

Summary
The global transcriptional regulator Hha of Escherichia coli controls biofilm formation and virulence. Previously, we showed that Hha decreases initial biofilm formation; here, we engineered Hha for two goals: to increase biofilm dispersal and to reduce biofilm formation. Using random mutagenesis, Hha variant Hha13D6 (D22V, L40R, V42I and D48A) was obtained that causes nearly complete biofilm dispersal (96%) by increasing apoptosis without affecting initial biofilm formation. Hha13D6 caused cell death probably by the activation of proteases since Hha‐mediated dispersal was dependent on protease HslV. Hha variant Hha24E9 (K62X) was also obtained that decreased biofilm formation by inducing gadW, glpT and phnF but that did not alter biofilm dispersal. Hence, Hha may be engineered to influence both biofilm dispersal and formation.
Introduction
Hha (high haemolysin activity, 72 aa) belongs to the Hha‐YmoA family of low‐molecular‐mass proteins (about 8 kDa) that regulate many genes in Gram‐negative bacteria (Madrid et al., 2007); Hha was identified as reducing haemolysin production by repressing the hly operon of Escherichia coli (Godessart et al., 1988). However, Hha does not bind specific DNA sequences; instead, Hha has a protein partner, H‐NS, and this complex binds specific sequences of the hly regulatory operon (Madrid et al., 2002) as well as 162 genes controlled by the Hha‐H‐NS complex (Baños et al., 2009). Hha also represses the pathogenicity locus of enterocyte effacement (LEE) set of operons in enterohemorrhagic E. coli by repressing transcription of ler, which encodes the activator of LEE (Sharma et al., 2005). We found that Hha is induced 30‐fold in E. coli biofilms relative to planktonic cells (Ren et al., 2004a) and that Hha decreases initial biofilm formation by repressing the transcription of rare codon tRNAs and by repressing transcription of fimbrial genes (García‐Contreras et al., 2008). Hha is also toxic and leads to cell lysis and biofilm dispersal due to activation of prophage lytic genes appY and rzpD of DLP12 and alpA and yfjZ of CP4‐57, and due to the induction of protease ClpXP (García‐Contreras et al., 2008). In addition, Hha induces excision of prophages CP4‐57 and DLP‐12 of E. coli (Wang et al., 2009). Hence, Hha is a global transcriptional regulator in modulating cell physiology.
Biofilms consist of a complex heterogeneity of cells (Stewart and Franklin, 2008) that are the result of diverse signals and regulatory networks during biofilm development (Prüßet al., 2006). The final stage of the biofilm cycle is the dispersal of cells from the biofilm into the environment (Kaplan, 2010). Like other stages of biofilm development, biofilm dispersal is a highly regulated process involving many sensory circuits (Karatan and Watnick, 2009). There have been several biofilm dispersal signals proposed including the auto‐inducing peptide of the agr quorum sensing system of Staphylococcus aureus that triggers biofilm detachment (Boles and Horswill, 2008) and changes in carbon sources (Sauer et al., 2004). To date, little is known about the intracellular molecular mechanisms of bacterial biofilm dispersal (Kaplan, 2010); however, phosphodiesterases can decrease concentrations of the secondary messenger cyclic diguanosine‐monophosphate (c‐di‐GMP) (Karatan and Watnick, 2009), which results in increased motility (Kaplan, 2010), and surfactants such as rhamnolipid produced by Pseudomonas aeruginosa can cause dispersal of biofilms (Boles et al., 2005). In addition, cell lysis may accompany the dispersal process; hence, biofilm dispersal in P. aeruginosa may be achieved through prophage‐mediated cell death (Webb et al., 2003) and in Pseudoalteromonas tunicata biofilm dispersal involves autolysis via AlpP (Mai‐Prochnow et al., 2006).
To control biofilm formation, nitric oxide was investigated as a dispersal agent not only for single‐species biofilms including Gram‐negative and Gram‐positive bacteria and yeasts but also for multi‐species biofilms of clinical and industrial relevance (Barraud et al., 2009). Also, T7 bacteriophage was engineered so that during bacteriophage infection dispersin B of Actinobacillus actinomycetemcomitans is produced to hydrolyse the glycosidic linkages of polymeric β‐1,6‐N‐acetyl‐d‐glucosamine found in the biofilm matrix (Lu and Collins, 2007).
Protein engineering and recombinant engineering are promising strategies to control biofilm formation, but they have not been applied for promoting biofilm dispersal. Previously, we created the first synthetic circuit for controlling biofilm formation via an external signal by manipulating indole concentrations via toluene o‐monooxygenase in a consortium of Pseudomonas fluorescens and E. coli (Lee et al., 2007a). In addition, we controlled E. coli biofilm formation by rewiring the quorum sensing regulator SdiA; upon addition of the extracellular signal N‐acylhomoserine lactone, biofilm formation was increased with SdiA variant 2D10, and SdiA variant 1E11 increased concentrations of the biofilm inhibitor indole and thereby reduced biofilm formation (Lee et al., 2009). Furthermore, we engineered global regulator H‐NS to control biofilm formation via prophage excision and cell death (Hong et al., 2010); however, the engineered H‐NS has no effect on biofilm dispersal. These results showed that bacterial biofilm formation may be controlled by manipulating key regulatory proteins and enzymes.
In this study, protein engineering was used to rewire the global transcriptional regulator Hha to control biofilms. The aims of this study were to engineer Hha to enhance biofilm dispersal, to engineer Hha to reduce biofilm formation, and to investigate the mechanism by which the Hha variants influence biofilm formation. Utilizing whole‐transcriptome analyses, random mutagenesis, saturation mutagenesis, site‐directed mutagenesis, double mutations, quantitative real‐time reverse transcription PCR (qRT‐PCR) and quantitative PCR (qPCR), we show Hha may be rewired to promote biofilm dispersal by interacting with protease HslV. We also show that Hha may be engineered to decrease biofilm formation.
Results
Random mutagenesis of Hha for biofilm dispersal
The E. coli BW25113 wild‐type strain undergoes natural biofilm dispersal in 96‐well plates after 31 h (results not shown), since the wild‐type strain produces Hha from its chromosome. Here, as expected, biofilm cells that lack Hha (hha/pCA24N) did not disperse at all time points tested (24, 32 and 48 h) (Fig. 1A). Therefore, Hha is important for biofilm dispersal as we showed previously (García‐Contreras et al., 2008); however, wild‐type Hha does not disperse biofilms after 32 and 48 h (Fig. 1A). Hence, we focused on creating Hha variants that could disperse biofilms from 24 to 48 h since wild‐type Hha induced biofilm dispersal somewhat in early biofilms but not in mature biofilms (Fig. 1A).
Figure 1.
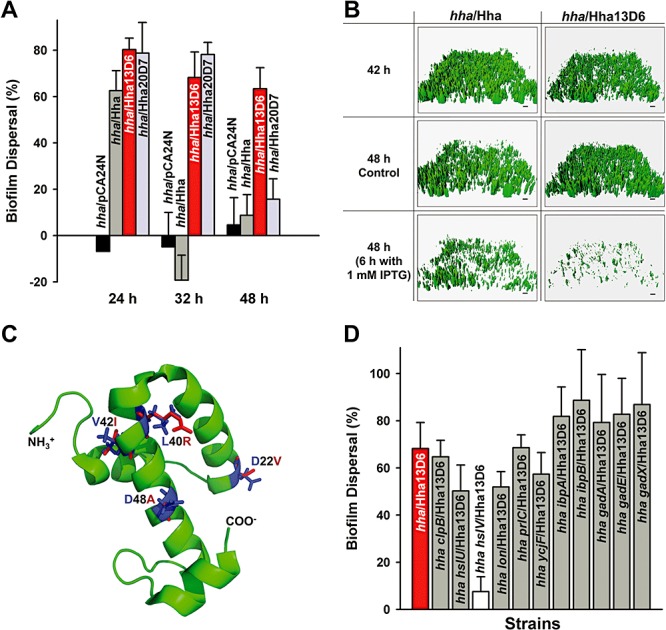
Biofilm dispersal with Hha13D6. Biofilm dispersal in 96‐well plates for BW25113 hha producing Hha13D6 and Hha20D7 in Luria‐Bertani (LB) glucose (0.2%) at 37°C (A). Biofilm dispersal was quantified by subtracting the normalized biofilm with Hha and its variants produced at 24, 32 and 48 h (8 h after adding 1 mM IPTG) from the normalized biofilm without Hha and its variants produced at 24, 32 and 48 h (no IPTG addition). Each data point is the average of at least two independent cultures, and one standard deviation is shown. Biofilm dispersal in flow‐cells for BW25113 hha producing Hha13D6 from pCA24N (B). Biofilms were formed on glass surfaces in flow‐cells for 42 h then 1 mM IPTG was added for 6 h to induce dispersal (control is no IPTG addition). Scale bar represents 10 µm. Modelled protein structure of Hha13D6 (C). Substituted residues of Hha13D6 (D22V, L40R, V42I and D48A) are shown in red, while the original residues were shown in blue. Impact of ClpB, HslU, HslV, Lon, PrlC, YcjF, IbpA, IbpB, GadA, GadE and GadX on Hha13D6‐mediated biofilm dispersal (D). Biofilm dispersal for cells producing Hha13D6 in LB glucose at 37°C after 32 h (8 h with 1 mM IPTG) in the following hosts: BW25113 hha (hha), BW25113 hha clpB (hha clpB), BW25113 hha hslU (hha hslU), BW25113 hha hslV (hha hslV), BW25113 hha lon (hha lon), BW25113 hha prlC (hha prlC), BW25113 hha ycjF (hha ycjF), BW25113 hha ibpA (hha ibpA), BW25113 hha ibpB (hha ibpB), BW25113 hha gadA (hha gadA), BW25113 hha gadE (hha gadE) and BW25113 hha gadX (hha gadX). Each data point is the average of at least two independent cultures, and one standard deviation is shown.
To engineer Hha for biofilm dispersal in the absence of effects related to wild‐type Hha and the quorum sensing signal indole (Lee et al., 2007b) produced by tryptophanase, we used a BW25113 hha tnaA double mutant for screening after error‐prone PCR (epPCR) of hha. The average error rate was determined to be 0.8% by sequencing three random hha genes. A total of 2256 colonies were screened in polystyrene 96‐well plates for robust biofilm formation in rich medium after 24 h followed by enhanced biofilm dispersal after 8 h of induction of hha with random mutations. Two Hha variants were identified with up to fivefold greater biofilm dispersal (wild‐type Hha had 10% biofilm dispersal under these conditions). Hha dispersal variant Hha13D6 had four amino acid replacements at D22V, L40R, V42I and D48A, and Hha20D7 had two replacements at D48G and R50C (Table 1). To eliminate any possible chromosomal mutation effects, all the pCA24N‐hha plasmids identified during the initial screens were re‐transformed into BW25113 hha tnaA, and the changes in biofilm dispersal were confirmed; hence, the phenotype changes in biofilm dispersal are due to the changes in the hha gene on the plasmids.
Table 1.
Protein sequences of the Hha variants from screening altered biofilm dispersal and biofilm formation.
| Hha variants | Replacements |
|---|---|
| Hha dispersal variants | |
| Hha13D6 | D22V, L40R, V42I, D48A |
| Hha20D7 | D48G, R50C |
| Hha saturation mutagenesis variant | |
| HhaSM2C1 | D22M, L40R, V42I, D48A |
| Hha biofilm variants | |
| Hha1D8 | L6I, E34V, N38E, K58E, K62X |
| Hha1D9 | E53X |
| Hha12H6 | V67E |
| Hha13B3 | Q19R, N57D |
| Hha24E9 | K62X |
| Hha dispersal‐biofilm combined variant | |
| Hha13D6‐24E9 | D22V, L40R, V42I, D48A, K62X |
X indicates termination.
The dispersal variants were transformed into BW25113 hha for further study in only an hha deletion background. Corroborating the results with BW25113 hha tnaA, Hha20D7 in BW25113 hha had enhanced biofilm dispersal at 24 and 32 h, and cells with Hha13D6 had enhanced biofilm dispersal at 24, 32 and 48 h, whereas wild‐type Hha caused little dispersal after 24 h (Fig. 1A). These results show that Hha may be rewired to enhance biofilm dispersal at both early and mature biofilms. Importantly, cells producing both the wild‐type Hha and engineered Hha13D6 formed similar biofilms at 24 h (Fig. 2A); therefore, Hha13D6 does not affect biofilm formation but instead controls only biofilm dispersal.
Figure 2.
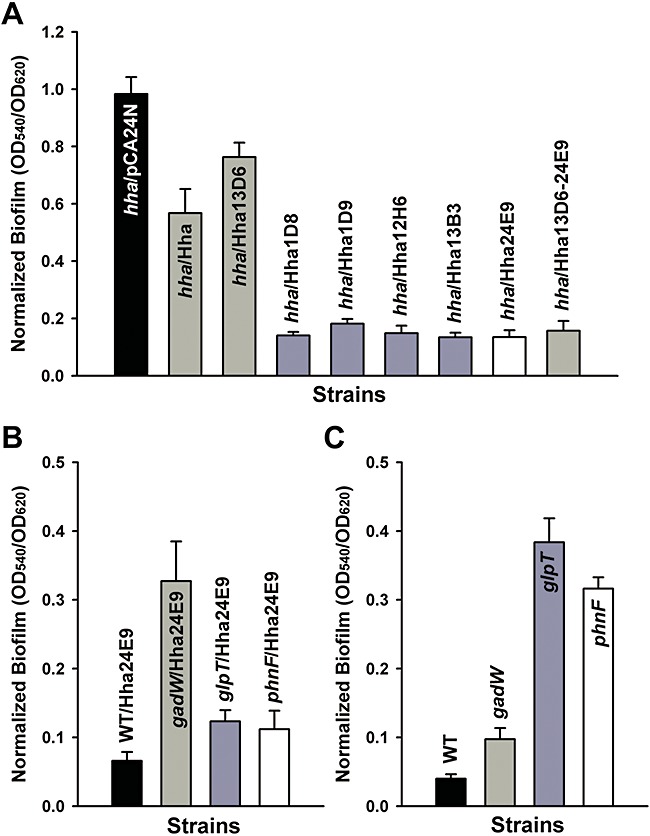
Engineering Hha for reduction in biofilm formation. Normalized biofilm formation for BW25113 hha (hha) producing Hha13D6, Hha1D8, Hha1D9, Hha12H6, Hha13B3, Hha24E9 and Hha13D6‐24E9 from pCA24N using 1 mM IPTG in LB glucose after 24 h at 37°C (A), and for BW25113 (WT), BW25113 gadW (gadW), BW25113 glpT (glpT), BW25113 phnF (phnF) producing Hha24E9 after 24 h (B). Normalized biofilm formation for BW25113 gadW, BW25113 glpT and BW25113 phnF after 7 h (C). Each data point is the average of at least 12 replicate wells from two independent cultures, and one standard deviation is shown.
To study biofilm dispersal by Hha13D6 in a more rigorous manner, we conducted flow‐cell biofilm dispersal experiments in which biofilms were formed for 42 h followed by induction of Hha for 6 h. We chose 42–48 h for the flow‐cell work since we found wild‐type BW25113 disperses naturally after 42 h (results not shown). As expected based on its enhanced biofilm dispersal in 96‐well plates, Hha13D6 induced more biofilm dispersal in the flow‐cell relative to wild‐type Hha (Fig. 1B). comstat statistical analysis (Table 2) shows that 97% of the biomass was removed with Hha13D6, and the mean thickness (96%) and the surface coverage (96%) were also reduced dramatically at 48 h with isopropyl‐β‐d‐thiogalactopyranoside (IPTG) addition compared with 48 h biofilms without IPTG. Overproducing wild‐type Hha removed 35% of the biofilms, and cells producing both Hha and Hha13D6 had similar amounts of biofilm initially at 42 h (Fig. 1B). Therefore, both static and flow‐cell biofilm experiments confirm that Hha13D6 was engineered to enhance biofilm dispersal dramatically.
Table 2.
comstat analysis for biofilms producing wild‐type Hha (BW25113 hha/pCA24N‐hha/pCM18) and Hha13D6 (BW25113 hha/pCA24N‐hha13D6/pCM18) in flow‐cells.
| comstat values | Time | hha/Hha | hha/Hha13D6 |
|---|---|---|---|
| Biomass (µm3 µm−2) | 42 h | 6 ± 1 | 5 ± 1 |
| 48 h | 4.0 ± 0.6 | 7 ± 3 | |
| 48 h (6 h with 1 mM IPTG) | 3 ± 3 | 0.2 ± 0.2 | |
| Surface coverage (%) | 42 h | 16 ± 3 | 15 ± 5 |
| 48 h | 13 ± 4 | 19 ± 6 | |
| 48 h (6 h with 1 mM IPTG) | 8 ± 7 | 0.8 ± 0.5 | |
| Mean thickness (µm) | 42 h | 10 ± 3 | 8 ± 2 |
| 48 h | 7 ± 1 | 11 ± 4 | |
| 48 h (6 h with 1 mM IPTG) | 5 ± 4 | 0.4 ± 0.4 | |
| Roughness coefficient | 42 h | 1.1 ± 0.2 | 1.2 ± 0.2 |
| 48 h | 1.4 ± 0.1 | 1.1 ± 0.3 | |
| 48 h (6 h with 1 mM IPTG) | 1.6 ± 0.3 | 1.95 ± 0.03 |
Biofilm parameters were measured in LB glucose (0.2%) medium after 48 h (6 h with 1 mM IPTG or no IPTG addition as a control) at 37°C.
Saturation mutagenesis at position D22, L40, V42 and D48 of Hha
Since Hha13D6 has four amino acid replacements (D22V, L40R, V42I and D48A) (Fig. 1C), each position was investigated for its role in the increased biofilm dispersal by substituting all possible amino acids via saturation mutagenesis using both wild‐type Hha and Hha13D6. After screening 2256 colonies (282 colonies × 4 positions × 2 proteins) to ensure with a 99% probability that all possible codons were utilized at four positions (Rui et al., 2004), we identified the D22M replacement (i.e. D22M along with L40R, V42I and D48A) that causes a 40 ± 10% increase in biofilm dispersal in the static biofilm assay compared with no IPTG addition (HhaSM2C1, Table 1); although this variant causes the cells to undergo less dispersal than Hha13D6. We did not obtain any interesting biofilm dispersal variants using wild‐type Hha. Therefore, the biofilm dispersal phenotype arises from the interaction of several amino acid replacements.
Hha13D6 increases cell lysis
Hha is toxic and is part of a putative toxin–antitoxin pair with antitoxin TomB (García‐Contreras et al., 2008). Therefore, we assayed the toxicity of the engineered Hha three ways and found Hha13D6 is substantially more toxic than wild‐type Hha. Hha13D6 repressed cell growth fourfold (Fig. 3A), decreased cell viability by 10 000‐fold (Fig. 3B), and increased cell lysis by an order of magnitude (Fig. 3C) at early times. Longer incubation times resulted in restoring both cell growth and viability for cells producing Hha13D6, which is probably due to an increase in resistance (Smith and Romesberg, 2007) to the Hha13D6 toxicity during the extended incubation.
Figure 3.
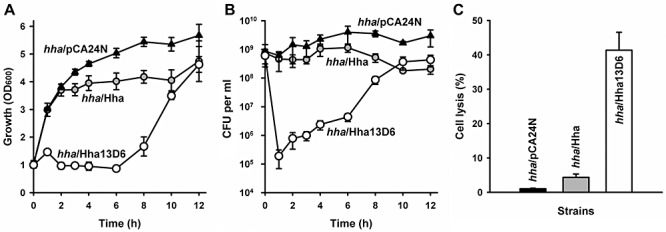
Hha13D6 causes cell lysis. Cell growth (A), CFU (B) and cell lysis (C) of Hha13D6 produced in BW25113 hha. Cells were grown in LB glucose at 37°C until a turbidity of 1, then 1 mM IPTG was added. Cell lysis was quantified by measuring extracellular DNA at 4 h after 1 mM IPTG addition. Two independent cultures were used.
HslV is important for biofilm dispersal with Hha13D6
Since whole‐transcriptome profiling yields insights into the mechanism of biofilm formation and dispersal (Wood, 2009), we investigated how overproducing Hha13D6 regulates gene expression during biofilm dispersal. Glass wool has been used by us as a substratum for biofilm formation for whole‐transcriptome analysis for E. coli (Ren et al., 2004a; Domka et al., 2007) and has been used for P. aeruginosa (Steyn et al., 2001; Ueda and Wood, 2009), since it provides a large surface to volume ratio which promotes a large amount of biofilm biomass. The results from whole‐transcriptome studies with glass wool have also been corroborated with 96‐well plate assays (Domka et al., 2007). Hence, in this experiment, biofilms were formed on glass wool for 16 h followed by 1 h of production of Hha13D6 and wild‐type Hha, and gene expression was compared.
Hha13D6 induced acid resistance genes (gadABCEX and hdeABD) and several heat‐shock genes including protease genes (clpB, hslOR, hslUV, htpG, ibpAB, lon, prlC and ycjF) compared with wild‐type Hha (Table S1). These results are consistent with our previous results with wild‐type Hha in that it increased production of the proteases ClpXP and Lon (García‐Contreras et al., 2008). Corroborating these results, qRT‐PCR confirmed that overproducing Hha13D6 induced the transcription of gadA by 1.6‐fold, gadE by 1.7‐fold, gadX by 1.5‐fold, clpB by 9.2‐fold, hslU by 3.3‐fold, hslV by 1.6‐fold, ibpA by 1.6‐fold, ibpB by 2.7‐fold, lon by 1.8‐fold, prlC by 3.2‐fold and ycjF by 1.6‐fold. ClpB refolds stress‐damaged proteins with DnaK (Hsp70) (Mogk et al., 2008) and has ATPase activity (Woo et al., 1992), and IbpAB facilitates refolding of denatured proteins by forming a functional triad with ClpB and DnaK (Mogk et al., 2003). ATP‐dependent proteases such as HslUV and Lon are responsible for the degradation of several regulatory proteins and abnormal proteins during stress, which controls many cellular processes (Schmidt et al., 2009). For example, overproduction of Lon inhibits translation in E. coli by degrading antitoxin YefM, which results in induction of the toxin gene yoeB (Christensen et al., 2004). PrlC cleaves peptides generated by proteases Lon, HslUV and ClpAP (Jain and Chan, 2007), and YcjF is a heat shock protein (Rasouly and Ron, 2009). Therefore, Hha13D6 may have more toxicity via the activation of heat shock (ClpB, HslUV, Lon, PrlC, YcjF, IbpAB) and acid resistance (GadAEX) systems.
To test this hypothesis, we assayed biofilm dispersal via production of Hha13D6 in hha hosts that also lack clpB, hslU, hslV, lon, prlC, ycjF, ibpA, ibpB, gadA, gadE and gadX (Fig. 1D). Deletion of hslV in the hha host reduced the level of biofilm dispersal by Hha13D6 by an order of magnitude, whereas deletion of clpB, lon, hslU, prlC or ycjF only partially decreased biofilm dispersal. In contrast, deletion of ibpA, ibpB, gadA, gadE or gadX did not reduce biofilm dispersal. Hence, HslV is important for the enhanced biofilm dispersal activity of Hha13D6. HslUV protease consists of two functional units, HslU ATPase and HslV peptidase, and although HslU stimulates proteolytic activity of HslV, HslV alone can digest certain unfolded proteins such as unfolded lactalbumin (Lee et al., 2007c). Hence, Hha13D6 enhances biofilm dispersal via proteases, primarily through HslV.
Hha13DD6 has little impact on c‐di‐GMP and swimming motility
Lower levels of c‐di‐GMP generally result in an increase in motility and induce biofilm dispersal (Kaplan, 2010). Since Hha13D6 induces biofilm dispersal, we measured swimming motility and c‐di‐GMP concentration using high‐performance liquid chromatography (HPLC) upon inducing wild‐type Hha and Hha13D6. Production of Hha13D6 reduced slightly the swimming motility of the host (halo diameter 4 ± 1 cm with Hha13D6 versus 5.6 ± 0.1 cm with wild‐type Hha). In addition, cellular c‐di‐GMP concentrations were decreased somewhat by Hha13D6 (c‐di‐GMP concentration 7 ± 3 nmol g−1 cells with Hha13D6 versus 13 ± 2 nmol g−1 cells with wild‐type Hha). Therefore, the decreased level of c‐di‐GMP by Hha13D6 may partially contribute to the greater biofilm dispersal, although Hha13D6 did not increase swimming motility. However, the small reduction in swimming motility with Hha13D6 confirms the whole‐transcriptome results that indicated the flagella operons (flgBCDEFG and fliACGHILN) were repressed (Table S1).
Random mutagenesis of Hha for biofilm formation
In an effort to identify Hha variants that cause reduced biofilm formation (rather than increased biofilm dispersal), 2656 colonies for altered biofilm formation were screened using polystyrene 96‐well plates, and five Hha variants were identified that decreased biofilm formation more than fourfold compared with wild‐type Hha (Table 1). Among the five Hha biofilm variants, three variants had truncations; hence, the amino‐terminal region of Hha is most important for controlling biofilm formation.
Among the five best biofilm variants, Hha24E9 (K62X) decreased biofilm formation fourfold compared with wild‐type Hha in BW25113 hha (Fig. 2A). To investigate how Hha24E9 represses biofilm formation, a whole‐transcriptome analysis was performed using biofilm cells of Hha24E9 versus wild‐type Hha in rich medium after 7 h (Table S2). Acid resistance (gadAEW), arginine metabolism (astABD), sulfate metabolism (cysHJ), glycerol‐3‐phosphate metabolism (glpABCKQT), phosphonate metabolism (phnACEFGJL) and outer membrane (ompW) genes were highly induced, and stress response (cspF, cpxP and bhsA) and glucose phosphotransferase system (ptsG) genes were repressed by Hha24E9. Although indole‐related genes (tnaABC) were induced, indole production does not occur in this medium due to catabolic repression. Corroborating the whole‐transcriptome results, Hha24E9 induced transcription of gadW by 26‐fold (ΔΔCT = −4.7 ± 0.2), glpT by 120‐fold (ΔΔCT = −6.9 ± 0.3) and phnF by 132‐fold (ΔΔCT = −7.0 ± 0.5).
To determine if some of the proteins encoded by these genes are related to the ability of Hha24E9 to reduce biofilm formation, we checked biofilm formation with Hha24E9 in gadW, glpT and phnF mutants and found Hha24E9 does not decrease biofilm formation in these mutants (Fig. 2B). Corroborating these results, deletion of gadW, glpT and phnF increased biofilm formation considerably after 7 h (Fig. 2C); hence, GadW, GlpT and PhnF decrease biofilm formation. Hence, the engineered Hha24E9 reduces biofilm formation by inducing acid resistance, glycerol‐3‐phosphate metabolism and phosphonate metabolism.
Combining the Hha13D6 and Hha24E9 amino acid replacements decreases biofilm formation but does not induce biofilm dispersal
Hha variants Hha13D6 (D22V, L40R, V42I and D48A) and Hha24E9 (K62X) were obtained with different phenotype screens; Hha24E9 does not induce biofilm dispersal (Fig. S1), and Hha13D6 does not decrease biofilm formation at 24 h (Fig. 2A), indicating that the Hha replacements necessary for biofilm dispersal are different from those that influence biofilm formation. To determine whether dispersal or biofilm formation could be enhanced by combining these amino acid replacements, truncation at K62 was introduced to Hha13D6 using site‐directed mutagenesis. Hha13D6‐24E9, the combined variant (Table 1), did not cause dispersal (Fig. S1) and reduced biofilm formation like Hha24E9 (Fig. 2A). These results indicate that the carboxy‐terminal region of Hha is necessary for biofilm dispersal and the amino‐terminal region is necessary for reducing biofilm formation.
Discussion
In this work, we show that the global transcriptional regulator Hha may be rewired to control biofilm dispersal, without affecting initial biofilm formation. In static biofilms, Hha13D6 removed 70% of the biofilm while there was little dispersal with wild‐type Hha (Fig. 1A), and in flow‐cells, the engineered Hha removed 96% of the biofilm (Fig. 1B), whereas wild‐type Hha removed 35% of the biofilm (Fig. 1B). Hha13D6 increased biofilm dispersal by causing apoptosis (Fig. 3C) by activating proteases (Table S1); hence, we have created one of the first protein switches to control cell lysis. Biofilm dispersal by the engineered Hha requires protease HslV since adding the hslV mutation reduced the dispersal activity of Hha13D6 (Fig. 1D).
Cell death in biofilms is an important mechanism of dispersal since it leads to the creation of voids inside the biofilm, which facilitates dispersal of the surviving cells (Webb et al., 2003). Hence, programmed cell death is a social (altruistic) activity (Wood, 2009). Cell death in biofilms is mediated by prophage (Webb et al., 2003) and autolysis proteins (Mai‐Prochnow et al., 2006). Since the biofilm matrix consists of structural proteins (Karatan and Watnick, 2009), protease activity is necessary for biofilm dispersal; for example, agr‐mediated detachment in Staphylococcus aureus biofilms does not occur in the presence of a protease inhibitor and is induced by extracellular proteases (Boles and Horswill, 2008). Also, wild‐type Hha disperses E. coli biofilms by inducing protease ClpXP that activates toxins by degrading antitoxins (García‐Contreras et al., 2008). Hence, induction of protease activity is a key mechanism of Hha13D6 to trigger biofilm dispersal by causing cell death and so by disrupting biofilm structure.
In addition to the evolution of Hha for biofilm dispersal, we also show that Hha may be reconfigured to control biofilm formation by inducing the activity of GadW, GlpT and PhnF (Fig. 2B). We showed previously that biofilm formation is connected to the acid resistance system, showing increased biofilm formation in the absence of acid resistance genes such as gadW, gadABC and hdeABD (Domka et al., 2007; Lee et al., 2007d), and GadW is dual regulator with GadX of the glutamate‐dependent decarboxylase acid‐resistance system of E. coli (Sayed et al., 2007). Hence, the engineered Hha24E9 uses the acid resistance regulatory system to inhibit biofilm formation.
We found the carboxy‐terminus of Hha is important for controlling biofilm dispersal but not for biofilm formation. Hha is a non‐specific DNA binding protein, but the Hha‐H‐NS complex allows specific binding to target DNA sequences to regulate transcription (Madrid et al., 2002). Hha consists of four α‐helices separated by a loop: helix 1 (8–16 aa), helix 2 (21–34 aa), helix 3 (37–55 aa) and helix 4 (65–69 aa), and all four helices interact with H‐NS to cause conformational changes in Hha at both the surface and in the hydrophobic core (Madrid et al., 2007). The dispersal variant Hha13D6 has amino acid replacements on helix 2 (D22V) and helix 3 (L40R, V42I and D48A) (Fig. 1C), and all three residues on helix 3 are perturbed by the N‐terminus H‐NS (García et al., 2005), suggesting that the replacements in Hha13D6 cause conformational change of Hha structure to interact with H‐NS and results in the enhanced biofilm dispersal. However, loss of C‐terminal 11 amino acids by truncation at K62 of Hha13D6 abolished the effect of the other four replacements. Since helix 4 also is highly affected by the binding of H‐NS (García et al., 2005), complete deletion of helix 4 of Hha may change the conformation of Hha and inhibit biofilm formation. In addition, whole‐transcriptome profiles of Hha13D6 (Table S1) and Hha24E9 (Table S2) support that gene regulation pathways of Hha13D6 are quite different from those of Hha24E9.
Biofilm dispersal is an essential process of biofilm cycles for disseminating cells into the environment for their survival (Kaplan, 2010), and engineering approaches to disperse biofilms, like using engineered phage (Lu and Collins, 2007), are promising (Kaplan, 2010). The current study demonstrates that genetic switches exist for both biofilm formation and dispersal, and once they are discerned, as in the case of Hha (Ren et al., 2004a; García‐Contreras et al., 2008), they may be manipulated to control bacterial activity. We envision that since biofilms are robust, i.e. biofilm cells withstand stress better than their planktonic counterparts (Singh et al., 2006), biofilms will be used for diverse applications and some of these applications will involve controlling biofilm formation including replacing some existing engineered biofilms with other engineered strains; this will require control of biofilm dispersal. This capability is important for patterning biofilms in microdevices as well as for creating sophisticated reactor systems that will be used to form bio‐refineries where various engineered strains produce a plethora of chemicals as function of position and depth in biofilms.
Experimental procedures
Bacterial strains and growth conditions
The bacterial strains and plasmids used in this study are listed in Table 3. We used the Keio collection (Baba et al., 2006) for isogenic mutants and the ASKA library (Kitagawa et al., 2005) for overexpressing genes. The gene deletions were confirmed by PCR using primers (Table S3). All strains were initially streaked from −80°C glycerol stocks on Luria–Bertani (LB) (Sambrook et al., 1989) agar plates and were cultured at 37°C in LB glucose (0.2%). Kanamycin (50 µg ml−1) was used for pre‐culturing isogenic knockout mutants, and chloramphenicol (30 µg ml−1) was used for maintaining pCA24N‐based plasmids. Genes were expressed from pCA24N using 1 mM IPTG (Sigma, St Louis, MO, USA).
Table 3.
Escherichia coli K‐12 strains and plasmids used in this study.
| Strains and plasmids | Genotype/relevant characteristics | Source |
|---|---|---|
| Strains | ||
| AG1 | recA1 endA1 gyrA96 thi‐1 hsdR17(rK‐mK+) supE44 relA1 | Kitagawa et al. (2005) |
| BW25113 | lacIqrrnBT14ΔlacZWJ16hsdR514 ΔaraBADAH33ΔrhaBADLD78 | Baba et al. (2006) |
| BW25113 hha | BW25113 ΔhhaΩ KmR | Baba et al. (2006) |
| BW25113 tnaA | BW25113 ΔtnaAΩ KmR | Baba et al. (2006) |
| BW25113 clpB | BW25113 ΔclpBΩ KmR | Baba et al. (2006) |
| BW25113 hslU | BW25113 ΔhslUΩ KmR | Baba et al. (2006) |
| BW25113 hslV | BW25113 ΔhslVΩ KmR | Baba et al. (2006) |
| BW25113 lon | BW25113 ΔlonΩ KmR | Baba et al. (2006) |
| BW25113 prlC | BW25113 ΔprlCΩ KmR | Baba et al. (2006) |
| BW25113 ycjF | BW25113 ΔycjFΩ KmR | Baba et al. (2006) |
| BW25113 gadA | BW25113 ΔgadAΩ KmR | Baba et al. (2006) |
| BW25113 gadE | BW25113 ΔgadEΩ KmR | Baba et al. (2006) |
| BW25113 gadX | BW25113 ΔgadXΩ KmR | Baba et al. (2006) |
| BW25113 ibpA | BW25113 ΔibpAΩ KmR | Baba et al. (2006) |
| BW25113 ibpB | BW25113 ΔibpBΩ KmR | Baba et al. (2006) |
| BW25113 gadW | BW25113 ΔgadWΩ KmR | Baba et al. (2006) |
| BW25113 glpT | BW25113 ΔglpTΩ KmR | Baba et al. (2006) |
| BW25113 phnF | BW25113 ΔphnFΩ KmR | Baba et al. (2006) |
| BW25113 hha tnaA | BW25113 ΔhhaΔtnaAΩ KmR | This study |
| BW25113 hha clpB | BW25113 ΔhhaΔclpBΩ KmR | This study |
| BW25113 hha hslU | BW25113 ΔhhaΔhslUΩ KmR | This study |
| BW25113 hha hslV | BW25113 ΔhhaΔhslVΩ KmR | This study |
| BW25113 hha lon | BW25113 ΔhhaΔlonΩ KmR | This study |
| BW25113 hha prlC | BW25113 ΔhhaΔprlCΩ KmR | This study |
| BW25113 hha ycjF | BW25113 ΔhhaΔycjFΩ KmR | This study |
| BW25113 hha gadA | BW25113 ΔhhaΔgadAΩ KmR | This study |
| BW25113 hha gadE | BW25113 ΔhhaΔgadEΩ KmR | This study |
| BW25113 hha gadX | BW25113 ΔhhaΔgadXΩ KmR | This study |
| BW25113 hha ibpA | BW25113 ΔhhaΔibpAΩ KmR | This study |
| BW25113 hha ibpB | BW25113 ΔhhaΔibpBΩ KmR | This study |
| Plasmids | ||
| pCA24N | CmR; lacIq, pCA24N | Kitagawa et al. (2005) |
| pCA24N‐hha | CmR; lacIq, pCA24N PT5‐lac::hha+ | Kitagawa et al. (2005) |
| pCA24N‐yddV | CmR; lacIq, pCA24N PT5‐lac::yddV+ | Kitagawa et al. (2005) |
| pCM18 | EmR, GFP+ | Hansen et al. (2001) |
| pCP20 | ApR and CmR plasmid with temperature‐sensitive replication and thermal induction of FLP synthesis | Cherepanov and Wackernagel (1995) |
KmR, CmR, EmR and ApR are kanamycin, chloramphenicol, erythromycin and ampicillin resistance respectively.
epPCR for random mutagenesis
hha from plasmid pCA24N‐hha under the control of T5‐lac promoter was mutated by epPCR as described previously (Fishman et al., 2004) using primers epPCR‐f and epPCR‐r (Table S3). The epPCR product was cloned into pCA24N‐hha using BseRI and HindIII after treating the plasmid with Antarctic phosphatase (New England Biolabs, Beverly, MA, USA). The ligation mixture was electroporated into BW25113 hha tnaA competent cells using the Gene Pulser/Pulse Controller (Bio‐Rad, Hercules, CA, USA) at 1.25 kV cm−1, 25 µF and 200 Ω.
Crystal violet biofilm assay to measure biofilm formation and biofilm dispersal
Biofilm formation was assayed in 96‐well polystyrene plates (Corning Costar, Cambridge, MA, USA) using 0.1% crystal violet staining (Fletcher, 1977). Briefly, each well was inoculated at an initial turbidity at 600 nm of 0.05 and grown without shaking for 24 h with 1 mM IPTG addition. The crystal violet dye stains both the air‐liquid interface and bottom liquid‐solid interface biofilm, and the total biofilm formation at both interfaces was measured at 540 nm, whereas cell growth was measured at 620 nm. Biofilm formation was normalized by the bacterial growth for each strain (turbidity at 620 nm) to reduce any growth effect. Similarly, for biofilm dispersal, biofilms were formed for 24 h without shaking. Then, 1 mM IPTG was added for 8 h to overproduce the Hha variants from the pCA24N vector, and biofilm formation was measured (total time 32 h). As a negative control, biofilm formation was also measured for no IPTG addition at 32 h. Biofilm dispersal was quantified by subtracting the normalized biofilm at 32 h (8 h after adding 1 mM IPTG) from the normalized biofilm at 32 h (no IPTG addition). Two independent cultures were used for each strain.
Biofilm and dispersal screening of Hha variants
In order to screen Hha variants for the altered biofilm formation, BW25113 hha tnaA strains producing Hha variants from pCA24N‐hha were grown overnight in 96‐well plates with 200 µl of medium at 250 r.p.m., the overnight cultures were diluted (1:100) in 300 µl of LB glucose, and biofilm formation with 1 mM IPTG was measured after 24 h through the crystal violet biofilm assay. As controls, BW25113 hha tnaA with empty pCA24N and pCA24N‐hha (wild‐type hha) were used. For dispersal screening of mutant hha alleles, biofilms were formed for 24 h without shaking. Then, 1 mM IPTG was added for 8 h to overexpress the hha alleles from the pCA24N, and no IPTG was added as negative control. Interesting hha alleles were re‐analysed by re‐streaking the colonies on fresh LB plates and by performing another biofilm or dispersal assay. Mutant hha alleles were sequenced using a primer seq‐f1 (Table S3). Each data point was averaged from more than 12 replicate wells (six wells from two independent cultures).
Flow‐cell biofilm experiments and image analysis
Strains were cultured with chloramphenicol (30 µg ml−1) to retain the pCA24N‐based plasmids and with erythromycin (300 µg ml−1) to maintain pCM18, which produces the green fluorescence protein (Hansen et al., 2001). Overnight cultures were diluted to a turbidity at 600 nm of 0.05 and pumped through the flow‐cell (BST model FC81, Biosurface Technologies, Bozeman, MT) at 10 ml h−1 for 2 h, and then fresh medium with chloramphenicol and erythromycin was pumped for 42 h, and 1 mM IPTG was added for 6 h to induce either wild‐type Hha or Hha13D6 for biofilm dispersal. As a negative control, no IPTG was added. The biofilms on the glass slides were visualized by exciting with an Ar laser at 488 nm (emission 510–530 nm) using a TCS SP5 scanning confocal laser microscope (Leica Microsystems, Wetzlar, Germany) with a 63× HCX PL FLUOTAR L dry objective. Nine random positions were chosen, and 25 images were taken for each position. Simulated three‐dimensional images were obtained using IMARIS software (BITplane, Zurich, Switzerland). Colour confocal flow‐cell images were converted to grey scale, and biomass, surface coverage, mean thickness and roughness coefficient were determined using comstat image‐processing software (Heydorn et al., 2000).
Saturation and site‐directed mutagenesis
Saturation mutagenesis was performed at the codons corresponding to positions D22, L40, V42 and D48 of Hha and Hha13D6. Saturation mutagenesis primers (Table S3) were designed to allow the replacement of all 20 amino acids using 32 possible codons with NNS (N is A, G, C or T, and S is G or C) (Leungsakul et al., 2006). Plasmid pCA24N‐hha and pCA24N‐hha13D6 were used as templates. PCR was performed using Pfu DNA polymerase at 95°C for 1 min, with 20 cycles of 95°C for 1 min, 55°C for 50 s and 68°C for 7 min, and a final extension of 68°C for 7 min. The constructed plasmids were electroporated into BW25113 hha after DpnI digestion of template plasmids. Similarly, site‐directed mutagenesis was performed to create a truncation at K62 of Hha13D6 to make Hha13D6‐24E9 using site‐directed mutagenesis primers (Table S3).
RNA isolation and whole‐transcriptome studies
For the whole‐transcriptome study of BW25113 hha/pCA24N‐hha13D6 versus BW25113 hha/pCA24N‐hha (biofilm dispersal), cells were grown in 250 ml for 16 h at 125 r.p.m. with 10 g of glass wool (Corning Glass Works, Corning, NY, USA) in 1 l Erlenmeyer flasks to form a robust biofilm (Ren et al., 2004a) and incubated an additional 1 h with 1 mM IPTG to induce wild‐type Hha and Hha13D6. Similarly, for the whole‐transcriptome study of BW25113 hha/pCA24N‐hha24E9 versus BW25113 hha/pCA24N‐hha (biofilm formation), cells were grown in 250 ml containing 1 mM IPTG for 7 h at 250 r.p.m. with 10 g of glass wool to form biofilms. Biofilm cells were obtained by rinsing and sonicating the glass wool in sterile 0.85% NaCl solution at 0°C, and RNALater buffer (Applied Biosystems, Foster City, CA, USA) was added to stabilize RNA during the RNA preparation steps. Total RNA was isolated from biofilm cells as described previously (Ren et al., 2004a) using a bead beater (Biospec, Bartlesville, OK, USA). cDNA synthesis, fragmentation and hybridizations to the E. coli GeneChip Genome 2.0 array (Affymetrix, Santa Clara, CA, USA; P/N 511302) were described previously (González Barrios et al., 2006). Genes were identified as differentially expressed if the expression ratio was higher than the standard deviation: 2.0‐fold (induced and repressed) cut‐off for the Hha13D6 DNA microarrays (standard deviation 1.3‐fold) and 10‐fold (induced) or fourfold (repressed) for the Hha24E9 DNA microarrays (standard deviation fourfold), and if the P‐value for comparing two chips was less than 0.05 (Ren et al., 2004b). The whole‐transcriptome data have been deposited in the NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/) and are accessible through accession number GSE21604.
qRT‐PCR
To corroborate the DNA microarray data, qRT‐PCR was used to quantify relative RNA concentrations using 100 ng as a template using the Power SYBR Green RNA‐to‐CT 1‐Step Kit (Applied Biosystems). The reaction and analysis was carried out by the StepOne Real‐Time PCR System (Applied Biosystems). The housekeeping gene rrsG was used to normalize the gene expression data. The annealing temperature was 60°C for all the genes in this study. Primers for qRT‐PCR are listed in Table S3.
Cell lysis assay
To quantify cell lysis, the DNA concentration in supernatants and in cells was measured by qPCR using primers for purA (encodes the subunit of adenylosuccinate synthetase) (Ma and Wood, 2009). BW25113 hha/pCA24N‐hha and BW25113 hha/pCA24N‐hha13D6 were grown until a turbidity of 1.0 at 600 nm and incubated 4 h with 1 mM IPTG to induce wild‐type Hha and Hha13D6. Then total DNA, which is all genomic DNA released by sonication, and extracellular DNA (eDNA) in the supernatant were isolated as described previously (Ma and Wood, 2009). The percentage of cell lysis was calculated as the ratio of eDNA relative to the total DNA. This experiment was performed with two independent cultures for each strain.
Quantification of c‐di‐GMP
c‐di‐GMP was quantified using HPLC as described previously (Ueda and Wood, 2009). BW25113 hha/pCA24N‐hha and BW25113 hha/pCA24N‐hha13D6 were grown from overnight cultures in 1 l of LB glucose (0.2%) medium with 30 µg ml−1 chloramphenicol until a turbidity of 1.0 at 600 nm and incubated 4 h with 1 mM IPTG to induce wild‐type Hha and Hha13D6. Briefly, formaldehyde (0.18%) was used to inactivate degradation of c‐di‐GMP, then cell pellets were boiled for 10 min, and nucleotides were extracted in 65% ethanol. HPLC (Waters 515 with a photodiode array detector, Milford, MA, USA) was used to detect nucleotides at 254 nm with a reverse‐phase column (Nova‐Pak C18 column; Waters, 3.9 × 150 mm, 4 µm). Synthetic c‐di‐GMP (BIOLOG Life Science Institute, Bremen, Germany) was used as the standard, and the c‐di‐GMP peak was verified by spiking the samples with the synthetic c‐di‐GMP. Escherichia coli AG1/pCA24N‐yddV, which has elevated c‐di‐GMP concentrations (Méndez‐Ortiz et al., 2006), was used as a positive control along with AG1/pCA24N.
Gene deletions
BW25113 hha tnaA, BW25113 hha clpB, BW25113 hha hslU, BW25113 hha hslV, BW25113 hha lon, BW25113 hha prlC, BW25113 hha ycjF, BW25113 hha ibpA, BW25113 hha ibpB, BW25113 hha gadA, BW25113 hha gadE and BW25113 hha gadX were constructed as described previously using the rapid gene knockout procedure with P1 transduction (Maeda et al., 2008) by using plasmid pCP20 (Cherepanov and Wackernagel, 1995) to eliminate the kanamycin resistance (KmR) gene. Mutations were confirmed by PCR with primers (Table S3).
Protein modelling
The amino acid sequences of the Hha13D6 were modelled into the known three‐dimensional structure of the E. coli Hha (residues 1–72; Protein Data Bank accession code 1jw2) (Yee et al., 2002). The three‐dimensional model was obtained using the SWISS‐MODEL server (http://swissmodel.expasy.org/) (Schwede et al., 2003). The molecular visualization program PyMOL (http://pymol.sourceforge.net/) was utilized to make the protein image of the molecular model.
Acknowledgments
This work was supported by the National Institutes of Health (R01 EB003872 and R01 GM089999). We thank the National Institute of Genetics in Japan for providing the Keio and ASKA strains.
Supporting Information
Additional Supporting Information may be found in the online version of this article:
Fig. S1. Biofilm dispersal with combined Hha13D6-24E9. Biofilm dispersal of BW25113 hha producing Hha13D6, Hha24E9 and Hha13D6-24E9 after 32 h (8 h with 1 mM IPTG) in LB glucose at 37°C. Each data point is the average of at least two independent cultures, and one standard deviation is shown.
Table S1. List of differentially expressed genes for biofilm dispersal of BW25113 hha/pCA24N-hha13D6 versus BW25113 hha/pCA24N-hha grown in LB glucose at 37°C for 16 h, and then 1 mM IPTG was added for 1 h. Raw data for the two DNA microarrays are available using GEO series accession number GSE21604.
Table S2. List of differentially expressed genes for biofilm formation of BW25113 hha/pCA24N-hha24E9 versus BW25113 hha/pCA24N-hha grown in LB glucose with 1 mM IPTG for 7 h at 37°C. Raw data for the two DNA microarrays are available using GEO series accession number GSE21604.
Table S3. Primers used for epPCR, DNA sequencing, saturation mutagenesis, site-directed mutagenesis, confirmation of the mutants, qPCR and qRT-PCR. Underlined bold text indicates the saturation mutation of the codon corresponding to D22, L40, V42 and D48 (5′-NNS) (N is A, G, C or T, and S is G or C). Underlined italic text indicates the site-directed mutation for the codon corresponding to truncation at K62 (5′-AAG to 5′-TAG for K62X) in hha-K62X-f and hha-K62X-r.
References
- Baba T., Ara T., Hasegawa M., Takai Y., Okumura Y., Baba M. Construction of Escherichia coli K‐12 in‐frame, single‐gene knockout mutants: the Keio collection. Mol Syst Biol. 2006;2:0008. doi: 10.1038/msb4100050. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baños R.C., Vivero A., Aznar S., García J., Pons M., Madrid C., Juárez A. Differential regulation of horizontally acquired and core genome genes by the bacterial modulator H‐NS. PLoS Genet. 2009;5:e1000513. doi: 10.1371/journal.pgen.1000513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barraud N., Storey M.V., Moore Z.P., Webb J.S., Rice S.A., Kjelleberg S. Nitric oxide‐mediated dispersal in single‐ and multi‐species biofilms of clinically and industrially relevant microorganisms. Microb Biotechnol. 2009;2:370–378. doi: 10.1111/j.1751-7915.2009.00098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boles B.R., Horswill A.R. agr‐mediated dispersal of Staphylococcus aureus biofilms. PLoS Pathog. 2008;4:e1000052. doi: 10.1371/journal.ppat.1000052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boles B.R., Thoendel M., Singh P.K. Rhamnolipids mediate detachment of Pseudomonas aeruginosa from biofilms. Mol Microbiol. 2005;57:1210–1223. doi: 10.1111/j.1365-2958.2005.04743.x. [DOI] [PubMed] [Google Scholar]
- Cherepanov P.P., Wackernagel W. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp‐catalyzed excision of the antibiotic‐resistance determinant. Gene. 1995;158:9–14. doi: 10.1016/0378-1119(95)00193-a. [DOI] [PubMed] [Google Scholar]
- Christensen S.K., Maenhaut‐Michel G., Mine N., Gottesman S., Gerdes K., Melderen L.V. Overproduction of the Lon protease triggers inhibition of translation in Escherichia coli: involvement of the yefM‐yoeB toxin‐antitoxin system. Mol Microbiol. 2004;51:1705–1717. doi: 10.1046/j.1365-2958.2003.03941.x. [DOI] [PubMed] [Google Scholar]
- Domka J., Lee J., Bansal T., Wood T.K. Temporal gene‐expression in Escherichia coli K‐12 biofilms. Environ Microbiol. 2007;9:332–346. doi: 10.1111/j.1462-2920.2006.01143.x. [DOI] [PubMed] [Google Scholar]
- Fishman A., Tao Y., Bentley W.E., Wood T.K. Protein engineering of toluene 4‐monooxygenase of Pseudomonas mendocina KR1 for synthesizing 4‐nitrocatechol from nitrobenzene. Biotechnol Bioeng. 2004;87:779–790. doi: 10.1002/bit.20185. [DOI] [PubMed] [Google Scholar]
- Fletcher M. The effects of culture concentration and age, time, and temperature on bacterial attachment to polystyrene. Can J Microbiol. 1977;23:1–6. [Google Scholar]
- García J., Cordeiro T.N., Nieto J.M., Pons I., Juárez A., Pons M. Interaction between the bacterial nucleoid associated proteins Hha and H‐NS involves a conformational change of Hha. Biochem J. 2005;388:755–762. doi: 10.1042/BJ20050002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García‐Contreras R., Zhang X.‐S., Kim Y., Wood T.K. Protein translation and cell death: the role of rare tRNAs in biofilm formation and in activating dormant phage killer genes. PLoS ONE. 2008;3:e2394. doi: 10.1371/journal.pone.0002394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godessart N., Muñoa F.J., Regue M., Juárez A. Chromosomal mutations that increase the production of a plasmid‐encoded haemolysin in Escherichia coli. J Gen Microbiol. 1988;134:2779–2787. doi: 10.1099/00221287-134-10-2779. [DOI] [PubMed] [Google Scholar]
- González Barrios A.F., Zuo R., Hashimoto Y., Yang L., Bentley W.E., Wood T.K. Autoinducer 2 controls biofilm formation in Escherichia coli through a novel motility quorum‐sensing regulator (MqsR, B3022) J Bacteriol. 2006;188:305–316. doi: 10.1128/JB.188.1.305-316.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen M.C., Palmer R.J., Jr, Udsen C., White D.C., Molin S. Assessment of GFP fluorescence in cells of Streptococcus gordonii under conditions of low pH and low oxygen concentration. Microbiology. 2001;147:1383–1391. doi: 10.1099/00221287-147-5-1383. [DOI] [PubMed] [Google Scholar]
- Heydorn A., Nielsen A.T., Hentzer M., Sternberg C., Givskov M., Ersbøll B.K., Molin S. Quantification of biofilm structures by the novel computer program COMSTAT. Microbiology. 2000;146:2395–2407. doi: 10.1099/00221287-146-10-2395. [DOI] [PubMed] [Google Scholar]
- Hong S.H., Wang X., Wood T.K. Controlling biofilm formation, prophage excision and cell death by rewiring global regulator H‐NS of Escherichia coli. Microb Biotechnol. 2010;3:344–356. doi: 10.1111/j.1751-7915.2010.00164.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain R., Chan M.K. Support for a potential role of E. coli oligopeptidase A in protein degradation. Biochem Biophys Res Commun. 2007;359:486–490. doi: 10.1016/j.bbrc.2007.05.142. [DOI] [PubMed] [Google Scholar]
- Kaplan J.B. Biofilm dispersal: mechanisms, clinical implications, and potential therapeutic uses. J Dent Res. 2010;89:205–218. doi: 10.1177/0022034509359403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karatan E., Watnick P. Signals, regulatory networks, and materials that build and break bacterial biofilms. Microbiol Mol Biol Rev. 2009;73:310–347. doi: 10.1128/MMBR.00041-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa M., Ara T., Arifuzzaman M., Ioka‐Nakamichi T., Inamoto E., Toyonaga H., Mori H. Complete set of ORF clones of Escherichia coli ASKA library (a complete set of E. coli K‐12 ORF archive): unique resources for biological research. DNA Res. 2005;12:291–299. doi: 10.1093/dnares/dsi012. [DOI] [PubMed] [Google Scholar]
- Lee J., Jayaraman A., Wood T. Indole is an inter‐species biofilm signal mediated by SdiA. BMC Microbiol. 2007a;7:42. doi: 10.1186/1471-2180-7-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J., Bansal T., Jayaraman A., Bentley W.E., Wood T.K. Enterohemorrhagic Escherichia coli biofilms are inhibited by 7‐hydroxyindole and stimulated by isatin. Appl Environ Microbiol. 2007b;73:4100–4109. doi: 10.1128/AEM.00360-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.W., Park E., Bang O., Eom S.‐H., Cheong G.‐W., Chung C.H., Seol J.H. Nucleotide triphosphates inhibit the degradation of unfolded proteins by HslV peptidase. Mol Cells. 2007c;23:252–257. [PubMed] [Google Scholar]
- Lee J., Page R., García‐Contreras R., Palermino J.‐M., Zhang X.‐S., Doshi O. Structure and function of the Escherichia coli protein YmgB: a protein critical for biofilm formation and acid‐resistance. J Mol Biol. 2007d;373:11–26. doi: 10.1016/j.jmb.2007.07.037. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J., Maeda T., Hong S.H., Wood T.K. Reconfiguring the quorum‐sensing regulator SdiA of Escherichia coli to control biofilm formation via indole and N‐acylhomoserine lactones. Appl Environ Microbiol. 2009;75:1703–1716. doi: 10.1128/AEM.02081-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leungsakul T., Johnson G.R., Wood T.K. Protein engineering of the 4‐methyl‐5‐nitrocatechol monooxygenase from Burkholderia sp. strain DNT for enhanced degradation of nitroaromatics. Appl Environ Microbiol. 2006;72:3933–3939. doi: 10.1128/AEM.02966-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu T.K., Collins J.J. Dispersing biofilms with engineered enzymatic bacteriophage. Proc Natl Acad Sci USA. 2007;104:11197–11202. doi: 10.1073/pnas.0704624104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q., Wood T.K. OmpA influences Escherichia coli biofilm formation by repressing cellulose production through the CpxRA two‐component system. Environ Microbiol. 2009;11:2735–2746. doi: 10.1111/j.1462-2920.2009.02000.x. [DOI] [PubMed] [Google Scholar]
- Madrid C., Nieto J.M., Paytubi S., Falconi M., Gualerzi C.O., Juárez A. Temperature‐ and H‐NS‐dependent regulation of a plasmid‐encoded virulence operon expressing Escherichia coli hemolysin. J Bacteriol. 2002;184:5058–5066. doi: 10.1128/JB.184.18.5058-5066.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madrid C., Balsalobre C., García J., Juárez A. The novel Hha/YmoA family of nucleoid‐associated proteins: use of structural mimicry to modulate the activity of the H‐NS family of proteins. Mol Microbiol. 2007;63:7–14. doi: 10.1111/j.1365-2958.2006.05497.x. [DOI] [PubMed] [Google Scholar]
- Maeda T., Sanchez‐Torres V., Wood T.K. Metabolic engineering to enhance bacterial hydrogen production. Microb Biotechnol. 2008;1:30–39. doi: 10.1111/j.1751-7915.2007.00003.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mai‐Prochnow A., Webb J.S., Ferrari B.C., Kjelleberg S. Ecological advantages of autolysis during the development and dispersal of Pseudoalteromonas tunicata biofilms. Appl Environ Microbiol. 2006;72:5414–5420. doi: 10.1128/AEM.00546-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Méndez‐Ortiz M.M., Hyodo M., Hayakawa Y., Membrillo‐Hernández J. Genome‐wide transcriptional profile of Escherichia coli in response to high levels of the second messenger 3′,5′‐cyclic diguanylic acid. J Biol Chem. 2006;281:8090–8099. doi: 10.1074/jbc.M510701200. [DOI] [PubMed] [Google Scholar]
- Mogk A., Deuerling E., Vorderwülbecke S., Vierling E., Bukau B. Small heat shock proteins, ClpB and the DnaK system form a functional triad in reversing protein aggregation. Mol Microbiol. 2003;50:585–595. doi: 10.1046/j.1365-2958.2003.03710.x. [DOI] [PubMed] [Google Scholar]
- Mogk A., Haslberger T., Tessarz P., Bukau B. Common and specific mechanisms of AAA+ proteins involved in protein quality control. Biochem Soc Trans. 2008;36:120–125. doi: 10.1042/BST0360120. [DOI] [PubMed] [Google Scholar]
- Prüß B.M., Besemann C., Denton A., Wolfe A.J. A complex transcription network controls the early stages of biofilm development by Escherichia coli. J Bacteriol. 2006;188:3731–3739. doi: 10.1128/JB.01780-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasouly A., Ron E.Z. Interplay between the heat shock response and translation in Escherichia coli. Res Microbiol. 2009;160:288–296. doi: 10.1016/j.resmic.2009.03.007. [DOI] [PubMed] [Google Scholar]
- Ren D., Bedzyk L.A., Thomas S.M., Ye R.W., Wood T.K. Gene expression in Escherichia coli biofilms. Appl Microbiol Biotechnol. 2004a;64:515–524. doi: 10.1007/s00253-003-1517-y. [DOI] [PubMed] [Google Scholar]
- Ren D., Bedzyk L.A., Ye R.W., Thomas S.M., Wood T.K. Differential gene expression shows natural brominated furanones interfere with the autoinducer‐2 bacterial signaling system of Escherichia coli. Biotechnol Bioeng. 2004b;88:630–642. doi: 10.1002/bit.20259. [DOI] [PubMed] [Google Scholar]
- Rui L., Kwon Y.M., Fishman A., Reardon K.F., Wood T.K. Saturation mutagenesis of toluene ortho‐monooxygenase of Burkholderia cepacia G4 for enhanced 1‐naphthol synthesis and chloroform degradation. Appl Environ Microbiol. 2004;70:3246–3252. doi: 10.1128/AEM.70.6.3246-3252.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J., Fritsch E.F., Maniatis T. Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Sauer K., Cullen M.C., Rickard A.H., Zeef L.A.H., Davies D.G., Gilbert P. Characterization of nutrient‐induced dispersion in Pseudomonas aeruginosa PAO1 biofilm. J Bacteriol. 2004;186:7312–7326. doi: 10.1128/JB.186.21.7312-7326.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sayed A.K., Odom C., Foster J.W. The Escherichia coli AraC‐family regulators GadX and GadW activate gadE, the central activator of glutamate‐dependent acid resistance. Microbiology. 2007;153:2584–2592. doi: 10.1099/mic.0.2007/007005-0. [DOI] [PubMed] [Google Scholar]
- Schmidt R., Bukau B., Mogk A. Principles of general and regulatory proteolysis by AAA+ proteases in Escherichia coli. Res Microbiol. 2009;160:629–636. doi: 10.1016/j.resmic.2009.08.018. [DOI] [PubMed] [Google Scholar]
- Schwede T., Kopp J., Guex N., Peitsch M.C. SWISS‐MODEL: an automated protein homology‐modeling server. Nucleic Acids Res. 2003;31:3381–3385. doi: 10.1093/nar/gkg520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma V.K., Carlson S.A., Casey T.A. Hyperadherence of an hha mutant of Escherichia coli O157:H7 is correlated with enhanced expression of LEE‐encoded adherence genes. FEMS Microbiol Lett. 2005;243:189–196. doi: 10.1016/j.femsle.2004.12.003. [DOI] [PubMed] [Google Scholar]
- Singh R., Paul D., Jain R.K. Biofilms: implications in bioremediation. Trends Microbiol. 2006;14:389–397. doi: 10.1016/j.tim.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Smith P.A., Romesberg F.E. Combating bacteria and drug resistance by inhibiting mechanisms of persistence and adaptation. Nat Chem Biol. 2007;3:549–556. doi: 10.1038/nchembio.2007.27. [DOI] [PubMed] [Google Scholar]
- Stewart P.S., Franklin M.J. Physiological heterogeneity in biofilms. Nat Rev Microbiol. 2008;6:199–210. doi: 10.1038/nrmicro1838. [DOI] [PubMed] [Google Scholar]
- Steyn B., Oosthuizen M.C., MacDonald R., Theron J., Brözel V.S. The use of glass wool as an attachment surface for studying phenotypic changes in Pseudomonas aeruginosa biofilms by two‐dimensional gel electrophoresis. Proteomics. 2001;1:871–879. doi: 10.1002/1615-9861(200107)1:7<871::AID-PROT871>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Ueda A., Wood T.K. Connecting quorum sensing, c‐di‐GMP, Pel polysaccharide, and biofilm formation in Pseudomonas aeruginosa through tyrosine phosphatase TpbA (PA3885) PLoS Pathog. 2009;5:e1000483. doi: 10.1371/journal.ppat.1000483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Kim Y., Wood T.K. Control and benefits of CP4‐57 prophage excision in Escherichia coli biofilms. ISME J. 2009;3:1164–1179. doi: 10.1038/ismej.2009.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb J.S., Thompson L.S., James S., Charlton T., Tolker‐Nielsen T., Koch B. Cell death in Pseudomonas aeruginosa biofilm development. J Bacteriol. 2003;185:4585–4592. doi: 10.1128/JB.185.15.4585-4592.2003. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo K.M., Kim K.I., Goldberg A.L., Ha D.B., Chung C.H. The heat‐shock protein ClpB in Escherichia coli is a protein‐activated ATPase. J Biol Chem. 1992;267:20429–20434. [PubMed] [Google Scholar]
- Wood T.K. Insights on Escherichia coli biofilm formation and inhibition from whole‐transcriptome profiling. Environ Microbiol. 2009;11:1–15. doi: 10.1111/j.1462-2920.2008.01768.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yee A., Chang X., Pineda‐Lucena A., Wu B., Semesi A., Le B. An NMR approach to structural proteomics. Proc Natl Acad Sci USA. 2002;99:1825–1830. doi: 10.1073/pnas.042684599. et al. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Biofilm dispersal with combined Hha13D6-24E9. Biofilm dispersal of BW25113 hha producing Hha13D6, Hha24E9 and Hha13D6-24E9 after 32 h (8 h with 1 mM IPTG) in LB glucose at 37°C. Each data point is the average of at least two independent cultures, and one standard deviation is shown.
Table S1. List of differentially expressed genes for biofilm dispersal of BW25113 hha/pCA24N-hha13D6 versus BW25113 hha/pCA24N-hha grown in LB glucose at 37°C for 16 h, and then 1 mM IPTG was added for 1 h. Raw data for the two DNA microarrays are available using GEO series accession number GSE21604.
Table S2. List of differentially expressed genes for biofilm formation of BW25113 hha/pCA24N-hha24E9 versus BW25113 hha/pCA24N-hha grown in LB glucose with 1 mM IPTG for 7 h at 37°C. Raw data for the two DNA microarrays are available using GEO series accession number GSE21604.
Table S3. Primers used for epPCR, DNA sequencing, saturation mutagenesis, site-directed mutagenesis, confirmation of the mutants, qPCR and qRT-PCR. Underlined bold text indicates the saturation mutation of the codon corresponding to D22, L40, V42 and D48 (5′-NNS) (N is A, G, C or T, and S is G or C). Underlined italic text indicates the site-directed mutation for the codon corresponding to truncation at K62 (5′-AAG to 5′-TAG for K62X) in hha-K62X-f and hha-K62X-r.