

Abstract
Fibrodysplasia Ossificans Progressiva (FOP) is a rare, autosomal dominant condition, classically characterised by heterotopic ossification beginning in childhood and congenital great toe malformations; occurring in response to a c.617 G>A ACVR1 mutation in the functionally important glycine/serine-rich domain of exon 6. Here we describe a novel c.587 T>C mutation in the glycine/serine-rich domain of ACVR1, associated with delayed onset of heterotopic ossification and an exceptionally mild clinical course. Absence of great toe malformations, the presence of early ossification of the cervical spine facets joints, plus mild bilateral camptodactyly of the 5th fingers, together with a novel ACVR1 mutation, are consistent with the ‘FOP-variant’ syndrome. The c.587 T>C mutation replaces a conserved leucine with proline at residue 196. Modelling of the mutant protein reveals a steric clash with the kinase domain that will weaken interactions with FKBP12 and induce exposure of the glycine/serine-rich repeat. The mutant receptor is predicted to be hypersensitive to ligand stimulation rather than being constitutively active, consistent with the mild clinical phenotype. This case extends our understanding of the ‘FOP-variant’ syndrome.
Keywords: Fibrodysplasia ossificans progressiva, FOP, ACVR1, Heterotopic ossification, Mutation
Introduction
Fibrodysplasia Ossificans Progressiva (FOP) (OMIM 135100) is a rare, autosomal dominant condition with complete penetrance [1]. However, most cases occur sporadically as reproductive capacity is challenged by the consequent disability. The global prevalence is estimated at 1 in 2 million, with no racial or gender associations [1,2]. Those affected develop heterotopic bone formation of tendons, ligaments, skeletal muscles and/or fascia, usually in childhood. Smooth and cardiac muscle is unaffected. Minor trauma and viral illness are sufficient to trigger painful flares of heterotopic ossification (HO); these can be unpredictable and spontaneous. Ossification is usually preceded by a period of soft tissue swelling and inflammation. Progressive extra-skeletal ankylosis restricts movement, often rendering patients’ wheelchair dependent by their 20’s. The earliest most recognizable feature of ‘classical’ FOP that may be observed at birth is congenital great toe malformations. Other additional features include short broad femoral necks, cervical spine fusion and development of osteochondromas.
In recent years, 8 activating single point mutations in the ACVR1 gene, coding for the transmembrane receptor Bone Morphogenetic Protein (BMP) type 1, have been identified in FOP patients (Table 1). Initially, following successful linkage analysis of 5 FOP families to 2q23-24 [1], the same heterozygous point mutation (617 G-A: R206H) was identified in 32 sporadic FOP cases [1]. Subsequently mutations at this same position have been identified in other FOP cases worldwide [3-5]. The non-synonymous 617 G-A mutation causes an R206H amino acid substitution at codon 206, which lies within a highly conserved glycine-serine (GS) activation domain close to the border with the protein kinase domain (Fig. 1A).
Table 1.
Summary of mutations in ACVR1 gene identified to date.
| ACVR1 mutation | ACVR1 Domain | Amino acid change |
Age of onset (years) |
Clinical Course | Additional features | Reference |
|---|---|---|---|---|---|---|
| c.617 G>A | GS | Arg206His | 0.3–14 | Moderate/severe | Classical FOP | [1,3-6] |
| c.605 G>T | GS | Arg202Ile | 14 | Moderate | Unilateral great toe malformation | [7] |
| c.590-592 delCTT | GS | Pro197-Phe198Leua | 11 | Moderate | Normal toes | [6] |
| c.619 C>G | GS | Gln207Glu | 0.5 | Severe | [6] | |
| c.983 G>A | Kinase | Gly328Glu | 2 | Slow | Cognitive impairment & extensive phalangeal deformities | [7,8] |
| Severe | Cognitive impairment & variable hair thinning | [2,6,9,10] | ||||
| c.982 G>A(orC) | Kinase | Gly328Argb | 21–26 | Slow | Variable great toe & thumb malformations | [6] |
| c.1067 G>A | Kinase | Gly356Asp | 10 | Slow | [11] | |
| c.774 G>C | Kinase | Arg258Ser | 4–14 | Slow | Variable great toe malformations | [9,12] |
| c.1124 G>C | Kinase | Arg375Pro | 14 | Slow | Normal toes | [6] |
| c.587 T>C | GS | Leu196Pro | 21 | Very slow | 5th finger camptodactyly, normal toes, cervical facet joint ossification |
NOVEL |
GS: Glycine/serine-rich domain; and kinase: protein kinase domain.
An in-frame3-base pair (CTT) heterozygous mutation replaces amino acids Proline (codon 197) and Phenylalanine (codon 198) with Leucine.
If Arg is replaced by Trp then onset 2–8 years & more severe clinical course [6].
Fig. 1.

Location of the novel L196P mutation. (A) Diagram illustrating the ACVR1 domain organisation with the L196P mutation shown in relation to the classical R206H FOP mutation. (B) Packing interactions of L196 in the wild-type ACVR1/FKBP12 structure. (C) The mutant L196 side chain would clash with I266 in the kinase domain (shown by a line of purple spheres). The GS and kinase domains are coloured orange and green, respectively. FKBP12 is coloured blue.
Ordinarily extracellular BMPs bind heterodimeric complexes of BMP type I and type II receptors, leading to GS domain phosphorylation and hence activation of the type I receptor, which in turn stimulates cytoplasmic Smad1/5/8 and p38MAPK signalling, altering gene transcription. In the presence of the R206H mutation, BMP type I receptors become mildly constitutively active, that is they are able to activate downstream signalling in the absence of ligand stimulation, leading to unregulated over active bone and cartilage formation in soft tissues [13-16].
More recently further new mutations in ACVR1 have been identified and the phenotype has been reported to vary prompting Shore & Kaplan to classify FOP syndromes into 3 groups, 1; the classical phenotype with progressive HO, malformed great toes and the c.617 G>A mutation (n=84), 2; the FOP-plus phenotype – HO, malformed great toes and unique additional features, either associated with c.617 G>A mutation or another ACVR1 mutation (n=8), 3; and the FOP-variant – HO, other digit deficits, variable additional features, other ACVR1 mutations (n=12) [6,14]. Here we report a novel coding mutation in ACVR1, with predicted changes in the GS domain protein structure causing an atypical phenotype with an exceptionally mild clinical course, consistent with a FOP-variant syndrome.
Clinical case
We report the case of a 45 year old woman with FOP; daughter of a Caucasian mother and Sikh father, with no family history of musculoskeletal disorder. Her 13 year old son remains asymptomatic and un-investigated.
Following an unremarkable childhood without major injury, she was involved in a motorbike accident at aged 21. Two months later she developed trismus requiring submandibular surgical exploration and release of the medial pterygoid muscle. Her trismus persisted and postoperative submandibular and epiglottic inflammation threatened her airway prompting a 3 week tracheostomy until the swelling subsided. CT imaging at the time confirmed pterygoid swelling and ossification. At presentation intramuscular analgesia had been given into her right thigh, where 3 weeks later, tender soft tissue swelling (18×13 cm) developed; a bone scan showed marked up-take in the thigh. Knee flexion was restricted, but was gradually alleviated by massage.
She remained asymptomatic until, aged 31 and 38 weeks through her first pregnancy, she required an emergency caesarean section for a placental abruption, requiring spinal anaesthetic and a 12 unit blood transfusion. Again, intramuscular thigh injections, this time on the left, prompted the development 4 days postpartum of tender inflammation within the left thigh. Prednisolone gave some relief; however thigh swelling and pain persisted with restricted hip and knee movement. X-rays showed mature soft tissue ossification within the right thigh, presumably related to the episode 10 years earlier (Fig. 2). MRI examination revealed soft tissue and intramuscular oedema of the left thigh. No ossification developed subsequently in her caesarean incision scar, or at the site of spinal injection.
Fig. 2.

Ossification within the soft tissues of the right thigh. The plain radiograph shows great strands and ribbons of ossification within the right thigh consistent with trauma from an intramuscular injection received 11 years earlier.
At the age of 42 she was involved in another car accident sustaining an impact from behind whilst stationary and wearing a seat belt. One week later she developed severe cervical, thoracic and lumbar spine pain, partly easing over 6 weeks. Cervical spine X-rays demonstrated cervical spondylosis and a suggestion of early ossification between the facets joints in the posterior aspect of cervical spine which had not been visible 11 years earlier. Seven months later she represented with abdominal wall, right flank, right shoulder and right breast pain. In 2006 a CT scan demonstrated diffuse swelling without ossification within her right rectus muscle, within her abdominal wall. It also showed, within the pelvis, the right obturator internus muscle to be abnormally enlarged with irregular areas of high density compatible with early ossification. Subsequent X-rays a year later showed ossification of the lumbar para-spinal muscles (Fig. 3) and ultrasound examination 2 years post injury also showed ossification within the right lower thoracic para-spinal muscles, with associated swelling and oedema within her skeletal muscles in the paralumbar and right breast areas. There was no ossification at the site of rectus muscle inflammation seen on the earlier CT in 2006. Following physiotherapy and hydrotherapy she now walks unaided for 600 m, swims 1500 m 3 times a week, drives a car, but remains unable to return to work in retail.
Fig. 3.
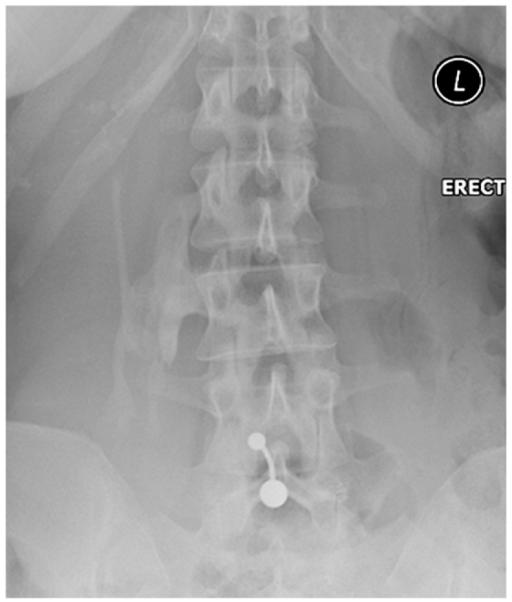
Ossification within the right para-spinal muscles. Plain radiograph of the lumbar spine demonstrates extensive ossification within the right para-spinal muscles.
On examination she is 165 cm, 64 kg and has 2 tattoos and 2 body-piercings without ossification. Her tracheostomy scar was not ossified, whilst the separate submandibular scar had associated ossification. She had restricted mouth opening. Her caesarean incision scar was not ossified; there were no palpable masses within the abdomen. She had bilateral 5th digit camptodactyly, but no other toe or finger abnormalities. There were non-tender bony masses palpable deep in both thighs in the region of the vastus lateralis muscles. Hip movements were full apart from moderately limited right hip internal rotation. She had an exaggerated spinal lumbar lordosis. Anterior flexion of the lumbar spine was restricted to 4 cm by anterior Schober’s and finger floor distance was 15 cm; lateral flexion normal. All other joints had full range of movement. The cervical spine was normal. A bony mass within the right lower back, within the lower erector spinae muscles, was just palpable, corresponding to the ossification seen on ultrasound. Hearing, visual acuity, cognition and scalp hair were normal.
Plain X-rays showed normal hand, feet and femoral neck morphology and no evidence of osteochondromas.
Methods
Informed consent was obtained for DNA testing. DNA was isolated from peripheral blood using the Flexigene™Kit (QiagenLtd, UK) according to the manufacturer’s instructions and re-suspended in the elution buffer. PCR primers (MWG Biotech AG, Edersberf, Germany) were designed to amplify exon 6 of ACVR1, which contains the c.617 G>A mutation. DNA was amplified using PCR with optimized MgCl2 concentrations and annealing temperatures using reagent from Sigma, UK. Amplified DNA was purified on filter plates (Millipore (UK) Ltd, Watford, UK) and sequenced in both forward and reverse directions with Big Dye version 3 using an automated sequencer (ABI3100, Applied Biosystems, Warrington, UK). Subsequently primers were designed for all 11 exons of ACVR1. To ensure that the mutation found in the patient was specifically associated with the condition, 100 healthy controls were also screened.
Results
Our patient was found to be heterozygous for a novel mutation c.587 T>C in exon 6 of the ACVR1 gene (Fig. 4). The c.587 T>C mutation corresponds to a residue change from leucine to proline at amino acid position 196 in the GS domain of the ACVR1 protein (L196P) (Fig. 1A). To determine the likely structural changes in the GS domain we modelled the mutant protein using internal coordinate modelling (ICM) [17] and the available crystal structure for the wild-type ACVR1/FKBP12 complex (PDB code 3h9r). The wild-type leucine side chain lies immediately downstream from the GS repeat and fills the hydrophobic core of the GS domain (Fig. 1B). Substitution of a proline side chain at this position is expected to result in a steric clash with the main-chain carboxyl of I266 (Fig. 1C) and force a moderate shift of the GS domain away from the kinase N-terminal lobe.
Fig. 4.

Heterozygosity for the novel mutation c.587 T>C in ACVR1. Direct DNA sequencing electropherogram showing patient heterozygosity for the novel mutation c.587 T>C in exon 6 of the ACVR1 gene and the resultant substitution of leucine at residue 196 in unaffected control with proline in the patient.
Discussion
We report a case of an FOP-variant phenotype with delayed clinical course, caused by a novel c.587 T>C mutation in exon 6 of the ACVR1 gene. Late onset of FOP, at 21 years of age, has only been described in three individuals and in these cases the mutation was located in the protein kinase domain rather than in the functionally important glycine/serine-rich domain where our novel mutation and the common R206H mutation are located [6] (Table 1). This case is atypical not only in respect of a delayed onset, but also her exceptionally mild clinical course, improved survival, reproductive capability, and unusually long intervals between traumatic triggers and ossification; whereas the intramuscular injections produced symptoms within 1–3 weeks, several months elapsed between road traffic injuries and symptom onset.
Classical FOP is characterised by two principle features; HO and congenital malformations of the great toes. In contrast to this our case has normal toes and instead only has mild bilateral camptodactyly of her 5th fingers — a common variant prevalent in 1–2% of the normal population [18]. Cervical spine facet joint ankylosis and degenerative changes have been reported in a majority of ‘FOP-plus’ cases [6]. Asymptomatic early ossification between the facets joints in the posterior aspect of her cervical spine [6] was the only variant FOP feature observed in this case; although consistent these cervical changes were milder in comparison with previous published reports [5-7]. Whilst attenuated, she still suffers from the unpredictable nature of FOP, characterised by her episodes of inflammation without subsequent ossification. Taken together this case fits with Shore & Kaplan’s definition of an FOP-variant syndrome [14]. However, this case has followed the most benign clinical course of any FOP case reported to date.
Recent evidence suggests that the R206H ACVR1 activating mutation responsible for classical FOP results from functional loss of the inhibitory activity of the negative regulator FK506 binding protein 12 kDa (FKBP12) [19]. FKBP12 binds type I receptors via the αGS2 helix interaction motif in the GS domain and inhibits signalling by shielding the serine and threonine residues from type II receptor-mediated phosphorylation [20-23]. The L196 position, affected in our patient, is conserved in all type I receptors and has been investigated by mutagenesis previously in TβR-I (L193). Substitutions of glycine or phenylalanine result in markedly reduced or normal FKBP12 binding, respectively [20], whereas alanine substitution produces an intermediate effect [24]. The novel mutation in our patient is also expected to reduce the effective affinity for FKBP12 as a result of a small steric shift of the GS domain, leading to enhanced ACVR1 activation. But as FKBP12 binding is not predicted to be completely attenuated, this might explain the relatively mild clinical phenotype seen in our case, in comparison with classic FOP.
Previous reports of a delayed FOP onset have been associated with mutations in the protein kinase domain [11]. The occurrence of a GS domain mutation with a slower and mild clinical course shows that the disease course is not related to a domain-specific effect, but rather depends upon the degree of structural perturbation within each mutant protein. In particular, our case suggests that GS domain mutations exist, which to a greater or lesser extent, diminish binding with the inhibitory protein FKBP12 and that the degree of this binding impairment may be reflected in the severity of the FOP phenotype. Knowledge gained regarding the structure and function of ACVR1 will hopefully help guide therapeutic developments for the future treatment of FOP.
Acknowledgments
The authors thank Dr J Pointon for her kind help with preparation of Fig. 4.
Role of the funding source
This work was supported by the University of Oxford FOP Research Fund; the Structural Genomics Consortium [to A.N.B], (registered charity number 1097737, that receives funds from the Canadian Institutes for Health Research; the Canadian Foundation for Innovation; Genome Canada through the Ontario Genomics Institute; GlaxoSmithKline; Karolinska Institute; the Knut and Alice Wallenberg Foundation; the Ontario Innovation Trust; the Ontario Ministry for Research and Innovation; Merck & Co Inc.; the Novartis Research Foundation; the Swedish Agency for Innovation Systems; the Swedish Foundation for Strategic Research; and The Wellcome Trust); and CLG is funded through a Wellcome Trust Clinical Research Training Fellowship [080280/Z/06/Z]. The funding sources had no involvement in the conduct of this research nor the preparation of the manuscript.
References
- [1].Shore EM, Xu M, Feldman GJ, Fenstermacher DA, Cho TJ, Choi IH, et al. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet. 2006;38:525–7. doi: 10.1038/ng1783. [DOI] [PubMed] [Google Scholar]
- [2].Connor JM, Evans DA. Fibrodysplasia ossificans progressiva. The clinical features and natural history of 34 patients. J Bone Joint Surg Br. 1982;64:76–83. doi: 10.1302/0301-620X.64B1.7068725. [DOI] [PubMed] [Google Scholar]
- [3].Lin GT, Chang HW, Liu CS, Huang PJ, Wang HC, Cheng YM. De novo 617 G-A nucleotide mutation in the ACVR1 gene in a Taiwanese patient with fibrodysplasia ossificans progressiva. J Hum Genet. 2006;51:1083–6. doi: 10.1007/s10038-006-0069-2. [DOI] [PubMed] [Google Scholar]
- [4].Nakajima M, Haga N, Takikawa K, Manabe N, Nishimura G, Ikegawa S. The ACVR1 617 G>A mutation is also recurrent in three Japanese patients with fibrodysplasia ossificans progressiva. J Hum Genet. 2007;52:473–5. doi: 10.1007/s10038-007-0128-3. [DOI] [PubMed] [Google Scholar]
- [5].Lee DY, Cho TJ, Lee HR, Park MS, Yoo WJ, Chung CY, et al. ACVR1 gene mutation in sporadic Korean patients with fibrodysplasia ossificans progressiva. J Korean Med Sci. 2009;24:433–7. doi: 10.3346/jkms.2009.24.3.433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kaplan FS, Xu M, Seemann P, Connor JM, Glaser DL, Carroll L, et al. Classic and atypical fibrodysplasia ossificans progressiva (FOP) phenotypes are caused by mutations in the bone morphogenetic protein (BMP) type I receptor ACVR1. Hum Mutat. 2009;30:379–90. doi: 10.1002/humu.20868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Petrie KA, Lee WH, Bullock AN, Pointon JJ, Smith R, Russell RG, et al. Novel mutations in ACVR1 result in atypical features in two fibrodysplasia ossificans progressiva patients. PLoS ONE. 2009;4:e5005. doi: 10.1371/journal.pone.0005005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Smith R, Russell RG, Woods CG. Myositis ossificans progressiva. Clinical features of eight patients and their response to treatment. J Bone Joint Surg Br. 1976;58:48–57. doi: 10.1302/0301-620X.58B1.818090. [DOI] [PubMed] [Google Scholar]
- [9].Bocciardi R, Bordo D, Di DM, Di RM, Ravazzolo R. Mutational analysis of the ACVR1 gene in Italian patients affected with fibrodysplasia ossificans progressiva: confirmations and advancements. Eur J Hum Genet. 2009;17:311–8. doi: 10.1038/ejhg.2008.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Carvalho DR, Navarro MM, Martins BJ, Coelho KE, Mello WD, Takata RI, et al. Mutational screening of ACVR1 gene in Brazilian fibrodysplasia ossificans progressiva patients. Clin Genet. 2010;77:171–6. doi: 10.1111/j.1399-0004.2009.01256.x. [DOI] [PubMed] [Google Scholar]
- [11].Furuya H, Ikezoe K, Wang L, Ohyagi Y, Motomura K, Fujii N, et al. A unique case of fibrodysplasia ossificans progressiva with an ACVR1 mutation, G356D, other than the common mutation (R206H) Am J Med Genet A. 2008;146A:459–63. doi: 10.1002/ajmg.a.32151. [DOI] [PubMed] [Google Scholar]
- [12].Ratbi I, Borcciadi R, Regragui A, Ravazzolo R, Sefiani A. Rarely occurring mutation of ACVR1 gene in Moroccan patient with fibrodysplasia ossificans progressiva. Clin Rheumatol. 2010;29:119–21. doi: 10.1007/s10067-009-1283-z. [DOI] [PubMed] [Google Scholar]
- [13].Shen Q, Little SC, Xu M, Haupt J, Ast C, Katagiri T, et al. The fibrodysplasia ossificans progressiva R206H ACVR1 mutation activates BMP-independent chondrogenesis and zebrafish embryo ventralization. J Clin Invest. 2009;119:3462–72. doi: 10.1172/JCI37412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Shore EM, Kaplan FS. Insights from a rare genetic disorder of extra-skeletal bone formation, fibrodysplasia ossificans progressiva (FOP) Bone. 2008;43:427–33. doi: 10.1016/j.bone.2008.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Fiori JL, Billings PC, DE LA Pena LS, Kaplan FS, Shore EM. Dysregulation of the BMP-p38 MAPK signaling pathway in cells from patients with fibrodysplasia ossificans progressiva (FOP) J Bone Miner Res. 2006;21:902–9. doi: 10.1359/jbmr.060215. [DOI] [PubMed] [Google Scholar]
- [16].Fukuda T, Kohda M, Kanomata K, Nojima J, Nakamura A, Kamizono J, et al. Constitutively activated ALK2 and increased SMAD1/5 cooperatively induce bone morphogenetic protein signaling in fibrodysplasia ossificans progressiva. J Biol Chem. 2009;284:7149–56. doi: 10.1074/jbc.M801681200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Abagyan RA, Totrov MM, Kuznetsov DA. ICM-A new method for protein modeling and design: applications to docking and structure prediction from the distorted native conformation. J Comp Chem. 1994;15:488–506. [Google Scholar]
- [18].Littman A, Yates JW, Treger A. Camptodactyly. A kindred study. JAMA. 1968;206:1565–7. doi: 10.1001/jama.206.7.1565. [DOI] [PubMed] [Google Scholar]
- [19].van Dinther M, Visser N, de Gorter DJ, Doorn J, Goumans MJ, de Boer J, et al. ALK2 R206H mutation linked to fibrodysplasia ossificans progressiva confers constitutive activity to the BMP type I receptor and sensitizes mesenchymal cells to BMP-induced osteoblast differentiation and bone formation. J Bone Miner Res. 2000:1–35. doi: 10.1359/jbmr.091110. [DOI] [PubMed] [Google Scholar]
- [20].Chen YG, Liu F, Massague J. Mechanism of TGFbeta receptor inhibition by FKBP12. EMBO J. 1997;16:3866–76. doi: 10.1093/emboj/16.13.3866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Huse M, Chen YG, Massague J, Kuriyan J. Crystal structure of the cytoplasmic domain of the type I TGF beta receptor in complex with FKBP12. Cell. 1999;96:425–36. doi: 10.1016/s0092-8674(00)80555-3. [DOI] [PubMed] [Google Scholar]
- [22].Okadome T, Oeda E, Saitoh M, Ichijo H, Moses HL, Miyazono K, et al. Characterization of the interaction of FKBP12 with the transforming growth factor-beta type I receptor in vivo. J Biol Chem. 1996;271:21687–90. doi: 10.1074/jbc.271.36.21687. [DOI] [PubMed] [Google Scholar]
- [23].Wang T, Donahoe PK. The immunophilin FKBP12: a molecular guardian of the TGF-beta family type I receptors. Front Biosci. 2004;9:619–31. doi: 10.2741/1095. [DOI] [PubMed] [Google Scholar]
- [24].Charng MJ, Kinnunen P, Hawker J, Brand T, Schneider MD. FKBP-12 recognition is dispensable for signal generation by type I transforming growth factor-beta receptors. J Biol Chem. 1996;271:22941–4. doi: 10.1074/jbc.271.38.22941. [DOI] [PubMed] [Google Scholar]