

A large number of recently launched pharmaceuticals and pharmaceutical candidates contain perfluoroalkyl groups because these moieties affect the physical properties and biological processing of organic molecules, while being stable to degradation.[1] For this reason, the introduction of trifluoromethyl groups into aryl halides by simple laboratory procedures is a major synthetic goal. Despite much effort to develop reactions that introduce trifluoromethyl groups into arene substrates, current methods are limited by some combination of high temperatures, high catalyst loadings, expensive reagents, catalysts, and ligands, low reactivity with electron-rich aromatic groups, and intolerance toward protic and electrophilic functional groups.[2–6] Methods to introduce longer perfluoroalkyl groups into aryl halides are even more limited.[7,8]
Recently, Amii and co-workers published a seminal report on copper-catalyzed trifluoromethylation of electron-deficient aryl iodides with triethyl(trifluoromethyl)silane (TESCF3) as reagent.[3] They suggested that the reaction proceeds through the phenanthroline-ligated copper(I) complex [(phen)CuCF3], but this complex had not been generated previously and was not isolated or detected spectroscopically in the catalytic system. The reaction was shown to occur with electron-poor aryl iodides, but no product was observed from reactions with electron-rich aryl iodides and the reaction of electron-neutral 4-butyl iodobenzene occurred in less than 50% yield. Furthermore, we found (see below) that the same reaction conducted with superstoichiometric amounts of copper and ligand gave the same modest yield of product from trifluoromethylation of 4-butyl iodobenzene.
As part of our studies on the structures and reactions of isolated copper complexes that are potential intermediates in cross-coupling reactions,[9] we sought to determine the structure and stability of the proposed [(phen)CuCF3] (1) and to determine the relative reactivity of this complex as a discrete species toward electron-poor and electron-rich iodoarenes. We report two routes to form [(phen)CuCF3] (1) and its higher perfluoroalkyl homologue [(phen)-CuCF2CF2CF3] (2) in high yield, one route that forms these two complexes as pure, isolated species and one that forms them in high yield in DMF. Most striking, isolated 1 and 2 react in high yield at room temperature with not only electron-poor iodoarenes, but also electron-neutral and electron-rich iodoarenes that possess a wide range of functional groups. Because these complexes are prepared from inexpensive reagents, complexes 1 and 2 represent practical reagents for the perfluoroalkylation of iodoarenes and selected bromoarenes. Mechanistic studies show that these complexes react without the intermediacy of aryl radicals.
Phen-ligated trifluoromethylcopper complex 1 was prepared by the reaction of [CuOtBu]4[10] with 1,10-phenanthroline, followed by the addition of TMSCF3. From this process, complex 1 was isolated as an orange-red solid in 96% yield. Perfluoropropylcopper complex 2 was synthesized in 97% yield by an analogous reaction starting with commercially available (perfluoropropyl)trimethylsilane (Scheme 1). Both complexes were characterized by 1H, 13C, and 19F NMR spectroscopy and elemental analysis. The only previously isolated trifluoromethyl complex was the NHC-ligated complex reported by Vicic et al.[11,12] No isolated higher perfluoroalkyl complexes have been reported.
Scheme 1.
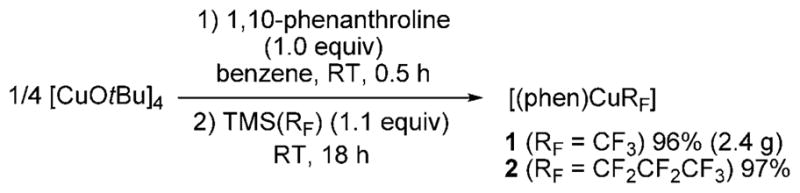
Synthesis of 1,10-phenanthroline-ligated (perfluoroalkyl)copper(I) complexes 1 and 2.
Complexes 1 and 2 are soluble in polar, aprotic solvents, such as DMF and DMSO, partially soluble in THF and CH2Cl2, and insoluble in less polar solvents, such as benzene and Et2O. Conductivity experiments on 1 indicate that this species adopts the ionic form [(phen)2Cu][Cu(CF3)2] in DMF (See Supporting Information for details of these measurements). The 19F NMR spectrum of 1 in DMF clearly shows two species; one component [(phen)2Cu][Cu(CF3)2] resonates at δ =−30.8 ppm, and the second component [(phen)CuCF3] resonates at δ =−22.6 ppm.[2e] Isolated 1 and 2 were stable at room temperature under nitrogen atmosphere for over one month without decomposition.
In contrast to the catalytic reactions proposed to occur through complex 1, isolated 1 reacts in high yields with electron-neutral and electron-rich aryl iodides. Complex 1 also reacts in high yield with aryl iodides containing a wide range of potentially reactive functional groups. The reactions of isolated 1 with excess aryl iodides and bromides are shown in Schemes 2 and 3. The reaction of 1 with 3a–3n gave trifluoromethylarenes 4a–4n in good yield. The reaction of 3a and 3b shows that compound 1 converts electron-neutral iodoarenes to trifluoromethylarenes in nearly quantitative yields, and the reactions of 3c and 3d show that this compound also converts electron-rich iodoarenes to trifluoromethylarenes in equally high yields. The reactions of 1 also tolerate a significant degree of steric hindrance. The combination of 1 and even 2,6-disubstituted aryl iodides 3l and 3m formed 4l and 4m in good yield. These results imply that iodoarenes react with an intermediate other than complex 1 in the previously reported catalytic reactions or that this complex is formed in only very small quantities under those reaction conditions.[3]
Scheme 2.

Reactivity of complex 1 with 5 equivalents aryl iodide 3 at room temperature. Yield was determined by 19F NMR analysis with 4-CF3OC6H4OMe as internal standard. The yield was reported as an average of two runs. Yield of 4b in parentheses was obtained from the reaction with complex 1 stored over 1 month under nitrogen atmosphere.
Scheme 3.
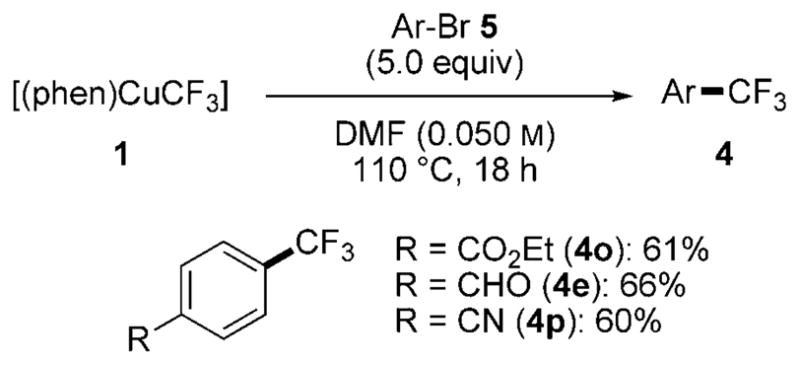
Reactivity of complex 1 with 5 equivalents aryl bromide 5 at 110°C.
Results of reactions of iodoarenes containing typically reactive functional groups, such as amino, alkoxy, hydroxyalkyl, halo, keto, and formyl groups, are also displayed in Scheme 2. Bromo- and chloro-substituted iodoarenes 3h and 3k reacted selectively at the iodide. In addition to the reactions of electron-rich iodoarenes noted above, the reactions with haloarenes containing aldehyde, alcohol, and ketone functionalities (3e, 3 f, and 3g) are particularly striking. Substrates containing these functional groups were not reported to undergo palladium- or copper-catalyzed trifluoromethylation reactions,[2a,c,3,4,6] but they do react with 1 to form the trifluoromethylarenes in high yield.
The reactivity of 1 with substrates 3e and 3g containing formyl and acetyl groups also contrasts with the reaction of these substrates groups with CF3 anions generated from Ruppert’s reagent in the presence of fluoride activators.[13] For example, conditions reported by Fuchikami and coworkers for the copper-mediated trifluoromethylation without added ligand (CuI + TESCF3 + KF) led to reaction of aryl iodide 3g to give a mixture of the desired cross-coupled product 4g and trifluoromethyl carbinols from nucleophilic addition to the ketone function.[8] In contrast, isolated 1 reacted with 3e and 3g to form the corresponding trifluoromethylarenes in high yield.
As shown in Scheme 3, reactions of electron-deficient bromides 5 also gave trifluoromethylarene products in good yield. Like reactions of aryl iodides, the reactions of aryl bromides tolerated electrophilic functional groups, such as ester, aldehyde, and nitrile moieties.
To gain insight into the differences between the reactivity of isolated 1 and the catalytic reactions proposed to occur through 1, we submitted several of the substrates in Scheme 2 to the recently reported catalytic reaction conditions.[3] In each case, we found that less than half the yield of the reaction of isolated 1 was observed from the reactions with catalytic amounts of 1.[14] In addition, the reaction of benzaldehyde 3e under published conditions involving catalytic amounts of phen and CuI formed the trifluoromethyl carbinol without any of the desired trifluoromethylarene, and reaction of acetophenone 3g under these conditions gave low conversion to a 1:1:2 mixture of 1-(4-(trifluoromethyl)phenyl)ethanone (4g), 1,1,1-trifluoro-2-(4-(trifluoromethyl)phenyl)propan-2-ol, and 1,1,1-trifluoro-2-(4-iodophenyl)propan-2-ol. This mixture of products is the same as that observed by Fuchikami and co-workers by copper-mediated trifluoromethylation without added ligand.[8] Moreover, reactions conducted with a copper and ligand loading of 100% under conditions otherwise identical to the catalytic conditions did not give product yields comparable to those observed with complex 1. Thus, the milder conditions and broader scope we observe for the reactions of complex 1 likely stems from the generation of 1 as a discrete species in high yield.
The unprecedented substrate scope displayed by 1 and the facile synthesis of this complex led us to appreciate that complex 1 itself represents a practical reagent for trifluoromethylation. For this complex to be useful, it must react with iodoarenes when the ratio of iodoarenes and copper complex 1 is nearly 1:1 and the iodoarene is the limiting reagent. Indeed, simply reducing the concentration of iodoarenes led to reactions of aryl iodides 3c, 3e, and 3g with 1.5 equiv compound 1 in yields similar to those obtained when using the copper complex as limiting reagent (Scheme 4). Simple aryl iodides, like 3t and 3q, and functionalized aryl iodides containing amide (3r), ester (3o and 3u), aldehyde (3u), bromide (3v), and heterocyclic (3w, 3x, 3y) functionality reacted with 1 to give trifluoromethylarenes 4 in good yields. Quinoline 3x containing an exposed basic nitrogen was converted to the corresponding trifluoromethyl heteroarene in greater than 90% yield at room temperature, and similar results were obtained for the purine and indole derivatives 3w and 3y. The functionalized aryl iodide 3z, which is available by metalation with Knochel’s LiCl-(TMP)MgX reagent and iodination of the arylmagnesium intermediate,[15] also gave product 4z in good yield. Finally, trifluoromethylation of the 3,3′-diiodo-binol derivative 3aa gave the MOM-protected 3,3′-(CF3)2-binol 4aa under mild conditions. Previous syntheses of 3,3′-(CF3)2-binol have required superstoichiometric amounts of Burton’s CuCF3 reagent or generate stoichiometric amounts of SO2 and MeI.[16,17]
Scheme 4.

Copper-mediated trifluoromethylation with isolated (trifluoromethyl)copper(I) complex 1. The reactions were conducted with 0.50 mmol aryl iodide 3 and 0.75 mmol 1 in DMF (0.25M) at 50°C; yields of isolated products are reported unless otherwise noted. The reactions of 3 c, 3 e and 3 bb were run on a 0.10 mmol scale. [a] The yield was determined by 19F NMR spectroscopy with 4-CF3OC6H4OMe as internal standard. [b] The reaction was performed with 1 that was weighted in air. [c] The reaction was performed at room temperature. [d] The reaction was performed with 1.2 equivalents 1 at room temperature. [e] 3.0 equivalents 1 was used.
Complex 1 that was stored for long periods under nitrogen displayed reactivity that is similar to that of freshly prepared complex. The reaction of 3b with 1 stored in glove box over one month gave 4b in 83% yield, which is comparable to the result with freshly prepared 1. Moreover, a glove box is unnecessary for conducting these reactions. The reaction of p-phenyl iodoarenes 3t with 1 that was weighed quickly in air formed the trifluoromethylarene product in 92% yield. Vinyl iodide 3bb reacted with 1 to afford the trifluoromethylalkene 4bb in 99% yield.
Having established a method to prepare 1 in isolated form, we sought a method to generate 1 as a discrete species in situ prior to addition of the iodoarenes to enable the use of 1 on larger scales. We prepared [(phen)CuOtBu] in situ in DMF from commercially available copper chloride, potassium tert-butoxide, and 1,10-phenanthroline, which were all weighed outside of the glove box. The solution of [(phen)-CuOtBu] was then treated with Ruppert’s reagent to afford phen-ligated trifluoromethylcopper 1. Subsequent addition of aryl iodides 3 to the mixture provided trifluoromethylarenes 4 in 83 to 99% yield. The use of copper chloride and potassium tert-butoxide, as well as formation of complex 1 prior to the addition of the aryl halide was important to achieve these high yields.
The results of reactions conducted under these conditions that generate 1 in situ are shown in Scheme 5. These reactions include those of aryl halides containing electron-donating groups, electron-withdrawing groups, potentially reactive functional groups, and both electron-rich and electron-poor heterocyclic compounds. Each of these reactions occurred in yields that were comparable to those of reactions of these iodoarenes with isolated 1. In addition, the reaction of benzyloxy-substituted 3q on a 10 mmol scale formed the trifluoromethylarene in quantitative yield after 24 h to give 2.5 g of the product 4q. Thus, the reactions of complex 1 generated in situ represent a procedure for trifluoromethylation of aryl iodides that occurs under mild conditions from inexpensive, commercially available reagents. The total cost of the reagents to generate 1 in situ is less than $1/mmol.
Scheme 5.

Copper-mediated trifluoromethylation with (trifluoromethyl)copper complex 1 generated in situ. All reactions were performed with 0.50 mmol haloarene as limiting agent without the use of a glove box unless otherwise noted. [a] The reaction was conducted with 1.5 equivalents 1 prepared in situ. [b] The reaction was conducted with 10 mmol iodoarene for 24 h. [c] The yield was determined by 19F NMR spectroscopy with 4-CF3OC6H4OMe as internal standard.
Prior to this work, one of the most general copper-mediated methods for arene trifluoromethylation involved the combination of catalytic CuI with methyl fluorosulfonyl-difluoroacetate FSO2CF2CO2Me. We compared the scope of this trifluoromethylation reaction under the reported conditions, but with the aryl halide as limiting reagent, to our method with complex 1 (Scheme 6).[18] An electron-rich and an electron-neutral aryl iodide were chosen to compare the reaction conditions. The yields of trifluoromethylarene products, as determined by 19F NMR analysis using 4-CF3OC6H4OMe as internal standard, were much higher under the reaction conditions with 1.5 equiv phen-ligated 1 than with catalytic CuI and 2.5 equiv FSO2CF2CO2Me.
Scheme 6.

Comparison of the reactions of aryl iodides 3. The yields were determined by 19F NMR spectroscopy with 4-CF3OC6H4OMe as internal standard.
The reactivity of 1 with aryl bromides as the limiting reagent was also examined and compared to the reaction of CuI and 5 equiv FSO2CF2CO2Me. (Scheme 7). Electron-poor aryl bromides, including those with nitro, cyano, and ethoxycarbonyl groups, reacted with 1 to give high yields of trifluoromethylarene products. In contrast, the reactions of these haloarenes with the combination of FSO2CF2CO2Me and CuI occurred in low yield.
Scheme 7.

Comparison of the reactions of aryl bromides 5. The yields were determined by 19F NMR spectroscopy with 4-CF3OC6H4OMe as internal standard.
Finally, these protocols were extended to the perfluoroalkylation[19,20] of iodoarenes to install fluoroalkyl groups that are longer than a per fluoromethyl group (Scheme 8). Similar to the trifluoromethylation described above, the perfluoroalkylation of iodoarenes with perfluoropropylcopper complex 2 formed perfluoropropylarenes 6 in good yields under mild conditions. These reactions occurred with electron-rich and electron-poor iodoarenes, as well as iodoarenes containing potentially reactive quinoline and nitroarene units. The yield of nitro-substituted 6c (88%) from the reaction of complex 2 was higher than that reported previously with the combination of CuI, KF, and TMSCF2CF2CF3 (41%).[8] The reaction of complex 2 prepared in situ outside of the glove box gave the product 6a in 79% yield after 18 h at 80°C. This transformation is milder and occurs in higher yields than previous perfluoroalkylations conducted by reductively coupling aryl iodides with perfluoroalkyl iodides.[7]
Scheme 8.
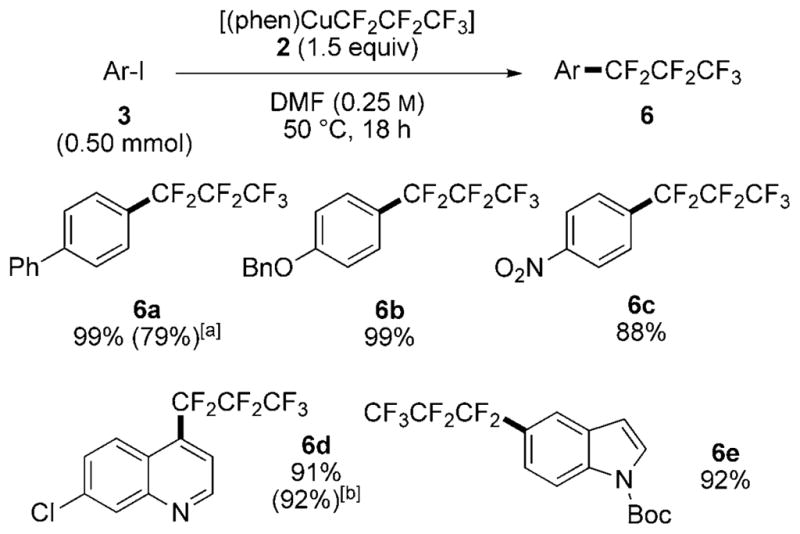
Perfluoroalkylation of aryl iodides 3. The reaction was performed with 1.5 equivalents 2 at 50°C unless otherwise noted. [a] The reaction was conducted at 80°C with perfluoropropylcopper complex 2 prepared in situ under the same conditions as for reactions in Scheme 5. [b] The reaction was performed at room temperature.
Preliminary data on the mechanism of the trifluoromethylation have been gained. To assess whether the reaction proceeds by free aryl radical intermediates,[21] we conducted the trifluoromethylation of 2-(allyloxy)iodobenzene 3cc. The aryl radical derived from this haloarene cyclizes with a rate constant of 1010 s−1.[22,23] Thus, if the trifluoromethylation occurs through an aryl radical, then cyclized product 7 should be observed (Scheme 9).[9b,c] Instead, the trifluoromethylarene 4cc was obtained in 91% yield, and cyclized product 7 was not detected by GC/MS analysis. This result implies that the copper-mediated trifluoromethylation reaction proceeds without the intermediacy of an aryl radical from electron transfer and expulsion of iodide.[24–26]
Scheme 9.
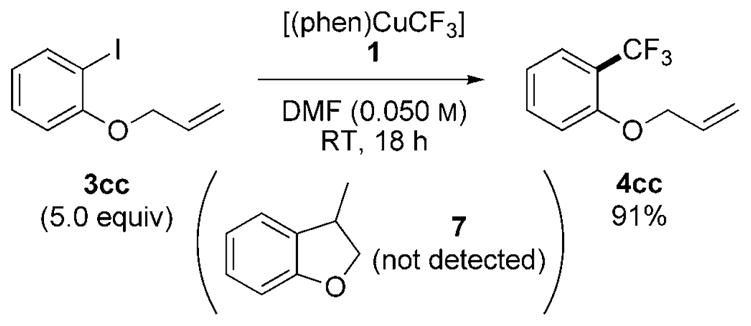
Trifluoromethylation of 2-(allyloxy)iodobenzene. Trifluoromethylarene 4 cc was obtained in 91% yield; cyclized product 7 was not detected by GC-MS analysis.
In summary, we have isolated a trifluoromethylcopper(I) reagent ligated by 1,10-phenanthroline that reacts with unprecedented range of aryl halides at room temperature to 50°C. In comparison to current alternative methods for trifluoromethylation of aryl halides, this system reacts under much milder conditions, tolerates a wider range of functional groups, tolerates basic heterocycles, reacts with more hindered substrates, can be extended to perfluoroalkylation, and occurs with a low total cost of goods. On a more fundamental level, the high reactivity of complexes 1 and 2 with a broad range of iodoarenes demonstrates that a general catalytic perfluoroalkylation of aryl iodides is not limited by the reactivity of the trifluoromethylcopper intermediate.
Experimental Section
Preparation of [(phen)CuCF3] (1): To an oven-dried 250 mL round-bottomed flask equipped with a stir bar were added [CuOtBu]4 (1.094 g, 2.00 mmol, 8.00 mmol for monomeric CuOtBu), 1,10-phenanthroline (1.442 g, 8.00 mmol, 1.00 equiv), and benzene (80 mL). The flask was sealed with a septum and the dark purple mixture was stirred at room temperature for 30 min, then TMSCF3 (1.31 mL, 8.80 mmol, 1.1 equiv) was added dropwise. The mixture was stirred at room temperature for 18 h to give a red-orange suspension. The suspension was filtered through a medium fritted funnel, and the solid was washed with Et2O (50 mL) and dried under vacuum to give 1 as an orange solid (2.397 g, 96% yield). 1H NMR (400 MHz, [D7]DMF): δ = 9.18 (d, J = 4.2 Hz, 2H), 8.89 (d, J = 8.0 Hz, 2H), 8.31 (s, 2H), 8.10 ppm (dd, J =4.2, 8.0 Hz, 2H). 13C{1H} NMR (100 MHz, [D7]DMF): δ = 150.4, 144.2, 138.3, 130.0, 127.8, 126.5 ppm (note that a carbon peak for CF3 was not observed due to 1) dynamic behavior of the complex, 2) broadening the peak through Cu–C coupling, and 3) splitting of the peak through C–F coupling). 19F NMR (376 MHz, [D7]DMF): δ = −22.6 (br), −30.9 ppm (s). Anal. calcd for C13H8CuN2F3: C 49.92, H 2.58, N 8.96, F 18.22; found: C 49.74, H 2.52, N 8.99, F 18.17.
General procedure for the reaction of isolated 1 with aryl iodides 3 as limiting agent: To a 20 mL vial equipped with a stir bar was added ArI 3 (if solid, 0.50 mmol), 1 (235 mg, 0.75 mmol, 1.5 equiv), and DMF (2.0 mL). Then ArI 3 (if liquid, 0.50 mmol) was added, and the mixture was stirred at the indicated temperature in Scheme 4 (room temperature or 50°C). After 18 h, the stirring was stopped, and the reaction mixture was diluted with Et2O and filtered through a pad of Celite. The Celite pad was washed with Et2O. The combined filtrate was washed with 1M aqueous HCl, saturated aqueous NaHCO3 solution and brine, and dried over Na2SO4. After filtration and evaporation of the solvent, the crude mixture was purified by flash silica gel column chromatography using pentane/Et2O or pentane as eluent to give ArCF3 4.
General procedure for the reaction of 1 generated in situ. General procedure outside glove box: To a 20 mL vial equipped with a stir bar and a Teflon-lined screw cap was added CuCl (99 mg, 1.0 mmol, 2.0 equiv). Then air in the vial was evacuated and dry nitrogen was refilled (once). KOtBu (112 mg, 1.0 mmol, 2.0 equiv) and 1,10-phenanthroline (180 mg, 1.0 mmol, 2.0 equiv) were added, then air in the vial was evacuated and dry nitrogen was refilled (twice). To the mixture was added DMF (2.0 mL), and the dark red mixture was stirred at room temperature for 30 min under nitrogen, then TMSCF3 (0.148 mL, 1.0 mmol, 2.0 equiv) was slowly added. The resulting mixture was further stirred at room temperature for 1 h and the stirring was stopped. Then the screw cap was removed and ArI 3 (0.50 mmol) was expeditiously added. While opening the cap, surface of the vial turned green, implying partial decomposition of the copper reagent. The cap of the vial was closed tightly and the vial was evacuated and refilled with dry nitrogen. The resulting mixture was stirred at 50°C for 18 h, then cooled, diluted with Et2O and filtered through a pad of Celite. The Celite pad was washed with Et2O and the combined organic layer was washed with 1M aqueous HCl, saturated aqueous NaHCO3 solution and brine, and dried over Na2SO4. After filtration and evaporation of the solvent, the crude mixture was purified by flash silica gel column chromatography using pentane/Et2O as eluent to give ArCF3 4.
Supplementary Material
Footnotes
The authors thank the NIH (GM-58108 to J.F.H. and 1F32M093540-01 to N.D.L.) and JSPS for a postdoctoral fellowship to H.M. We thank Dr. Ramesh Giri for reagents, and Dr. Vera V. Mainz for helpful discussion concerning NMR experiments. T.T. was supported in part by Global COE Program (Chemistry Innovation through Cooperation of Science and Engineering), MEXT (Japan).
Supporting information for this article is available on the WWW under http://dx.doi.org/10.1002/anie.201100633.
References
- 1.Purser S, Moore PR, Swallow S, Gouverneur V. Chem Soc Rev. 2008;37:320–330. doi: 10.1039/b610213c. [DOI] [PubMed] [Google Scholar]
- 2.For approaches to trifluoromethylation of aryl halides, see the following and reference [3]: Cho EJ, Senecal TD, Kinzel T, Zhang Y, Watson DA, Buchwald SL. Science. 2010;328:1679–1681. doi: 10.1126/science.1190524.Grushin VV, Marshall WJ. J Am Chem Soc. 2006;128:12644–12645. doi: 10.1021/ja064935c.Kitazume T, Ishikawa N. Chem Lett. 1982:137–140.Monnier F, Taillefer M. Angew Chem. 2009;121:7088–7105. doi: 10.1002/anie.200804497.Angew Chem Int Ed. 2009;48:6954–6971. doi: 10.1002/anie.200804497.Wiemers DM, Burton DJ. J Am Chem Soc. 1986;108:832–834.Cottet F, Schlosser M. Eur J Org Chem. 2002:327–330.Kütt A, Movchun V, Rodima T, Dansauer T, Rusanov EB, Leito I, Kaljurand I, Koppel J, Pihl V, Koppel I, Ovsjannikov G, Toom L, Mishima M, Medebielle M, Lork E, Roschenthaler GV, Koppel IA, Kolomeitsev AA. J Org Chem. 2008;73:2607–2620. doi: 10.1021/jo702513w.Dubinina GG, Furutachi H, Vicic DA. J Am Chem Soc. 2008;130:8600–8601. doi: 10.1021/ja802946s.Dubinina GG, Ogikubo J, Vicic DA. Organometallics. 2008;27:6233–6235.
- 3.Oishi M, Kondo H, Amii H. Chem Commun. 2009:1909–1911. doi: 10.1039/b823249k. [DOI] [PubMed] [Google Scholar]
- 4.For recent developments in trifluoromethylation of arenes, see: Ball ND, Kampf JW, Sanford MS. J Am Chem Soc. 2010;132:2878–2879. doi: 10.1021/ja100955x.Wang XS, Truesdale L, Yu JQ. J Am Chem Soc. 2010;132:3648–3649. doi: 10.1021/ja909522s.
- 5.For recent developments in trifluoromethylation of aryl boronic acids, see: Senecal TD, Parsons AT, Buchwald SL. J Org Chem. 2011;76:1174–1176. doi: 10.1021/jo1023377.Chu L, Qing FL. Org Lett. 2010;12:5060–5063. doi: 10.1021/ol1023135.
- 6.For a copper-catalyzed trifluoromethylation of aryl halides that appeared during the processing of this manuscript, see: Knauber T, Arikan F, Röschenthaler G-V, Gooßen LJ. Chem Eur J. 2011;17:2689–2697. doi: 10.1002/chem.201002749.
- 7.Burton DJ, Yang ZY. Tetrahedron. 1992;48:189–275. [Google Scholar]
- 8.Urata H, Fuchikami T. Tetrahedron Lett. 1991;32:91–94. [Google Scholar]
- 9.a) Giri R, Hartwig JF. J Am Chem Soc. 2010;132:15860–15863. doi: 10.1021/ja105695s. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tye JW, Weng Z, Johns AM, Incarvito CD, Hartwig JF. J Am Chem Soc. 2008;130:9971–9983. doi: 10.1021/ja076668w. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Tye JW, Weng ZQ, Giri R, Hartwig JF. Angew Chem. 2010;122:2231–2235. doi: 10.1002/anie.200902245. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew Chem Int Ed. 2010;49:2185–2189. doi: 10.1002/anie.200902245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsuda T, Hashimoto T, Saegusa T. J Am Chem Soc. 1972;94:658–659. [Google Scholar]
- 11.For isolation of the only prior ligated copper-trifluoromethyl compound, see references [2h] and [2i].
- 12.Burton and co-workers reported the observation of a ligandless CuCF3 species by NMR spectroscopy in reference [2e], but this compound has not been isolated and fully characterized.
- 13.Krishnamurti R, Bellew DR, Prakash GKS. J Org Chem. 1991;56:984–989. [Google Scholar]
- 14.Yields were obtained by the reaction of 10% CuI, 10% 1,10- phenanthroline, 2 equiv KF, 2 equiv TESCF3 in a 1:1 mixture of NMP:DMF at 60°C for 24 h, as described by Amii et al.[3] Yields were determined by 19F NMR spectroscopy with 4- CF3OC6H4OMe as internal standard. Substrate 3b gave 4b in 36% yield. Substrate 3c gave 4c in 43% yield. Substrate 3g gave 4g in approximately 10% yield with the trifluoromethyl carbinols as the major products. Substrate 3e gave none of 4e and formed exclusively the trifluoromethyl carbinol product. 3p gave 4p in 41% yield; 3o gave 4o in 43% yield. Substrates 3l and 3m gave 4l and 4m in 17 and 39% yield.
- 15.Lin WW, Baron O, Knochel P. Org Lett. 2006;8:5673–5676. doi: 10.1021/ol0625536. [DOI] [PubMed] [Google Scholar]
- 16.Kobayashi S, Ishitani H. 6476250 B1. Patent US. 2002
- 17.Wu TR, Shen LX, Chong JM. Org Lett. 2004;6:2701–2704. doi: 10.1021/ol0490882. [DOI] [PubMed] [Google Scholar]
- 18.Chen QY, Wu SW. J Chem Soc Chem Commun. 1989:705–706. [Google Scholar]
- 19.Carr GE, Chambers RD, Holmes TF, Parker DG. J Chem Soc Perkin Trans. 1988;1:921–926. [Google Scholar]
- 20.Xiao JC, Ye CF, Shreeve JM. Org Lett. 2005;7:1963–1965. doi: 10.1021/ol050426o. [DOI] [PubMed] [Google Scholar]
- 21.Jones GO, Liu P, Houk KN, Buchwald SL. J Am Chem Soc. 2010;132:6205–6213. doi: 10.1021/ja100739h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Annunziata A, Galli C, Marinelli M, Pau T. Eur J Org Chem. 2001:1323–1329. [Google Scholar]
- 23.Abeywickrema AN, Beckwith ALJ. J Chem Soc Chem Commun. 1986:464–465. [Google Scholar]
- 24.Enemærke RJ, Christensen TB, Jensen H, Daasbjerg K. J Chem Soc Perkin Trans. 2001;2:1620–1630. [Google Scholar]
- 25.Takeda N, Poliakov PV, Cook AR, Miller JR. J Am Chem Soc. 2004;126:4301–4309. doi: 10.1021/ja0389671. [DOI] [PubMed] [Google Scholar]
- 26.Andrieux CP, Saveant JM, Zann D. New J Chem. 1984;8:107–116. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.