

Abstract
Vascular dementia is, in its current conceptual form, a distinct type of dementia with a spectrum of specific clinical and pathophysiological features. However, in a very large majority of cases, these alterations occur in an already aged brain, characterized by a milieu of cellular and molecular events common for different neurodegenerative diseases. The cell signaling defects and molecular dyshomeostasis might lead to neuronal malfunction prior to the death of neurons and the alteration of neuronal networks. In the present paper, we explore some of the molecular mechanisms underlying brain malfunction triggered by cerebrovascular disease and risk factors. We suggest that, in the age of genetic investigation and molecular diagnosis, the concept of vascular dementia needs a new approach.
1. Vascular Dementia—Historical Considerations
Just how far back in time should one go when searching data of vascular dementia (VaD)? In 1549, Jason Pratensis published De Cerebri Morbi, linking dementia to stroke [1], and in 1658, Johann Jakof Wepfer theorized that a broken brain blood vessel may cause apoplexy (stroke) [2]. The correlation between atherosclerotic disease and dementia was clearly identified only at the beginning of the 20th century by two well-known contributors to the field of neurodegeneration: Alois Alzheimer and Otto Binswanger [3]. The modern era of vascular dementia began in the 1960s, under the leadership of the Newcastle College of Medicine [4]. The concept of VaD was ever since under permanent scrutiny and revision, in light of new clinical, pathological, and imagery data (Figure 1). In the early 1970's, multiple infarct dementia was recognized as a major type of dementia, apart from Alzheimer's disease, characterized not by “neuronal atrophy” but by atherosclerotic burden. In 1975, Vladimir Hachinski defined the “ischemic score,” later used for the clinical diagnostic of vascular dementias [5]. However, the concept of VaD soon became controversial due to an increased discrepancy between the incidence of cognitive disorders and that of the “strategic stroke.” Furthermore, the early prevention of multi-infarct dementia (MID), the aging of the general population, and an arising need to define “normal aging” versus “pathological aging” [6] added to this controversy. The struggle to identify preventable and treatable factors widened the pathogenic spectrum of VaD [7]. Several epidemiological studies reported associations of hypertension, type 2 diabetes, obesity, and inflammation with VAD and, in some cases, AD. These all coincide with those of stroke, which in turn is an established factor for cognitive decline and VAD [8] and underlines furthermore the need for a new classification of dementia types [9]. During the last two decades, there was a switch of exploration from classical pathology to new imaging techniques at the molecular level. Therefore, new pathogenic pathways were identified, which greatly increased the complexity of mechanisms of neuronal loss due to cerebral vascular injury [10].
Figure 1.

Evolution throughout time of vascular dementia concept.
In each stage of clinical and imaging research, new attempts were made to define VaD as an individual, self-standing class of dementia. Mayer-Gross et al. presented in the late 60s a set a criteria including dementia with focal signs and symptoms consistent with stroke, a fluctuating course, preservation of intellectual powers, and personality until late in the disease. Importantly, definition included vascular risk factors such as hypertension [11].
The “multi-infarct dementia” (MID), first described by Vladimir Hachinski, was characterized by a number of small ischemic strokes that may not result in focal neurologic deficits, but in time, by cumulative damage, would lead to cognitive decline. Later, the Hachinski ischemic scale, used for MID diagnosis, was modified by Loeb and Gandolfo to include CT scan criteria [12].
In the 1990s, as acknowledgment of overlapping features of various types of dementia, VaD criteria included clinical and imaging features of probable and possible disease. Criteria for definite VaD would require histopathological evidence from biopsy or autopsy [13].
Currently, the most widely used criteria for VaD include the Diagnostic and Statistical Manual of Mental Disorders (DSM), Alzheimer's Disease Diagnostic and Treatment Centers (ADDTC), International Statistical Classification of Diseases (ICD), and National Institute of Neurological Disorders and Stroke—Association Internationale pour la Recherche et l'Enseignement en Neurosciences (NINDS-AIREN) criteria [14].
2. The Concept of Brain Ageing
The concept of brain ageing stated at first that cell death might be responsible for the progressive deterioration of different physiological functions. Studies on aged animals [15] from over two decades ago reported neuronal loss with aging, with or without cortical thinning (depending on the type of method used for quantification), but with diminution of the total volume of gray matter. By the end of the 1980s, reports of preserved neuronal number, despite cortical thinning in human brain [16], started to challenge the previous data and were followed by confirmatory studies on animals [17–20]. This controversy was solved by modern imaging investigational methods, starting with computer tomographic analysis in the early 1980's [21] and continuing with recent PET and MRI analyses [22, 23]. These techniques demonstrated that brain atrophy does occur with age in the healthy, nondemented elderly, involving both gray and white matter, but the loss is rather of neuronal connections, not of neurons. Furthermore, quantitation of neurons showed that, despite frontal and medial temporal cortical thinning, the number of neurons is preserved in healthy adults. Freeman et al. reported that, in frontal and temporal neocortical regions, the neuronal count remained relatively constant over a 50-year age range, suggesting that the atrophy is a reflection of the 3D neuronal network loosening rather than perikaryal loss [24]. The prefrontal cortical neurons seem to be particularly vulnerable to ageing, as a decrease in dendritic branching has been reported in neocortex of both rat [25] and human brains [26–28]. By contrast, there is no significant change in dendritic length of hippocampal granule cells, nor a reduction in spine density in the dentate gyrus of aged humans [29] or rats [30].
White matter reduction is also a consistent finding in the aged human brain, possibly as an indicator of defective myelination (although oligodendrocyte number seems to increase). White matter loss is strongly correlated with vascular risk factors, particularly hypertension and stroke [31], two pathologies included in the broad spectrum of VaD risk factors. However, the involvement of white matter abnormalities and the presence of lacunae yielded contradictory results in terms of functional integrity and cognitive impairment [32].
At the molecular level, aging is a “decrease in homeostatic reserve” [33] which interferes with neuronal ability to limit and buffer the increase of reactive oxygen species (ROS) production, to sustain a protective response to cytotoxic stimuli or to limit vicious cycles such as inflammatory environments. DNA damage increases with age (some of which is ROS related), somatic mutation in human lymphocytes being nine times more frequent in aged human subjects than in neonates [34], and mitochondrial DNA being even more sensitive than nuclear DNA. Mitochondrial aging brings its share of vulnerability to stress in aged cells, with decreased ATP reserves [35] along with affected cellular calcium removal systems and low buffering capacity [36]. Moreover, one should take into account the fact that, in the brain, these processes affect, at different rates, different cell types that share a homeostatic balance. On the other hand, understanding aging of the nervous tissue, as compared to other tissues, could be a more challenging task due to a more complex regulation, signaling, and intercellular interactions.
3. VaD from a Molecular Perspective
The molecular perspective on VaD is rather limited; the general concept of this type of cognitive impairment has derived from clinical and imaging findings and is correlated, at the cellular level, with neuronal death and the sudden interruption of neuronal networks. The main pathological changes leading to different forms of vascular dementia take place in both large (atherosclerosis and thrombosis) and small (lipohyalinosis and fibrosis) cerebral vessels, secondary to common vascular risk factors, such as hypertension, diabetes mellitus, and dyslipidemia. The reduction in cerebral blood flow (CBF) starts early during vascular disease [37] and, therefore, a major vascular event can be preceded by a variable period of chronic hypoxia. As a result, the brain cellular microenvironment might change and adaptive processes may lead to cellular malfunction, rather than cellular death.
3.1. Cerebral Blood Flow and Ischemia-Triggered Molecular Events
Normal aging is associated with low cerebral flow and velocity at rest [38] and an attenuation of responsivity to hypoxia, hypercapnia, or blood pressure alterations [39]. These modifications may appear due to either histological alterations of the vessel wall (thickening of basement membrane, loss of pericytes, and an overall reduction in cortical vascular bed) or lower metabolic demand. The same changes in blood flow, but at a higher rate, were documented in subcortical ischemic VaD patients by PET studies, with some groups reporting a preferential decrease in frontal lobe regions [40]. In laboratory rats, chronic hypoxia increases the CBF for several days, after which a decrease towards the baseline is noted, probably due to compensatory mechanisms such as increased hematocrit and decreased metabolic needs [41]. Hypoxia inducible factor-1 (HIF-1) was used by Ritz et al. as a marker of hypoxia in the cortex of young (2 months) and old (9 months) spontaneously hypertensive rats (SHR) and stroke-prone SHR, in their study on hypoxic alterations of nonneuronal populations [42]. Interestingly, the increase in HIF1α was documented only in aged animals, along with an imbalance between microvessels and astrocytes at the level of the neurovascular unit. In hypoxic conditions, HIF-1α is upregulated, dimerizes with HIF-1β (the constitutively expressed subunit of HIF-1), translocates into the nucleus, and binds to hypoxia-responsive elements (HREs) of target genes, such as vascular endothelial growth factor (VEGF), glucose transporter-1 (GLUT1), lactate dehydrogenase (LDH), erythropoietin (Epo), and nitric oxide synthase (NOS).
3.2. Inflammatory Cytokines, Adhesion Molecules, and Endothelial Malfunction
Endothelial malfunction is considered to be a first step in the development of atherosclerosis, and may be objectified by overexpression of inflammatory cytokines and adhesion molecules, leading to monocyte recruitment in the nascent atherosclerotic plaque and overproduction of reactive oxygen species (ROS), as a sign of mitochondrial, peroxisomal, and lysosomal alteration.
Measurements of plasma markers in VaD patients showed increased levels of proinflammatory cytokines (IL1, IL6, TNFα) as well as anti-inflammatory cytokines (IL-10) [43]. IL-6 and TNFα levels increase with aging in animals and humans, and IL-6 transgenic mice also show progressive proliferative cerebellar angiopathy and blood-barrier (BBB) breakdown. These events indicate the endothelium as one of the main targets of proinflammatory cytokine IL-6 [44]. The same transgenic strains indicated for the first time a causative relationship between local production of IL-6 in the brain and the age-related decline in learning and cognitive function, demonstrating dendritic vacuolization, stripping of dendritic spines, decreased synaptic density, and loss of GABA-producing neurons in the hippocampus. In association with neurodegenerative changes, a diffuse nonproliferative gliosis with marked activation of astrocytes and microglia was identified in GFAP-IL6 mice [45]. Furthermore, studies in transgenic mice overexpressing TNF and/or its receptors (p55 and p75NTR) demonstrated that IL-6 is a potent microglial activator and, depending on the receptor it activates, (i) an endothelial activator (via p75NTR), leading to increased expression of adhesion molecules, BBB disruption, and CNS leukocyte infiltration or (ii) a demyelinating agent and oligodendrocyte apoptosis inducer via p55 [46]. According to Batti and O'Connor, although TNFα has no effect on synaptic transmission or long-term potentation (LTP) under basal conditions, it severely impairs the recovery of postsynaptic transmission after hypoxic exposure [47]. They also showed that the TNFα effect is p38/MAPK mediated, a signaling pathway involved as well in hypoxic neuronal death in the CA1 region of the hippocampus. But, in addition to its neurotoxic nature, TNFα may also exert neuroprotective effects [48, 49] in selected signaling contexts.
Suggested to be another marker of chronic inflammation [50], E-selectin is an endothelial adhesion molecule, that is involved in weak linking of circulating leukocytes. Its expression is upregulated by IL-1 and TNFα. Elevated levels of E-selectin have been previously linked to experimental and clinical brain ischemia [51], and high levels of soluble selectin (sE-selectin) have been correlated with severe cerebrovascular disease [50]. Generating immune tolerance against E selectin by repeated low-dose mucosal administration in lab rats had a protective effect against hemorrhagic strokes in HRS rats and against VCI development in Wistar rats, as shown by Wakita et al. [52].
3.3. Oxidative Stress
The impact of ROS on cognitive function is elegantly demonstrated by studies of superoxide dismutase (SOD) isoenzyme transgenic mice. Overexpression of mitochondrial SOD has a neuroprotective role against drug-induced neurotoxicity, overexpression of cytoplasmic SOD improves age-related impairments in LTP, and overexpression of extracellular SOD is correlated with better spatial memory in laboratory rats [53]. Following cerebral ischemia, the production of free radicals was increased in aged rats and human endothelial cells, mainly by overproduction in the monocyte/macrophage system, especially when stimulated by inflammatory mediators [54].
3.4. Effect of VaD Molecular Alterations on Neuronal and Glial Populations
Hypoxia is associated with increased expression of all NO synthase isoforms, including neuronal (nNOS), astrocyte and microglia-inducible isoform (iNOS), and endothelial isoform (eNOS) [55], which are involved in neuronal death through inhibition of mitochondrial respiration and NMDA/Ca2+-induced exotoxicity [56, 57]. Brain cells are particularly sensitive to ROS aggression due to their high content of polyunsaturated fatty acids, which constitute a substrate for lipid peroxidation. Exposure of brain cells to oxidative stress increases the accumulation of cholesterol in cell membranes [58], leading to decreased fluidity and impaired transmembrane transport.
Hypoxia also upregulates the expression of BDNF—a neurotrophic factor with important roles in neuroplasticity and hippocampus-related learning. This might serve as a protective mechanism against a paucity of hippocampal BDNF mRNA and BDNF plasma levels at older ages [59]. BDNF is further reduced by vascular risk factors such as hypertension and poor glucose metabolism [60]. However, hypoxic upregulation of BDNF is not accompanied by upregulation of its high-affinity receptor Trk-B, but of its low-affinity receptor p75NTR, a TNF superfamily receptor. The p75NTR expression is upregulated by hypoxic conditions and is correlated with an increase in caspase-3 activation in cortical and hippocampal neurons, leading to apoptosis [61]. The upregulation of p75NTR is linked to NOS stimulation and to Ca-mediated regulation of expression, suggesting a complex transformation of the pattern of molecular expression in chronic ischemia and VaD.
4. Mixed versus Pure Dementia
“Mixed” dementia is, by the very definition of Vladimir Hachinski himself, “Alzheimer's disease and cerebral infarcts contributing to the dementia” [6], but other coexisting pathologies are also common in dementia such as Parkinson disease (in about 20% of patients with AD) and dementia with Lewy bodies (up to 50%) [62].
Many data suggest that “pure” vascular dementia is rare and is the exception, rather than the rule [63–65]. Vasculopathy as a trigger of AD neuropathological features has been proposed repeatedly before [66–68], and it is very likely that a patient with late-onset AD may already have a vascular burden and shares with VaD vascular risk factors. Moreover, Zhang et al. demonstrated that the low-oxygen dependent increase in HIF1α expression was accompanied by an increase of BACE1 protein levels and a secondary increase in Aβ production [69]. These data suggest that restoration of normal oxygen levels to hypoxic tissues, for example, by the use of small molecules that lower the affinity of oxygen for hemoglobin, could be an interesting issue for research [70, 71].
Activation of inflammation is a consistent finding in AD, as shown in cell culture models [72, 73], animal models [74, 75], and postmortem studies on AD brains [76–78].
Inflammation was related to the onset of cognitive decline and also correlated with disease progression by measurements of serum TNFα and the TNFα/IL1-β ratio. Patients with AD show elevated levels of TGF-β that are correlated with low expression of TGF-R in the affected brain areas, especially around cerebral vessels with CAA [45]. Furthermore, inflammation is associated with ROS production, and oxidative stress has a dual relationship with Aβ peptide: (i) it favors the aggregation of Aβ into a fibrillar form and (ii) it mediates the toxic effect of Aβ on neuronal cells, as shown by the protective effect of antioxidants and free radical scavengers [79]. In turn, some Aβ peptides (such as the 25–35 form) have an intrinsic lipoperoxidizing effect, as established on neocortex-derived synaptosomes [80]. Oxidative stress is demonstrated by the increased amount of 4-hydroxynonenal (HNE), which was shown to interfere with plasmalemmal ATPases and transporters, including Ca2+ shifters, further increasing metabolic imbalance in AD.
Downstream Aβ production and accumulation results in secondary endothelial malfunction through: (i) amyloid angiopathy; (ii) NOS inhibition [50]; (iii) atherogenesis correlated with endothelial activation and overexpression of inflammatory cytokines and adhesion molecules, even before Aβ deposition [81]; (iv) lipid peroxidation in the frontal cortex in AD brains [82]; (v) BBB alteration [83].
To conclude, there is an overlap of events between chronic hypoxia and AD on several levels, such as hypoxic-triggered cellular pathways, inflammatory environment, growth factor signalling, and calcium homeostasis (Figure 2). Thus, from the molecular level perspective, the diagnostic criteria for neurodegenerative diseases have become ill defined or insufficient and there is a true need for redefinition.
Figure 2.
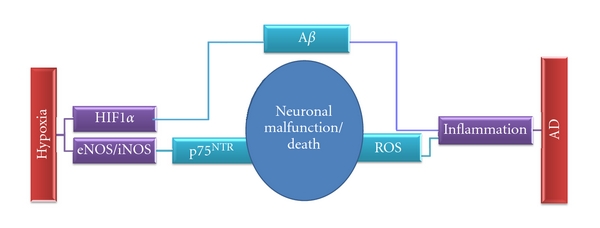
Some mechanisms converging towards neuronal malfunction in two major types of dementia.
5. Overlapping of Normal Aging and Neurodegenerative Diseases at Cellular and Molecular Level
Normal aging and various types of neurodegeneration share common molecular events (Table 1), such as alteration of cerebral blood flow, neuroinflammatory environment, and endothelial malfunction.
Table 1.
Comparison between normal aging and neurodegenerative diseases from a molecular perspective.
| Parameter | Normal aging | Vascular dementia | Alzheimer's disease | Other neurodegenerative disorders |
|---|---|---|---|---|
| CBF | Diminished with lower velocity, but with preserved dynamic adaptability [84] | Diminished in parietal and frontal lobes, some authors reported also a decrement in superior temporal gyri, thalami, anterior cingulate gyri [85] | Diminished only in parietal cortices and later in advanced disease in frontal lobes [86] | Diminished in preoccipital and occipital regions in PD [87] and LBD [88] |
|
| ||||
| VEGF -A | Low basal levels produced by astrocytes [89] | Upregulation of VEGF and VEGF R2 in astrocytes [90] | Low serum levels and decreased secretion by peripheral immune cells [91] | FTLD—associated with VEGF gene promoter polymorphism in selected populations [92] |
|
| ||||
| Inflammatory cytokines | ||||
| IL-6 | Increased mRNA compared to young subjects [93] | High blood levels, associated with high CRP may be associated with high risk [94] | Positive immunoreactivity in amyloid plaques and increased concentration in AD brain, compared to age-matched subjects [95] | Increased in cerebral and cerebellar cortex of Huntington patients [96] |
| TNFα | Increased basal levels in aged laboratory animals with week induction injury response [97] | Modulates neuronal cell loss in cerebral ischemia [98] | Increased expression in AD brain, along with TNF-R1 [99] | Increased in plasma [100], CSF of PD patients and in PD brains, especially in areas with greatest loss of dopaminergic neurons [101] |
| TGFβ1 | Detected at low levels in CSF and produced in CNS at low levels by neuronal cells [102] | Increased in CNS and CSF after stroke [103] | Increased in areas with amyloid burden [104] | CAA—directly related to amyloid vascular deposition [105] |
|
| ||||
| Adhesion molecules | sVCAM increased [106] | sVCAM increased in atherosclerotic disease [107]; sE-selectin increased in severe cerebrovascular disease [108] | sVCAM elevated in late onset AD [50] | sVCAM increased in Down Syndrome [100] |
|
| ||||
| ROS | Increased accumulation with aging [109] | Increased in ischemia animal models and stroke patients [110] | Increased: Aβ-related ROS generation and MAOS [111] | Increased in PD in vitro models [112] and animal models [113] |
|
| ||||
| Lipid metabolism | Accumulation of ceramides and free cholesterol in cerebral cortex [114] | Hypercholesterolemia is a known risk factor for VaD | Increased levels of cholesterol, and activation of cholesterol biosynthesis pathway [115] | PD dementia does not correlate with apoE polymorphism or lipid profile [116] |
| GLUT 1 | Altered structure and function of GLUT-1 [117] | Downregulated in prolonged hypoxia [118] | Low expression in AD hippocampus and double transgenic APP/PS1 animal model Learning increases expression in mouse brain [119] | Insufficiently investigated in neurodegeneration, but involved in “Glut-1 deficiency syndrome”— a treatment-resistant form of epilepsy [120] |
|
| ||||
| BDNF | Decreased mRNA in human plasma and hippocampus [121] | Increased expression following hypoxic stress in cell cultures [122, 123] and lab animals [123] | Decreased expression in hippocampus temporal and frontal cortex [124] | Reduced BDNF expression in the caudate and putamen in HD patients [96] Reduced mRNA BDNF expression [125] and protein [126] in striatal neurons in PD patients |
|
| ||||
| Calcium | Reduced homeostatic reserve [33] | Involved in ischemia-induced excitotoxicity [127] | Aβ disrupts Ca homeostasis in cortical neuronal cell cultures [117] | Excitotoxicity and excessive Ca2+-mediated nitric oxide production are believed to contribute to the death of dopaminergic neurons in PD [118]; Huntingtin transgenic mice express mitochondrial Ca overload upon glutamate stimulation [119] |
MAOS: membrane-associated oxidative stress VDCC: voltage dependent calcium channels, FTLD: frontotemporal lobar dementia, LBD: Lewy body dementia, and HD: Huntington disease.
Aging favors the production of proinflammatory cytokines, mostly through microglial and astrocytic activation [54]. Aging has also been associated, at the cellular level, with increased production of reactive oxygen species (ROS) [109]. Oxidative alteration of enzymes and the subsequent loss of enzymatic activity is a trait of the aging brain, particularly, in the anterior frontal lobe [49]. Oxidative stress leads to the accumulation of free cholesterol [79], along with ceramides, lipid peroxides, and derived aldehydes (such as HNE), that covalently bind to membrane proteins, altering their functions.
Oxidative stress is involved as well in the disruption of Ca2+ homeostasis, an effect studied especially in neurons, where Ca2+ is a vital mediator of neuronal signaling. It appears that, in aged neurons, several Ca2+ homeostatic systems are affected [33] and there is impairment in the maintenance of a nontoxic Ca2+ overload [120].
Although it seems that levels of nNOS and eNOS do not change with age, still there is an increase in NOS activity in aged rat cortex. These two NOS isoforms are Ca2+ induced, which correlates with the above-mentioned impairment of aged cells to deal with Ca2+ overload. Furthermore, consistent with the Ca2+-independent nature of iNOS, there are several reports underlining its absence in the normal aged cortex of lab rats [15, 60, 105].
6. Conclusions
Instead of considering VaD a pure result of neuronal death and the interruption of neuronal networks that support cognitive function, we hypothesize that early brain malfunction is induced by vascular risk factors and chronic hypoxia. A reduction of CBF and a series of molecular events precede the major ischemic events in vascular cognitive impairment. Based on these subtle changes, intervention at early stages could prevent the full-blown development of dementia, which might represent a “point of no return” for the neurovascular units and neuronal networks with few chances for effective treatment.
Acknowledgments
This paper was supported by the Sectorial Operational Programme Human Resources Development (SOP HRD), financed from the European Social Fund and by the Romanian Government under contract number POSDRU/89/1.5/S/64109, and by the Executive Unit for Financing Higher Education, Research, Development and Innovation—Romania (UEFISCDI), Program 4 (Partnerships in Priority Domains), Grant no. 41-013/2007.
Abbreviations
- Aβ:
Amyloid beta peptide
- p75NTR:
Low affinity receptor for tumor necrosis factor α
- HIF 1α:
Hypoxia Inducible Factor 1α
- ROS:
Reactive oxygen species
- eNOS:
Endothelial nitric oxide synthase
- iNOS:
Inducible nitric oxide synthase.
References
- 1.Iemolo F, Duro G, Rizzo C, Castiglia L, Hachinski V, Caruso C. Pathophysiology of vascular dementia. Immunity & Ageing. 2009;6:p. 13. doi: 10.1186/1742-4933-6-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pearce JM. Johann Jakob Wepfer (1620-95) and cerebral haemorrhage. Journal of Neurology, Neurosurgery and Psychiatry. 1997;62(4):p. 387. doi: 10.1136/jnnp.62.4.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mast H, Tatemichi TK, Mohr JP. Chronic brain ischemia: the contributions of Otto Binswanger and Alois Alzheimer to the mechanisms of vascular dementia. Journal of the Neurological Sciences. 1995;132(supplement 1):4–10. doi: 10.1016/0022-510x(95)00116-j. [DOI] [PubMed] [Google Scholar]
- 4.Román G. Vascular dementia: a historical background. International Psychogeriatrics. 2003;15(1):11–13. doi: 10.1017/S1041610203008901. [DOI] [PubMed] [Google Scholar]
- 5.Hachinski VC, Iliff LD, Zilhka E, et al. Cerebral blood flow in dementia. Archives of Neurology. 1975;32(9):632–637. doi: 10.1001/archneur.1975.00490510088009. [DOI] [PubMed] [Google Scholar]
- 6.Hachinski VC. The decline and resurgence of vascular dementia. Canadian Medical Association Journal. 1990;142(2):107–111. [PMC free article] [PubMed] [Google Scholar]
- 7.Konno S, Sterling Meyer J, Terayama Y, Margishvili GM, Mortel KF. Classification, diagnosis and treatment of vascular dementia. Drugs and Aging. 1997;11(5):361–373. doi: 10.2165/00002512-199711050-00004. [DOI] [PubMed] [Google Scholar]
- 8.Moretti A, Gorini A, Villa RF. Pharmacotherapy and prevention of vascular dementia. CNS & Neurological Disorders—Drug Targets. 2011;10(3):370–390. doi: 10.2174/187152711794653832. [DOI] [PubMed] [Google Scholar]
- 9.Stewart R. Vascular dementia: a diagnosis running out of time. British Journal of Psychiatry. 2002;180:152–156. doi: 10.1192/bjp.180.2.152. [DOI] [PubMed] [Google Scholar]
- 10.Cacabelos R, Fernandez-Novoa L, Perez-Trullen JM, Franco-Maside A, Alvarez XA. Serum histamine in Alzheimer’s disease and multi-infarct dementia. Methods and Findings in Experimental and Clinical Pharmacology. 1992;14(9):711–715. [PubMed] [Google Scholar]
- 11.Schneck MJ. Vascular dementia. Topics in Stroke Rehabilitation. 2008;15(1):22–26. doi: 10.1310/tsr1501-22. [DOI] [PubMed] [Google Scholar]
- 12.Loeb C, Gandolfo C. Diagnostic evaluation of degenerative and vascular dementia. Stroke. 1983;14(3):399–401. doi: 10.1161/01.str.14.3.399. [DOI] [PubMed] [Google Scholar]
- 13.Roman GC, Tatemichi TK, Erkinjuntti T, et al. Vascular dementia: diagnostic criteria for research studies: report of the NINDS-AIREN International Workshop. Neurology. 1993;43(2):250–260. doi: 10.1212/wnl.43.2.250. [DOI] [PubMed] [Google Scholar]
- 14.Pohjasvaara T, Mantyla R, Ylikoski R, Kaste M, Erkinjuntti T. Comparison of different clinical criteria (DSM-III, ADDTC, ICD-10, NINDS-AIREN, DSM-IV) for the diagnosis of vascular dementia. National Institute of Neurological Disorders and Stroke. Stroke. 2000;31(12):2952–2957. doi: 10.1161/01.str.31.12.2952. [DOI] [PubMed] [Google Scholar]
- 15.Sturrock RR. A quantitative lifespan study of changes in cell number, cell division and cell death in various regions of the mouse forebrain. Neuropathology and Applied Neurobiology. 1979;5(6):433–456. doi: 10.1111/j.1365-2990.1979.tb00642.x. [DOI] [PubMed] [Google Scholar]
- 16.Terry RD, DeTeresa R, Hansen LA. Neocortical cell counts in normal human adult aging. Annals of Neurology. 1987;21(6):530–539. doi: 10.1002/ana.410210603. [DOI] [PubMed] [Google Scholar]
- 17.Vincent SL, Peters A, Tigges J. Effects of aging on the neurons within area 17 of rhesus monkey cerebral cortex. Anatomical Record. 1989;223(3):329–341. doi: 10.1002/ar.1092230312. [DOI] [PubMed] [Google Scholar]
- 18.Merrill DA, Roberts JA, Tuszynski MH. Conservation of neuron number and size in entorhinal cortex layers II, III, and V/VI of aged primates. Journal of Comparative Neurology. 2000;422(3):396–401. doi: 10.1002/1096-9861(20000703)422:3<396::aid-cne6>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 19.Peters A, Leahu D, Moss MB, McNally KJ. The effects of aging on area 46 of the frontal cortex of the rhesus monkey. Cerebral Cortex. 1994;4(6):621–635. doi: 10.1093/cercor/4.6.621. [DOI] [PubMed] [Google Scholar]
- 20.Gazzaley AH, Thakker MM, Hof PR, Morrison JH. Preserved number of entorhinal cortex layer II neurons in aged macaque monkeys. Neurobiology of Aging. 1997;18(5):549–553. doi: 10.1016/s0197-4580(97)00112-7. [DOI] [PubMed] [Google Scholar]
- 21.Soininen H, Puranen M, Riekkinen PJ. Computed tomography findings in senile dementia and normal aging. Journal of Neurology Neurosurgery and Psychiatry. 1982;45(1):50–54. doi: 10.1136/jnnp.45.1.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scahill RI, Frost C, Jenkins R, Whitwell JL, Rossor MN, Fox NC. A longitudinal study of brain volume changes in normal aging using serial registered magnetic resonance imaging. Archives of Neurology. 2003;60(7):989–994. doi: 10.1001/archneur.60.7.989. [DOI] [PubMed] [Google Scholar]
- 23.Mungas D, Harvey D, Reed BR, et al. Longitudinal volumetric MRI change and rate of cognitive decline. Neurology. 2005;65(4):565–571. doi: 10.1212/01.wnl.0000172913.88973.0d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Freeman SH, Kandel R, Cruz L, et al. Preservation of neuronal number despite age-related cortical brain atrophy in elderly subjects without Alzheimer disease. Journal of Neuropathology and Experimental Neurology. 2008;67(12):1205–1212. doi: 10.1097/NEN.0b013e31818fc72f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grill JD, Riddle DR. Age-related and laminar-specific dendritic changes in the medial frontal cortex of the rat. Brain Research. 2002;937(1-2):8–21. doi: 10.1016/s0006-8993(02)02457-5. [DOI] [PubMed] [Google Scholar]
- 26.de Brabander JM, Kramers RJ, Uylings HB. Layer-specific dendritic regression of pyramidal cells with ageing in the human prefrontal cortex. European Journal of Neuroscience. 1998;10(4):1261–1269. doi: 10.1046/j.1460-9568.1998.00137.x. [DOI] [PubMed] [Google Scholar]
- 27.Uylings HBM, De Brabander JM. Neuronal changes in normal human aging and Alzheimer’s disease. Brain and Cognition. 2002;49(3):268–276. doi: 10.1006/brcg.2001.1500. [DOI] [PubMed] [Google Scholar]
- 28.Dickstein DL, Kabaso D, Rocher AB, Luebke JI, Wearne SL, Hof PR. Changes in the structural complexity of the aged brain. Aging Cell. 2007;6(3):275–284. doi: 10.1111/j.1474-9726.2007.00289.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Burke SN, Barnes CA. Neural plasticity in the ageing brain. Nature Reviews Neuroscience. 2006;7(1):30–40. doi: 10.1038/nrn1809. [DOI] [PubMed] [Google Scholar]
- 30.Rapp PR, Gallagher M. Preserved neuron number in the hippocampus of aged rats with spatial learning deficits. Proceedings of the National Academy of Sciences of the United States of America. 1996;93(18):9926–9930. doi: 10.1073/pnas.93.18.9926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kennedy KM, Raz N. Pattern of normal age-related regional differences in white matter microstructure is modified by vascular risk. Brain Research. 2009;1297:41–56. doi: 10.1016/j.brainres.2009.08.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kramer JH, Mungas D, Reed BR, et al. Longitudinal MRI and cognitive change in healthy elderly. Neuropsychology. 2007;21(4):412–418. doi: 10.1037/0894-4105.21.4.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Toescu EC. Normal brain ageing: models and mechanisms. Philosophical Transactions of the Royal Society B. 2005;360(1464):2347–2354. doi: 10.1098/rstb.2005.1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ames BN, Shigenaga MK, Hagen TM. Oxidants, antioxidants, and the degenerative diseases of aging. Proceedings of the National Academy of Sciences of the United States of America. 1993;90(17):7915–7922. doi: 10.1073/pnas.90.17.7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brunk UT, Terman A. The mitochondrial-lysosomal axis theory of aging: accumulation of damaged mitochondria as a result of imperfect autophagocytosis. European Journal of Biochemistry. 2002;269(8):1996–2002. doi: 10.1046/j.1432-1033.2002.02869.x. [DOI] [PubMed] [Google Scholar]
- 36.Toescu EC, Verkhratsky A. Parameters of calcium homeostasis in normal neuronal ageing. Journal of Anatomy. 2000;197(4):563–569. doi: 10.1046/j.1469-7580.2000.19740563.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cohen RA. Hypertension and cerebral blood flow: implications for the development of vascular cognitive impairment in the elderly. Stroke. 2007;38(6):1715–1717. doi: 10.1161/STROKEAHA.107.487165. [DOI] [PubMed] [Google Scholar]
- 38.Matsuda H, Maeda T, Yamada M. Age-matched normal values and topographic maps for regional cerebral blood flow measurements by Xe-133 inhalation. Stroke. 1984;15(2):336–342. doi: 10.1161/01.str.15.2.336. [DOI] [PubMed] [Google Scholar]
- 39.Iadecola C, Park L, Capone C. Threats to the mind: aging, amyloid, and hypertension. Stroke. 2009;40(3):S40–S44. doi: 10.1161/STROKEAHA.108.533638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pirson AS, Vander Borght T, Van Laere K. Age and gender effects on normal regional cerebral blood flow. American Journal of Neuroradiology. 2006;27(6):1161–1162. [PMC free article] [PubMed] [Google Scholar]
- 41.Zhou H, Saidel GM, Lamanna JC. Cerebral blood flow adaptation to chronic hypoxia. Advances in Experimental Medicine and Biology. 2008;614:371–377. doi: 10.1007/978-0-387-74911-2_41. [DOI] [PubMed] [Google Scholar]
- 42.Ritz MF, Fluri F, Engelter ST, Schaeren-Wiemers N, Lyrer PA. Cortical and putamen age-related changes in the microvessel density and astrocyte deficiency in spontaneously hypertensive and stroke-prone spontaneously hypertensive rats. Current Neurovascular Research. 2009;6(4):279–287. doi: 10.2174/156720209789630311. [DOI] [PubMed] [Google Scholar]
- 43.Angelopoulos P, Agouridaki H, Vaiopoulos H, et al. Cytokines in Alzheimer’s disease and vascular dementia. International Journal of Neuroscience. 2008;118(12):1659–1672. doi: 10.1080/00207450701392068. [DOI] [PubMed] [Google Scholar]
- 44.Campbell IL, Hofer MJ, Pagenstecher A. Transgenic models for cytokine-induced neurological disease. Biochimica et Biophysica Acta. 2009;1802(10):903–917. doi: 10.1016/j.bbadis.2009.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Campbell IL, Stalder AK, Akwa Y, Pagenstecher A, Asensio VC. Transgenic models to study the actions of cytokines in the central nervous system. NeuroImmunoModulation. 1998;5(3-4):126–135. doi: 10.1159/000026329. [DOI] [PubMed] [Google Scholar]
- 46.Akassoglou K, Bauer J, Kassiotis G, et al. Oligodendrocyte apoptosis and primary demyelination induced by local TNF/p55TNF receptor signaling in the central nervous system of transgenic mice: models for multiple sclerosis with primary oligodendrogliopathy. American Journal of Pathology. 1998;153(3):801–813. doi: 10.1016/S0002-9440(10)65622-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Batti L, O’Connor JJ. Tumor necrosis factor-α impairs the recovery of synaptic transmission from hypoxia in rat hippocampal slices. Journal of Neuroimmunology. 2010;218(1-2):21–27. doi: 10.1016/j.jneuroim.2009.11.002. [DOI] [PubMed] [Google Scholar]
- 48.Ginis I, Hallenbeck JM, Liu J, Spatz M, Jaiswal R, Shohami E. Tumor necrosis factor and reactive oxygen species cooperative cytotoxicity is mediated via inhibition of NF-kappaB. Molecular Medicine. 2000;6(12):1028–1041. [PMC free article] [PubMed] [Google Scholar]
- 49.Farfara D, Lifshitz V, Frenkel D. Neuroprotective and neurotoxic properties of glial cells in the pathogenesis of Alzheimer’s disease: Alzheimer’s Review Series. Journal of Cellular and Molecular Medicine. 2008;12(3):762–780. doi: 10.1111/j.1582-4934.2008.00314.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zuliani G, Cavalieri M, Galvani M, et al. Markers of endothelial dysfunction in older subjects with late onset Alzheimer’s disease or vascular dementia. Journal of the Neurological Sciences. 2008;272(1-2):164–170. doi: 10.1016/j.jns.2008.05.020. [DOI] [PubMed] [Google Scholar]
- 51.Zhang RL, Chopp M, Zhang ZG, et al. E-selectin in focal cerebral ischemia and reperfusion in the rat. Journal of Cerebral Blood Flow and Metabolism. 1996;16(6):1126–1136. doi: 10.1097/00004647-199611000-00006. [DOI] [PubMed] [Google Scholar]
- 52.Wakita H, Ruetzler C, Illoh KO, et al. Mucosal tolerization to E-selectin protects against memory dysfunction and white matter damage in a vascular cognitive impairment model. Journal of Cerebral Blood Flow and Metabolism. 2008;28(2):341–353. doi: 10.1038/sj.jcbfm.9600528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hu D, Serrano F, Oury TD, Klann E. Aging-dependent alterations in synaptic plasticity and memory in mice that overexpress extracellular superoxide dismutase. Journal of Neuroscience. 2006;26(15):3933–3941. doi: 10.1523/JNEUROSCI.5566-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Popa-Wagner A, Buga AM, Kokaia Z. Perturbed cellular response to brain injury during aging. Ageing Research Reviews. 2011;10(1):71–79. doi: 10.1016/j.arr.2009.10.008. [DOI] [PubMed] [Google Scholar]
- 55.Canuelo A, Siles E, Martinez-Romero R, Peinado MA, Martinez-Lara E. The nitric oxide system response to hypoxia/reoxygenation in the aged cerebral cortex. Experimental Gerontology. 2007;42(12):1137–1145. doi: 10.1016/j.exger.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 56.Mander P, Brown GC. Nitric oxide, hypoxia and brain inflammation. Biochemical Society Transactions. 2004;32(6):1068–1069. doi: 10.1042/BST0321068. [DOI] [PubMed] [Google Scholar]
- 57.Scorziello A, Pellegrini C, Secondo A, et al. Neuronal NOS activation during oxygen and glucose deprivation triggers cerebellar granule cell death in the later reoxygenation phase. Journal of Neuroscience Research. 2004;76(6):812–821. doi: 10.1002/jnr.20096. [DOI] [PubMed] [Google Scholar]
- 58.Adibhatla RM, Hatcher JF. Altered lipid metabolism in brain injury and disorders. Subcellular Biochemistry. 2008;49:241–268. doi: 10.1007/978-1-4020-8831-5_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tapia-Arancibia L, Aliaga E, Silhol M, Arancibia S. New insights into brain BDNF function in normal aging and Alzheimer disease. Brain Research Reviews. 2008;59(1):201–220. doi: 10.1016/j.brainresrev.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 60.Kennedy KM, Rodrigue KM, Land SJ, Raz N. BDNF Val66Met polymorphism influences age differences in microstructure of the Corpus Callosum. Frontiers in Human Neuroscience. 2009;3:p. 19. doi: 10.3389/neuro.09.019.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hota SK, Barhwal K, Singh SB, Ilavazhagan G. Chronic hypobaric hypoxia induced apoptosis in CA1 region of hippocampus: a possible role of NMDAR mediated p75NTR upregulation. Experimental Neurology. 2008;212(1):5–13. doi: 10.1016/j.expneurol.2008.01.030. [DOI] [PubMed] [Google Scholar]
- 62.Langa KM, Foster NL, Larson EB. Mixed dementia: emerging concepts and therapeutic implications. Journal of the American Medical Association. 2004;292(23):2901–2908. doi: 10.1001/jama.292.23.2901. [DOI] [PubMed] [Google Scholar]
- 63.Korczyn AD. Mixed dementia—the most common cause of dementia. Annals of the New York Academy of Sciences. 2002;977:129–134. doi: 10.1111/j.1749-6632.2002.tb04807.x. [DOI] [PubMed] [Google Scholar]
- 64.Jellinger KA, Attems J. Neuropathological evaluation of mixed dementia. Journal of the Neurological Sciences. 2007;257(1-2):80–87. doi: 10.1016/j.jns.2007.01.045. [DOI] [PubMed] [Google Scholar]
- 65.Jellinger KA, Attems J. Is there pure vascular dementia in old age? Journal of the Neurological Sciences. 2010;299(1-2):150–154. doi: 10.1016/j.jns.2010.08.038. [DOI] [PubMed] [Google Scholar]
- 66.Scheibel AB, Duong T, Jacobs R. Alzheimer’s disease as a capillary dementia. Annals of Medicine. 1989;21(2):103–107. doi: 10.3109/07853898909149194. [DOI] [PubMed] [Google Scholar]
- 67.De La Torre JC. Is Alzheimer’s disease a neurodegenerative or a vascular disorder? Data, dogma, and dialectics. Lancet Neurology. 2004;3(3):184–190. doi: 10.1016/S1474-4422(04)00683-0. [DOI] [PubMed] [Google Scholar]
- 68.Pluta R, Ułamek M. Brain ischemia and ischemic blood-brain barier as etiological factors in sporadic Alzheimer’s disease. Neuropsychiatric Disease and Treatment. 2008;4(5):855–864. doi: 10.2147/ndt.s3739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang X, Zhou K, Wang R, et al. Hypoxia-inducible factor 1α (HIF-1α)-mediated hypoxia increases BACE1 expression and β-amyloid generation. Journal of Biological Chemistry. 2007;282(15):10873–10880. doi: 10.1074/jbc.M608856200. [DOI] [PubMed] [Google Scholar]
- 70.Kieda C, Greferath R, Da Silva CC, Fylaktakidou KC, Lehn JM, Nicolau C. Suppression of hypoxia-induced HIF-1α and of angiogenesis in endothelial cells by myo-inositol trispyrophosphate-treated erythrocytes. Proceedings of the National Academy of Sciences of the United States of America. 2006;103(42):15576–15581. doi: 10.1073/pnas.0607109103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Biolo A, Greferath R, Siwik DA, et al. Enhanced exercise capacity in mice with severe heart failure treated with an allosteric effector of hemoglobin, myo-inositol trispyrophosphate. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(6):1926–1929. doi: 10.1073/pnas.0812381106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lukiw WJ, Zhao Y, Jian GC. An NF-κB-sensitive micro RNA-146a-mediated inflammatory circuit in alzheimer disease and in stressed human brain cells. Journal of Biological Chemistry. 2008;283(46):31315–31322. doi: 10.1074/jbc.M805371200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.von Bernhardi R, Ramirez G, Toro R, Eugenin J. Pro-inflammatory conditions promote neuronal damage mediated by Amyloid Precursor Protein and decrease its phagocytosis and degradation by microglial cells in culture. Neurobiology of Disease. 2007;26(1):153–164. doi: 10.1016/j.nbd.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 74.Cole GM, Morihara T, Lim GP, Yang F, Begum A, Frautschy SA. NSAID and antioxidant prevention of Alzheimer’s disease: lessons from in vitro and animal models. Annals of the New York Academy of Sciences. 2004;1035:68–84. doi: 10.1196/annals.1332.005. [DOI] [PubMed] [Google Scholar]
- 75.Munoz L, Ranaivo H, Roy SM, et al. A novel p38α MAPK inhibitor suppresses brain proinflammatory cytokine up-regulation and attenuates synaptic dysfunction and behavioral deficits in an Alzheimer’s disease mouse model. Journal of Neuroinflammation. 2007;4, article no. 21 doi: 10.1186/1742-2094-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lue LF, Brachova L, Civin WH, Rogers J. Inflammation, Aβ deposition, and neurofibrillary tangle formation as correlates of Alzheimer’s disease neurodegeneration. Journal of Neuropathology and Experimental Neurology. 1996;55(10):1083–1088. [PubMed] [Google Scholar]
- 77.Lanzrein AS, Johnston CM, Perry VH, Jobst KA, King EM, Smith AD. Longitudinal study of inflammatory factors in serum, cerebrospinal fluid, and brain tissue in Alzheimer disease: interleukin-1β, interleukin-6, interleukin-1 receptor antagonist, tumor necrosis factor-α, the soluble tumor necrosis factor receptors I and II, and α1-antichymotrypsin. Alzheimer Disease and Associated Disorders. 1998;12(3):215–227. doi: 10.1097/00002093-199809000-00016. [DOI] [PubMed] [Google Scholar]
- 78.Shepherd CE, Thiel E, McCann H, Harding AJ, Halliday GM. Cortical inflammation in Alzheimer disease but not dementia with Lewy bodies. Archives of Neurology. 2000;57(6):817–822. doi: 10.1001/archneur.57.6.817. [DOI] [PubMed] [Google Scholar]
- 79.Cutler RG, Kelly J, Storie K, et al. Involvement of oxidative stress-induced abnormalities in ceramide and cholesterol metabolism in brain aging and Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2004;101(7):2070–2075. doi: 10.1073/pnas.0305799101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Christen Y. Oxidative stress and Alzheimer disease. American Journal of Clinical Nutrition. 2000;71(2) doi: 10.1093/ajcn/71.2.621s. [DOI] [PubMed] [Google Scholar]
- 81.Tibolla G, Norata GD, Meda C, et al. Increased atherosclerosis and vascular inflammation in APP transgenic mice with apolipoprotein E deficiency. Atherosclerosis. 2010;210(1):78–87. doi: 10.1016/j.atherosclerosis.2009.10.040. [DOI] [PubMed] [Google Scholar]
- 82.Santpere G, Puig B, Ferrer I. Oxidative damage of 14-3-3 zeta and gamma isoforms in Alzheimer’s disease and cerebral amyloid angiopathy. Neuroscience. 2007;146(4):1640–1651. doi: 10.1016/j.neuroscience.2007.03.013. [DOI] [PubMed] [Google Scholar]
- 83.Bowman GL, Quinn JF. Alzheimer’s disease and the blood-brain barrier: past, present and future. Aging Health. 2008;4(1):47–57. doi: 10.2217/1745509X.4.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Carey BJ, Eames PJ, Blake MJ, Panerai RB, Potter JF. Dynamic cerebral autoregulation is unaffected by aging. Stroke. 2000;31(12):2895–2900. doi: 10.1161/01.str.31.12.2895. [DOI] [PubMed] [Google Scholar]
- 85.Shim YS, Yang DW, Kim BS, Shon YM, Chung YA. Comparison of regional cerebral blood flow in two subsets of subcortical ischemic vascular dementia: statistical parametric mapping analysis of SPECT. Journal of the Neurological Sciences. 2006;250(1-2):85–91. doi: 10.1016/j.jns.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 86.Schuff N, Matsumoto S, Kmiecik J, et al. Cerebral blood flow in ischemic vascular dementia and Alzheimer’s disease, measured by arterial spin-labeling magnetic resonance imaging. Alzheimer’s and Dementia. 2009;5(6):454–462. doi: 10.1016/j.jalz.2009.04.1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Palhagen SE, Ekberg S, Walinder J, Granerus AK, Granerus G. HMPAO SPECT in Parkinson’s disease (PD) with major depression (MD) before and after antidepressant treatment. Journal of Neurology. 2009;256(9):1510–1518. doi: 10.1007/s00415-009-5155-x. [DOI] [PubMed] [Google Scholar]
- 88.Shimizu S, Hanyu H, Hirao K, Sato T, Iwamoto T, Koizumi K. Value of analyzing deep gray matter and occipital lobe perfusion to differentiate dementia with Lewy bodies from Alzheimer’s disease. Annals of Nuclear Medicine. 2008;22(10):911–916. doi: 10.1007/s12149-008-0193-5. [DOI] [PubMed] [Google Scholar]
- 89.Matsuda T, Abe T, Wu JL, Fujiki M, Kobayashi H. Hypoxia-inducible factor-1α DNA induced angiogenesis in a rat cerebral ischemia model. Neurological Research. 2005;27(5):503–508. doi: 10.1179/016164105X25144. [DOI] [PubMed] [Google Scholar]
- 90.Patel NS, Mathura VS, Bachmeier C, et al. Alzheimer’s beta-amyloid peptide blocks vascular endothelial growth factor mediated signaling via direct interaction with VEGFR-2. Journal of Neurochemistry. 2009;112(1):66–76. doi: 10.1111/j.1471-4159.2009.06426.x. [DOI] [PubMed] [Google Scholar]
- 91.Solerte SB, Ferrari E, Cuzzoni G, et al. Decreased release of the angiogenic peptide vascular endothelial growth factor in alzheimer’s disease: recovering effect with insulin and DHEA sulfate. Dementia and Geriatric Cognitive Disorders. 2005;19(1):1–10. doi: 10.1159/000080963. [DOI] [PubMed] [Google Scholar]
- 92.Borroni B, Ghezzi S, Agosti C, et al. Preliminary evidence that VEGF genetic variability confers susceptibility to frontotemporal lobar degeneration. Rejuvenation Research. 2008;11(4):773–780. doi: 10.1089/rej.2008.0711. [DOI] [PubMed] [Google Scholar]
- 93.Campbell IL. Structural and functional impact of the transgenic expression of cytokines in the CNS. Annals of the New York Academy of Sciences. 1998;840:83–96. doi: 10.1111/j.1749-6632.1998.tb09552.x. [DOI] [PubMed] [Google Scholar]
- 94.Ravaglia G, Forti P, Maioli F, et al. Blood inflammatory markers and risk of dementia: the Conselice Study of Brain Aging. Neurobiology of Aging. 2007;28(12):1810–1820. doi: 10.1016/j.neurobiolaging.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 95.Hüll M, Strauss S, Berger M, Volk B, Bauer J. The participation of interleukin-6, a stress-inducible cytokine, in the pathogenesis of Alzheimer’s disease. Behavioural Brain Research. 1996;78(1):37–41. doi: 10.1016/0166-4328(95)00213-8. [DOI] [PubMed] [Google Scholar]
- 96.Silvestroni A, Faull RLM, Strand AD, Möllera T. Distinct neuroinflammatory profile in post-mortem human Huntington’s disease. NeuroReport. 2009;20(12):1098–1103. doi: 10.1097/WNR.0b013e32832e34ee. [DOI] [PubMed] [Google Scholar]
- 97.Campuzano O, Castillo-Ruiz MM, Acarin L, Castellano B, Gonzalez B. Increased levels of proinflammatory cytokines in the aged rat brain attenuate injury-induced cytokine response after excitotoxic damage. Journal of Neuroscience Research. 2009;87(11):2484–2497. doi: 10.1002/jnr.22074. [DOI] [PubMed] [Google Scholar]
- 98.Pettigrew LC, Kindy MS, Scheff S, et al. Focal cerebral ischemia in the TNFalpha-transgenic rat. Journal of Neuroinflammation. 2008;5, article no. 47 doi: 10.1186/1742-2094-5-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhao M, Cribbs DH, Anderson AJ, et al. The induction of the TNFα death domain signaling pathway in Alzheimer’s disease brain. Neurochemical Research. 2003;28(2):307–318. doi: 10.1023/a:1022337519035. [DOI] [PubMed] [Google Scholar]
- 100.Licastro F, Chiappelli M, Ruscica M, Carnelli V, Corsi MM. Altered cytokine and acute phase response protein levels in the blood of children with Downs syndrome, relationship with dementia of Alzheimer’s type. International Journal of Immunopathology and Pharmacology. 2005;18(1):165–172. doi: 10.1177/039463200501800117. [DOI] [PubMed] [Google Scholar]
- 101.Mogi M, Harada M, Riederer P, Narabayashi H, Fujita K, Nagatsu T. Tumor necrosis factor-α (TNF-α) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neuroscience Letters. 1994;165(1-2):208–210. doi: 10.1016/0304-3940(94)90746-3. [DOI] [PubMed] [Google Scholar]
- 102.Brionne TC, Tesseur I, Masliah E, Wyss-Coray T. Loss of TGF-β1 leads to increased neuronal cell death and microgliosis in mouse brain. Neuron. 2003;40(6):1133–1145. doi: 10.1016/s0896-6273(03)00766-9. [DOI] [PubMed] [Google Scholar]
- 103.Krupinski J, Kumar P, Kumar S, Kaluza J. Increased expression of TGF-β1 in brain tissue after ischemic stroke in humans. Stroke. 1996;27(5):852–857. doi: 10.1161/01.str.27.5.852. [DOI] [PubMed] [Google Scholar]
- 104.Blasko I, Stampfer-Kountchev M, Robatscher P, Veerhuis R, Eikelenboom P, Grubeck-Loebenstein B. How chronic inflammation can affect the brain and support the development of Alzheimer’s disease in old age: the role of microglia and astrocytes. Aging Cell. 2004;3(4):169–176. doi: 10.1111/j.1474-9728.2004.00101.x. [DOI] [PubMed] [Google Scholar]
- 105.Wyss-Coray T, Masliah E, Mallory M, et al. Amyloidogenic role of cytokine TGF-β1 in transgenic mice and in Alzheimer’s disease. Nature. 1997;389(6651):603–606. doi: 10.1038/39321. [DOI] [PubMed] [Google Scholar]
- 106.Ewers M, Mielke MM, Hampel H. Blood-based biomarkers of microvascular pathology in Alzheimer’s disease. Experimental Gerontology. 2009;45(1):75–79. doi: 10.1016/j.exger.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Peter K, Nawroth P, Conradt C, et al. Circulating vascular cell adhesion molecule-1 correlates with the extent of human atherosclerosis in contrast to circulating intercellular adhesion molecule-1, E-selectin, P-selectin, and thrombomodulin. Arteriosclerosis, Thrombosis, and Vascular Biology. 1997;17(3):505–512. doi: 10.1161/01.atv.17.3.505. [DOI] [PubMed] [Google Scholar]
- 108.Fassbender K, Bertsch T, Mielke O, Mühlhauser; F, Hennerici M. Adhesion molecules in cerebrovascular diseases: evidence for an inflammatory endothelial activation in cerebral large- and small-vessel disease. Stroke. 1999;30(8):1647–1650. doi: 10.1161/01.str.30.8.1647. [DOI] [PubMed] [Google Scholar]
- 109.Smith CD, Carney JM, Starke-Reed PE, et al. Excess brain protein oxidation and enzyme dysfunction in normal aging and in Alzheimer disease. Proceedings of the National Academy of Sciences of the United States of America. 1991;88(23):10540–10543. doi: 10.1073/pnas.88.23.10540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Choi K, Kim J, Kim GW, Chulhee C. Oxidative stress-induced necrotic cell death via mitochondira-dependent burst of reactive oxygen species. Current Neurovascular Research. 2009;6(4):213–222. doi: 10.2174/156720209789630375. [DOI] [PubMed] [Google Scholar]
- 111.Shi Q, Gibson GE. Oxidative stress and transcriptional regulation in Alzheimer disease. Alzheimer Disease and Associated Disorders. 2007;21(4):276–291. doi: 10.1097/WAD.0b013e31815721c3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Esteves AR, Arduino DM, Swerdlow RH, Oliveira CR, Cardoso SM. Oxidative stress involvement in alpha-synuclein oligomerization in Parkinson’s disease cybrids. Antioxidants and Redox Signaling. 2009;11(3):439–448. doi: 10.1089/ars.2008.2247. [DOI] [PubMed] [Google Scholar]
- 113.Miller RL, James-Kracke M, Sun GY, Sun AY. Oxidative and inflammatory pathways in Parkinson’s disease. Neurochemical Research. 2009;34(1):55–65. doi: 10.1007/s11064-008-9656-2. [DOI] [PubMed] [Google Scholar]
- 114.Yehuda S, Rabinovitz S, Carasso RL, Mostofsky DI. The role of polyunsaturated fatty acids in restoring the aging neuronal membrane. Neurobiology of Aging. 2002;23(5):843–853. doi: 10.1016/s0197-4580(02)00074-x. [DOI] [PubMed] [Google Scholar]
- 115.Recuero M, Vicente MC, Martínez-García A, et al. A free radical-generating system induces the cholesterol biosynthesis pathway: a role in Alzheimer’s disease. Aging Cell. 2009;8(2):128–139. doi: 10.1111/j.1474-9726.2009.00457.x. [DOI] [PubMed] [Google Scholar]
- 116.Jasinska-Myga B, Opala G, Goetz CG, et al. Apolipoprotein E gene polymorphism, total plasma cholesterol level, and Parkinson disease dementia. Archives of Neurology. 2007;64(2):261–265. doi: 10.1001/archneur.64.2.261. [DOI] [PubMed] [Google Scholar]
- 117.Ferreiro E, Resende R, Costa R, Oliveira CR, Pereira CMF. An endoplasmic-reticulum-specific apoptotic pathway is involved in prion and amyloid-beta peptides neurotoxicity. Neurobiology of Disease. 2006;23(3):669–678. doi: 10.1016/j.nbd.2006.05.011. [DOI] [PubMed] [Google Scholar]
- 118.Beal MF. Excitotoxicity and nitric oxide in Parkinson’s disease pathogenesis. Annals of Neurology. 1998;44(3 supplement 1):S110–S114. doi: 10.1002/ana.410440716. [DOI] [PubMed] [Google Scholar]
- 119.Tang TS, Slow E, Lupu V, et al. Disturbed Ca2+ signalling and apoptosis of medium spiny neurons in Huntington’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(7):2602–2607. doi: 10.1073/pnas.0409402102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Raza M, Deshpande LS, Blair RE, Carter DS, Sombati S, DeLorenzo RJ. Aging is associated with elevated intracellular calcium levels and altered calcium homeostatic mechanisms in hippocampal neurons. Neuroscience Letters. 2007;418(1):77–81. doi: 10.1016/j.neulet.2007.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Webster MJ, Herman MM, Kleinman JE, Shannon Weickert C. BDNF and trkB mRNA expression in the hippocampus and temporal cortex during the human lifespan. Gene Expression Patterns. 2006;6(8):941–951. doi: 10.1016/j.modgep.2006.03.009. [DOI] [PubMed] [Google Scholar]
- 122.Marini AM, Choi J, Labutta R. Synaptic deprivation and age-related vulnerability to hypoxic-ischemic neuronal injury: a hypothesis. Annals of the New York Academy of Sciences. 2001;939:238–253. doi: 10.1111/j.1749-6632.2001.tb03631.x. [DOI] [PubMed] [Google Scholar]
- 123.Walton M, Connor B, Lawlor P, et al. Neuronal death and survival in two models of hypoxic-ischemic brain damage. Brain Research Reviews. 1999;29(2-3):137–168. doi: 10.1016/s0165-0173(98)00053-8. [DOI] [PubMed] [Google Scholar]
- 124.Phillips HS, Hains JM, Armanini M, Laramee GR, Johnson SA, Winslow JW. BDNF mRNA is decreased in the hippocampus of individuals with Alzheimer’s disease. Neuron. 1991;7(5):695–702. doi: 10.1016/0896-6273(91)90273-3. [DOI] [PubMed] [Google Scholar]
- 125.Howells DW, Porritt MJ, Wong JYF, et al. Reduced BDNF mRNA expression in the Parkinson’s disease substantia nigra. Experimental Neurology. 2000;166(1):127–135. doi: 10.1006/exnr.2000.7483. [DOI] [PubMed] [Google Scholar]
- 126.Parain K, Murer MG, Yan Q, et al. Reduced expression of brain-derived neurotrophic factor protein in Parkinson’s disease substantia nigra. NeuroReport. 1999;10(3):557–561. doi: 10.1097/00001756-199902250-00021. [DOI] [PubMed] [Google Scholar]
- 127.Bull R, Finkelstein JP, Galvez J, et al. Ischemia enhances activation by Ca2+ and redox modification of ryanodine receptor channels from rat brain cortex. The Journal of Neuroscience. 2008;28(38):9463–9472. doi: 10.1523/JNEUROSCI.2286-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]