, Cook HT, Pusey CD. Experimental autoimmune vasculitis: an animal model of anti-neutrophil cytoplasmic autoantibody-associated systemic vasculitis. Am J Pathol. 2009;174:1212–1220. doi: 10.2353/ajpath.2009.080458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura Y, Zhou L, Miwa T, Song WC. Genetic and therapeutic targeting of properdin in mice prevents complement-mediated tissue injury. J Clin Invest. 2010;120:3545–3554. doi: 10.1172/JCI41782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thurman JM, Royer PA, Ljubanovic D, Dursun B, Lenderink AM, Edelstein CL, Holers VM. Treatment with an inhibitory monoclonal antibody to mouse factor B protects mice from induction of apoptosis and renal ischemia/reperfusion injury. J Am Soc Nephrol. 2006;17:707–715. doi: 10.1681/ASN.2005070698. [DOI] [PubMed] [Google Scholar]