

Abstract
The roots of Krameria lappacea are used traditionally against oropharyngeal inflammation. So far, the astringent and antimicrobial properties of its proanthocyanidin constituents are considered to account for the anti-inflammatory effect. The aim of the present study was to characterize pharmacologically a lipophilic extract of K. lappacea roots and several isolated lignan derivatives (1–11) in terms of their putative anti-inflammatory activity. The dichloromethane extract (ID50 77 μg/cm2) as well compounds 1–11 (ID50 0.31–0.60 μmol/cm2) exhibited topical antiedematous properties comparable to those of indomethacin (ID50 0.29 μmol/cm2) in a mouse ear in vivo model. Two of the most potent compounds, 2-(2-hydroxy-4-methoxyphenyl)-5-(3-hydroxypropyl)benzofuran (5) and (+)-conocarpan (7), were studied regarding their time-dependent edema development and leukocyte infiltration up to 48 h after croton oil-induced dermatitis induction, and they showed activity profiles similar to that of hydrocortisone. In vitro studies of the isolated lignan derivatives demonstrated the inhibition of NF-κB, cyclooxygenase-1 and -2, 5-lipoxygenase, and microsomal prostaglandin E2 synthase-1 as well as antioxidant properties, as mechanisms possibly contributing to the observed in vivo effects. The present findings not only support the ethnopharmacological use of K. lappacea roots but also reveal that the isolated lignan derivatives contribute strongly to the anti-inflammatory activity of this herbal drug.
The roots of Krameria lappacea (Dombey) Burdet et Simpson (syn. K. triandra Ruiz et Pavon; Krameriaceae) (Ratanhiae Radix; rhatany) are used traditionally in South America as chewing sticks for cleaning and strengthening teeth, for reducing blood flow, and as an astringent against diarrhea and mouth ulcers.(1) At the end of the 18th century, this species was introduced into European medicine as a remedy against stomachache, diarrhea, menstrual problems, nose bleeds, and oropharyngeal inflammation.2,3 Today, the drug is listed in the European Pharmacopoeia and is the subject of an ESCOP monograph. Its therapeutic indications comprise mild incidences of inflammation of the mouth and throat such as stomatitis, gingivitis, and pharyngitis.(4) So far, various phenolic constituents have been reported as secondary metabolites of K. lappacea, ranging from medium to high molecular weight oligomeric proanthocyanidins(5) as major constituents (approximately 10%), to phlobaphenes and a series of lignan derivatives. Extracts of the roots, as well as some of the pure constituents, were shown to possess antimicrobial, antioxidant, and photoprotective activities.3,6−9 For the use of the drug against inflammation, vegetable tannins having astringent and antimicrobial properties are considered as the active components.(5) Lignan derivatives represent a group of constituents in the roots of K. lappacea, and previous studies have shown that members of this compound class may have the potential to act as anti-inflammatory agents.10,11 Thus, the aim of the present investigation was to examine whether lignans from K. lappacea may block inflammation. From the dichloromethane extract of K. lappacea roots, 11 lignan derivatives were isolated and identified. Since rhatany preparations are used mainly topically against infections and inflammation, as a first step a potential topical anti-inflammatory effect in vivo was assessed using a croton oil-induced mouse ear dermatitis model. Since the inflammatory response is a complex process, influenced by a number of different mediators, various molecular targets were evaluated to gain insight into the mode of action of the isolated secondary metabolites. Their effects were investigated on classical anti-inflammatory targets such as the glucocorticoid receptor (GR), cyclooxygenase-1 and -2 (COX-1 and -2), 5-lipoxygenase (5-LO), and nuclear factor kappa B (NF-κB), as well as on more recently studied therapeutic targets including the peroxisome proliferator-activated receptors (PPAR)-α and -γ and the inducible microsomal prostaglandin E2 synthase (mPGES)-1.
Results and Discussion
Isolation of Lignan Derivatives
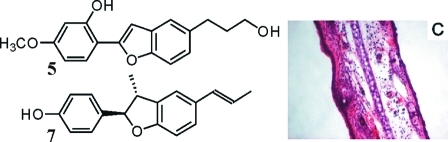
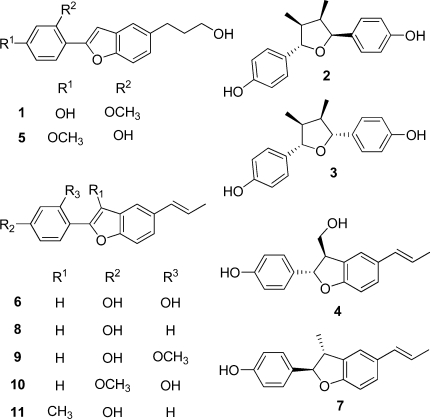
Preliminary tests revealed CH2Cl2 as a suitable extraction solvent for the roots of K. lappacea in order to maximize the yield of lignan derivatives and to minimize the vegetable tannin content in the resultant extract. Eleven previously known lignan derivatives, comprising neolignans, norneolignans, and 7,7′-epoxy lignans, were isolated using different chromatographic techniques. Identity in each case was verified by determination of the optical activity, 1D- and 2D-NMR experiments, and mass spectrometry, by comparison with published data,7,12−16 as 5-(3-hydroxypropyl)-2-(2-methoxy-4-hydroxyphenyl)benzofuran (1), (−)-larreatricin (2), meso-3,3′-didemethoxynectandrin B (3), (2S,3S)-2,3-dihydro-3-hydroxymethyl-2-(4-hydroxyphenyl)-5-(E)-propenylbenzofuran (4), 2-(2-hydroxy-4-methoxyphenyl)-5-(3-hydroxypropyl)benzofuran (5), 2-(2,4-dihydroxyphenyl)-5-(E)-propenylbenzofuran (6), (+)-conocarpan (7), 2-(4-hydroxyphenyl)-5-(E)-propenylbenzofuran (8), rataniaphenol III (9), rataniaphenol I (10), and rataniaphenol II (11).
In Vivo Antiedema Activities
The topical anti-inflammatory effect of the CH2Cl2 extract of K. lappacea roots as well as of the isolated lignan derivatives was determined as antiedema activity 6 h (the maximum of edema formation in control mice) after induction of dermatitis.(17) The extract exhibited a potent dose-dependent inhibition of edema, which ranged from 24% at the lowest dose (30 μg/cm2) to 86% for the highest administration (300 μg/cm2). All isolated lignan derivatives significantly reduced the edematous response from about 15% (0.1 μmol/cm2) to about 80% (1.0 μmol/cm2), in a dose-dependent manner. The same doses of the nonsteroidal anti-inflammatory drug (NSAID) indomethacin reduced the induced edema formation between 25% and 80%, while the glucocorticoid hydrocortisone showed edema inhibition from 29% to 77%, at 10 times lower doses (Table 1). To evaluate the anti-inflammatory potency of the extract as well as of the isolated compounds, ID50 values were assessed. The ID50 value of the extract was determined as 77 μg/cm2. The lignan derivatives showed ID50 values in the range 0.31–0.60 μmol/cm2, corresponding to 80–160 μg/cm2, which were comparable to indomethacin (ID50 0.29 μmol/cm2, corresponding to 104 μg/cm2) and about 10 to 20 times less potent compared to hydrocortisone (ID50 0.03 μmol/cm2, corresponding to 11 μg/cm2) (Table 1).
Table 1. Topical Anti-inflammatory Activity of the CH2Cl2 Extract and Lignan Derivatives (1–11) from K. lappacea.
| test substance | dose (μmol/cm2)/(μg/cm2) | edema (mg) mean ± SE | edema inhibition (%) | ID50 (μmol/cm2)/(μg/cm2) | ||
|---|---|---|---|---|---|---|
| control | 6.9 ± 0.2 | |||||
| CH2Cl2 ext. | 30 | 5.3 ± 0.3a | 24 | 77 | ||
| 100 | 2.9 ± 0.5a | 58 | ||||
| 300 | 1.0 ± 0.2a | 86 | ||||
| 1 | 0.10 | 30 | 6.0 ± 0.3a | 13 | 0.45 | 134 |
| 0.30 | 89 | 4.7 ± 0.3a | 32 | |||
| 1.00 | 298 | 1.7 ± 0.1a | 75 | |||
| 2 | 0.10 | 28 | 5.6 ± 0.3a | 19 | 0.46 | 131 |
| 0.30 | 85 | 4.2 ± 0.4a | 39 | |||
| 1.00 | 284 | 2.3 ± 0.2a | 67 | |||
| 3 | 0.10 | 28 | 5.9 ± 0.3a | 14 | 0.36 | 102 |
| 0.30 | 85 | 3.3 ± 0.3a | 52 | |||
| 1.00 | 284 | 1.9 ± 0.2a | 72 | |||
| 4 | 0.10 | 28 | 5.7 ± 0.4a | 17 | 0.42 | 118 |
| 0.30 | 85 | 4.4 ± 0.3a | 36 | |||
| 1.00 | 282 | 1.7 ± 0.2a | 75 | |||
| 5 | 0.10 | 30 | 5.2 ± 0.3a | 25 | 0.36 | 107 |
| 0.30 | 89 | 3.4 ± 0.2a | 50 | |||
| 1.00 | 298 | 2.3 ± 0.2a | 67 | |||
| 6 | 0.10 | 27 | 5.9 ± 0.3a | 15 | 0.60 | 160 |
| 0.30 | 80 | 4.9 ± 0.3a | 29 | |||
| 1.00 | 266 | 2.5 ± 0.3a | 64 | |||
| 7 | 0.10 | 27 | 5.2 ± 0.4a | 25 | 0.31 | 82 |
| 0.30 | 80 | 3.8 ± 0.2a | 45 | |||
| 1.00 | 266 | 1.3 ± 0.1a | 81 | |||
| 8 | 0.10 | 25 | 5.9 ± 0.3a | 14 | 0.44 | 110 |
| 0.30 | 75 | 4.5 ± 0.3a | 35 | |||
| 1.00 | 250 | 1.8 ± 0.3a | 74 | |||
| 9 | 0.10 | 28 | 6.1 ± 0.2 | 12 | 0.48 | 134 |
| 0.30 | 84 | 4.4 ± 0.3a | 35 | |||
| 1.00 | 280 | 2.1 ± 0.3a | 70 | |||
| 10 | 0.10 | 28 | 6.2 ± 0.3 | 9 | 0.57 | 160 |
| 0.30 | 84 | 4.6 ± 0.3a | 33 | |||
| 1.00 | 280 | 2.5 ± 0.3a | 64 | |||
| 11 | 0.10 | 26 | 6.0 ± 0.4 | 13 | 0.40 | 106 |
| 0.30 | 79 | 4.0 ± 0.2a | 41 | |||
| 1.00 | 264 | 1.7 ± 0.2a | 75 | |||
| indomethacin | 0.10 | 36 | 5.2 ± 0.4a | 25 | 0.29 | 104 |
| 0.30 | 107 | 3.4 ± 0.3a | 51 | |||
| 1.00 | 358 | 1.4 ± 0.2a | 80 | |||
| hydrocortisone | 0.01 | 4 | 4.9 ± 0.3a | 29 | 0.03 | 11 |
| 0.03 | 11 | 3.1 ± 0.3a | 55 | |||
| 0.10 | 36 | 1.6 ± 0.2a | 77 | |||
p < 0.05 in the analysis of variance, as compared with controls.
The anti-inflammatory activities of the most promising compounds, 5 and 7, at 0.4 μmol/cm2, a dose leading to about 50% edema reduction at 6 h, were investigated further with regard to both edema development and leukocyte infiltration up to 48 h after dermatitis induction and were compared to indomethacin (0.4 μmol/cm2) and hydrocortisone (0.04 μmol/cm2).
Edematous Response
The time-dependent effect of the test compounds on edema formation is represented in Figure 1. Control animals developed an edematous response still measurable after 48 h, with a maximum 6 h after croton oil application, followed by a progressive decrease. Compounds 5 and 7 exerted a significant inhibitory activity at each observation time, showing reductions in the ranges of 28–89% and 25–61%, respectively. Interestingly, despite the similar activity profile, compound 5 provoked maximum edema reduction after 3 h (89%), which declined to 29% reduction at 48 h. In contrast, (+)-conocarpan (7) exhibited a long-lasting steady anti-inflammatory effect, with a maximum response after 6 h (61%) and a still-pronounced activity at 48 h with edema reduction by 41%. The effect of an equimolar dose of indomethacin was significant only 3 and 6 h after the induction of dermatitis, when it reduced the edematous response by 84% and 76%, respectively. Indomethacin then lost its antiedematous effect substantially, as previously observed.(17) Hydrocortisone (0.04 μmol/cm2) reduced edema formation at all observation times significantly, from 79% (6 h) to 60% (12 h) (Figure 1).
Figure 1.

Effect of compounds 5 and 7 and the reference drugs, indomethacin and hydrocortisone, on the time course of the edematous response up to 48 h (● controls; ◼ 0.4 μmol/cm2 compound 5; ◻ 0.4 μmol/cm2 compound 7; ▲ 0.4 μmol/cm2 indomethacin; Δ 0.04 μmol/cm2 hydrocortisone); *p < 0.05 in the analysis of variance, as compared to controls. Each point represents the mean of the results from 10 mice.
The activity profile of the two benzofuran derivatives 5 and 7 and of the reference drugs on the whole edematous response up to 48 h was quantified by calculating the ratio between the AUCs for mice treated with these compounds and the AUCs of control animals. Compounds 5 and 7 reduced the global edematous response by the same extent (47%, and 45%, respectively), exerting an effect 2-fold more potent than that of indomethacin (24% reduction) but significantly lower compared to hydrocortisone (69% reduction) (see Table S1, Supporting Information).
Cellular Infiltration
The effect of the test compounds on leukocyte infiltration up to 48 h is represented in Figure 2. The recruitment of leukocytes in the inflamed ear tissue of control animals, measured as myeloperoxidase activity, was already detectable 3 h after induction of dermatitis. It increased up to 24 h and slightly decreased until 48 h. Compounds 5 and 7 caused a significant reduction of leukocyte infiltration at all observation times, ranging from 24% to 35% and 27% to 44% inhibition, respectively. Indomethacin and hydrocortisone exerted comparable effects (27–49% and 35–56% reductions, respectively), which were significant at each observation time (Figure 2).
Figure 2.

Effect of compounds 5 and 7 and the reference drugs, indomethacin and hydrocortisone, on the time-course of leukocyte infiltration measured as myeloperoxidase activity up to 48 h (● controls; ◼ 0.4 μmol/cm2 compound 5; ◻ 0.4 μmol/cm2 compound 7; ▲ 0.4 μmol/cm2 indomethacin; Δ 0.04 μmol/cm2 hydrocortisone); *p < 0.05 in the analysis of variance, as compared to controls. Each point represents the mean of the results from 10 mice.
The effects of the benzofurans on the global granulocyte infiltrate calculated from the AUCs represented in Figure 2 (32% and 37% reduction) were comparable to those of the reference drugs indomethacin (42% reduction) and hydrocortisone (51% reduction) (see Table S1, Supporting Information).
Histological Analysis
The anti-inflammatory effects of compounds 5 and 7 and of the reference compounds were evaluated additionally by the histological examination of ear tissues. Ear dermal tissue of the control mice showed degranulated mast cells visible as soon as 3 h after dermatitis induction (see Figure S1B, Supporting Information). Dilated blood vessels and dermal swelling were also observable, becoming more evident after 6 h (Figure 3B) and progressively attenuating after 9 h. Moreover, an increased number of infiltrated neutrophilic granulocytes was visible after 6 h, then increasing up to 24 h (see Figure S2B, Supporting Information) and being still sustained after 48 h. Ears of mice treated with compound 5 or compound 7 (0.4 μmol/cm2) showed a general reduction of these vascular and cellular changes due to inflammation, including the presence of mast cells preserved from degranulation (Figures 3C, S1C, S2C; 3D, S1D, S2D, Supporting Information). Similarly, ear tissues from mice treated with indomethacin (0.4 μmol/cm2) showed reduced mast cell degranulation and leukocyte infiltration at all observation times. However, after 9 h the dermal swelling was comparable to that of control mice (Figure 3E, Figures S1E, and S2E, Supporting Information). Ear biopsies from animals treated with hydrocortisone (0.04 μmol/cm2) revealed an attenuation of all the vascular and cellular signs of inflammation (Figure 3F, Figures S1F, and S2F, Supporting Information).
Figure 3.
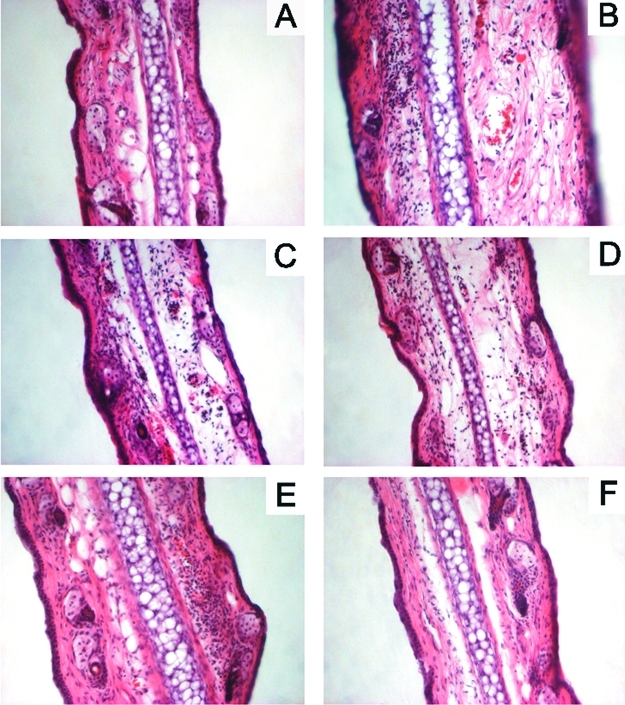
Sections of mouse ears 6 h after the induction of the croton oil dermatitis (A: untreated ear; B: control; C: 0.4 μmol/cm2 compound 5; D: 0.4 μmol/cm2 compound 7; E: 0.4 μmol/cm2 indomethacin; F: 0.04 μmol/cm2 hydrocortisone). Hematoxylin and eosin staining, 25× magnification.
Inhibition of NF-κB Activity
To obtain insight into the molecular mechanism(s) of the observed in vivo effects, the K. lappacea root CH2Cl2 extract was tested initially for its potential to inhibit NF-κB transactivation activity in TNF-α-stimulated HEK-293 cells stably transfected with a NF-κB-driven luciferase reporter gene. Since this crude extract showed a pronounced activity (>50% inhibition) at a concentration of 10 μg/mL, IC50 values for all isolated lignan derivatives (1–11) were determined. Interestingly, all compounds significantly reduced NF-κB-dependent luciferase activity in a concentration-dependent manner. Four compounds (5, 6, 8, and 11) were able to inhibit NF-κB activation in the low micromolar range, comparably to the positive control, parthenolide (IC50 1.4 μM), with IC50 values ranging from 1.4 to 6.4 μM. Another four isolates (1, 2, 4, 10) showed IC50 values between 11.6 and 14.7 μM. Only three compounds (3, 7, 9) exhibited low NF-κB inhibition with IC50 values higher than 20 μM (Table 2). Although the compounds tested show different substitution patterns, no clear structure–activity relationship conclusions could be deduced. Since luciferase reporter gene expression reflects a downstream event of the NF-κB signaling cascade that did not permit conclusions to be made regarding direct targets of the respective test compounds, all compounds were tested also for their potential to inhibit IKK2 (inhibitor of NF-κB kinase subunit beta) activity, as previously described.(18) However, none of the compounds inhibited IKK2 at a concentration of 10 μM (data not shown), thereby excluding this kinase as a direct target of the lignans. For comparison, the reference compound used as a positive control in the IKK2 test, IKK2 inhibitor IV, is known to have an IC50 in the low nM range and in our hands completely suppressed IKK2 enzymatic activity to the background level when applied at a concentration of 400 nM. Since GR as well as PPARα and -γ are known to have NF-κB-antagonist action, the potential of all lignan derivatives to activate these receptors using luciferase reporter gene assays specific for PPARα, PPARγ, and GR was assessed.19,20 Given that none of the compounds tested showed activities in any of the three assays (data not shown) at 10 μM, effects at these three nuclear receptors can be excluded. For comparison, the reference compound used for GR activation, dexamethasone, induced around a 10-fold signal induction at 2.5 μM. Troglitazone used as a reference PPARγ activator induced a 6-fold activation with an EC50 of 0.4 μM, and the positive control for PPARα activation, GW7647, induced a 2.8-fold activation, with an EC50 of 0.6 nM.
Table 2. IC50 Values of the Isolated Lignan Derivatives (1–11) in NF-κB, COX-1, COX-2, 5-LO, mPGES-1, and DPPH Assays (values in μM).
| compound | inhibition of NF-κB | inhibition of COX 1 | inhibition of COX 2 | inhibition of mPGES-1 | inhibition of 5-LO | radical scavenging activity |
|---|---|---|---|---|---|---|
| parthenolide | 1.4 ± 0.3 | |||||
| indomethacin | 2.1 ± 1.3 | |||||
| NS-398 | 2.1 ± 1.1 | |||||
| MK-886 | 2.1 ± 0.4 | |||||
| zileuton | 6.3 ± 1.4 | |||||
| ascorbic acid | 24.2 ± 5.1 | |||||
| 1 | 13.1 ± 3.7 | >50 | >50 | >50 | 41.4 ± 7.5 | 37.7 ± 4.4 |
| 2 | 11.6 ± 1.8 | >50 | >50 | >50 | >50 | |
| 3 | >20 | >50 | >50 | >50 | >50 | |
| 4 | 14.2 ± 2.3 | >50 | >50 | 39.5 ± 4.9 | >50 | |
| 5 | 5.9 ± 1.2 | >50 | >50 | 42.0 ± 7.4 | 27.2 ± 8.8 | 42.3 ± 2.6 |
| 6 | 3.4 ± 1.1 | 3.8 ± 0.8 | 7.1 ± 1.0 | 7.4 ± 1.7 | >50 | 22.1 ± 5.2 |
| 7 | >20 | >50 | >50 | 19.5 ± 0.8 | 18.4 ± 3.4 | |
| 8 | 1.4 ± 0.3 | 18.3 ± 4.3 | 2.0 ± 0.6 | 5.3 ± 1.3 | >50 | |
| 9 | >20 | 2.7 ± 2.1 | 7.6 ± 1.8 | 13.5 ± 2.8 | >50 | 36.3 ± 6.7 |
| 10 | 14.7 ± 3.2 | >50 | >50 | 13.0 ± 2.2 | >50 | 24.2 ± 5.8 |
| 11 | 6.4 ± 1.1 | 3.0 ± 0.7 | 1.7 ± 0.4 | 11.0 ± 1.6 | >50 |
Free-Radical-Scavenging Activity
Reactive oxygen species are known to be involved in the activation of NF-κB. To check the potential radical-scavenging activity of the lignan derivatives, a DPPH assay was performed. The K. lappacea root extract significantly inhibited free-radical formation with an IC50 value of 42.4 ± 6.3 μg/mL. Compounds 2–4, 7, 8, and 11 had no effect in this assay up to 100 μM. Five compounds (1, 5, 6, 9, and 10) showed free-radical scavenging activity in a concentration-dependent manner with IC50 values ranging from 22 to 42 μM. A structure–activity relationship comparison revealed that all the compounds active in the DPPH assay have a methoxy (1 and 9) or hydroxy (5, 6, and 10) group located at the ortho-position in relation to the benzofuran scaffold. Although these values are in the same range as the radical-scavenging activity of the positive control ascorbic acid (IC50 24.2 ± 5.1 μM) (Table 2), there was no apparent correlation between the DPPH-radical-scavenging potential of the compounds and their NF-κB inhibitory potential.
Targets of the Arachidonic Acid Cascade
To clarify the impact of the K. lappacea lignans on further targets, the isolated constituents were investigated for their potential to interfere with another crucial pathway in the inflammatory response, namely, the arachidonic acid cascade, which is responsible for the formation of pro-inflammatory prostaglandins and leukotrienes. Important enzymes are COX-1 and -2 and mPGES-1, catalyzing the formation of PGG2 and PGE2, and 5-LO, which is responsible for the production of leukotrienes via the intermediate molecule 5-HPETE.
Inhibition of PGE2 Formation via COX-1 and COX-2
Initial screening experiments of the CH2Cl2-soluble K. lappacea root extract (50 μg/mL) revealed inhibition of COX-1 and COX-2, with inhibition rates of 82.5 ± 8.9% and 83.9 ± 5.8%. Four (6, 8, 9, 11) of the 11 isolated lignan constituents (50 μM) inhibited these enzymes to an extent of 57.2% and 83.3% (data not shown). IC50 value determination of the most potent substances revealed that compounds 6, 9, and 11 had no isoform-specificity since they showed almost identical IC50 values, 2.7–3.8 μM for COX-1 and 1.7–7.6 μM for COX-2, comparable to the reference drugs indomethacin (IC50 value COX-1, 2.1 μM) and NS-398 (IC50 value COX-2, 2.1 μM). In contrast, compound 8 inhibited preferentially COX-2 (IC50 value COX-2, 2.0 μM; IC50 value COX-1, 18.3 μM) (Table 2). Accordingly, the 5-(E)-propenylbenzofuran moiety with an unsaturated furan ring and a para-hydroxy group seems to be essential for this activity.
Inhibition of 5-LO-Mediated LTB4 Formation
Investigation of the plant CH2Cl2 extract (50 μg/mL) and the 11 pure compounds (50 μM) revealed a significant inhibition of leukotriene formation by the extract (69.5 ± 4.1%) as well as by three of the 11 lignan derivatives (1, 5, 7; 80.2% to 94.6% inhibition, data not shown). IC50 values were determined for the three most active constituents. (+)-Conocarpan (7) showed the strongest activity, with an IC50 value of 18.4 μM, followed by compounds 5 (IC50 27.2 μM) and 1 (IC50 41.4 μM). Compared to the positive control zileuton (IC50 6.3 μM), only compound 7 showed a relevant activity in the ex vivo–in vitro setup (Table 2).
Inhibition of mPGES-1
In addition to the previous experiments, the isolated lignan derivatives were assessed for their ability to interfere with mPGES-1 in a cell-free assay. Therefore, an initial screening experiment with all pure compounds at a concentration of 10 μM was performed, resulting in the identification of compounds 6 and 8 as potent inhibitors of mPGES-1. Both compounds blocked PGE2 formation in a concentration-dependent manner, with IC50 values of 7.4 and 5.3 μM, respectively (Table 2). As a reference, the IC50 value of compound MK-886 was 2.1 μM (Table 2). Compounds 4, 5, 7, and 9–11 showed moderate inhibitions, with IC50 values ranging from 11.0 to 42.0 μM. The 7,7′-epoxy lignans (2, 3) as well as compound 1 were regarded as inactive in this model (IC50 values >50 μM).
Taken together it has been shown that several of the lignan derivatives investigated possess a pronounced topical anti-inflammatory activity in vivo, comparable to that of indomethacin, and about 7 times less compared to hydrocortisone. An attempt to clarify the mode of action of the pure compounds revealed two relevant pathways, namely, the NF-κB-pathway, where almost all investigated compounds showed activity, and selected enzymes of the arachidonic acid cascade, which were influenced by only some of the benzofuran derivatives. Since the in vivo and in vitro effects determined, especially for the most in vivo active compounds, 5 and 7, did not always correspond, it can be concluded that additional inflammatory mediators might contribute to the anti-inflammatory activities observed. The elucidation of the responsible target(s) within the NF-κB pathway as well as the possible impact on other inflammatory cascades is part of an ongoing study. The present findings support the medicinal use of the roots of K. lappacea in the treatment of oropharyngeal inflammation as well as the contribution of lignan derivatives to the anti-inflammatory activity of this herbal product. The actions of the pure compounds isolated on multiple targets may explain the promising anti-inflammatory activity of the CH2Cl2-soluble extract of K. lappacea roots. Considering its potency (higher than indomethacin), this extract may be able to be developed as a pharmaceutical agent for the treatment of topical inflammatory processes, after suitable standardization and clinical safety evaluations.
Experimental Section
General Experimental Procedures
The optical rotations were measured in MeOH using a Perkin-Elmer 341 polarimeter (Wellesley, MA) at 25 °C. 1D and 2D NMR experiments were recorded on a Bruker DRX 300 (Bruker Biospin Rheinstetten, Germany) operating at 300.13 MHz (1H) and 75.47 MHz (13C) at 300 K in acetone-d6 with 0.03% TMS (Eurisotop, Gif-sur-Yvette, France), which was used as internal standard. ESIMS were obtained on an Esquire 3000plus mass spectrometer (Bruker Daltonics, Bremen, Germany), using the following parameters: split, 1:5; alternating mode; spray voltage, 4.5 kV, 350 °C; dry gas, 10.0 L/min; nebulizer 40 psi; full scan mode, m/z 100–1500. For fractionation a P.C. Inc. (Potomac; MD; model HSCCC multilayer (triple) coil, ser. 690) HSCCC instrument with a Gilson 302/803 C pump system model 302 (Villiers-la-Bel, France) as well as a Dionex system with a P580 pump, ASI-100 autosampler, UVD 170U detector, and a Gilson 206 fraction collector (semipreparative HPLC) were used.
All solvents used for isolation were purchased from VWR International (Darmstadt, Germany). Solvents for HPLC were obtained from Merck (Darmstadt, Germany). Ultrapure water was produced by a Sartorius Arium 611 UV water purification system (Göttingen, Germany). Croton oil, indomethacin, hydrocortisone, tetramethylbenzidine (TMB), hexadecylammonium bromide (HTAB), and 96-well microtiter plates were purchased from Sigma-Aldrich (Milan, Italy). Ketamine hydrochloride (Inoketam 100) was purchased from Virbac srl (Milan, Italy). Other chemicals used for in vivo experiments were of analytical grade and purchased from Carlo Erba (Milan, Italy). Dulbecco’s modified Eagle’s medium (DMEM) containing 4.5 g/L glucose was purchased from Lonza Group AG (Basel, Switzerland). Fetal bovine serum was from Invitrogen (Lofer, Austria). Chemicals used to measure NF-κB transactivation activity were purchased from Sigma-Aldrich (Vienna, Austria). A stable NF-κB luciferase 293 reporter cell line (HEK-293/NFκB-luc) was purchased from Panomics (Redwood City, CA), and the plasmid pEGFP-C1 was from Clontech (St-Quentin-en-Yvelines, France). Parthenolide was obtained from Alexis Biochemicals/Enzo Life Sciences (Lörrach, Germany). Prostaglandin H synthase 1 and 2 and NS-398 were purchased from Cayman Chemical Company (Ann Arbor, MI), while the competitive PGE2 EIA kit was purchased from Assay Designs Inc. (Ann Arbor, MI). Indomethacin was purchased from ICN (Aurora, OH), and zileuton was a product from Sequoia (Oxford, UK). Dexamethasone, 2,2-diphenyl-1-picrylhydrazyl (DPPH), and ascorbic acid were from Sigma-Aldrich (Vienna, Austria). GW7647 and troglitazone were purchased from Cayman Europe (Tallinn, Estonia). MK-886 (3-[1-(4-chlorobenzyl)-3-tert-butylthio-5-isopropylindol-2-yl]-2,2-dimethylpropanoic acid) was purchased from Cayman Chemical Company (Ann Arbor, MI), and A549 cells were from Dr. Thorsten Maier, University of Frankfurt.
Plant Material
Dried, ground roots of K. lappacea (500 g; KL 6269) were purchased from Mag. Pharm. Kottas-Heldenberg & Sohn, Vienna, Austria. Identity as well as quality was in accordance with the monograph of the European Pharmacopoeia. A voucher specimen (KL 6269) authenticated by Dr. Daniel Remias, University of Innsbruck, Austria, is deposited at the Institute of Pharmacy/Pharmacognosy, University of Innsbruck (Austria).
Extraction and Isolation
Ground roots (300 g) were exhaustively extracted with CH2Cl2 in a Soxhlet apparatus for five days. The extract was evaporated to dryness, yielding 16.3 g. A portion (15.5 g) of the obtained extract was separated by flash silica gel 60 (40–63 μm, Merck, Darmstadt, Germany; 270 g, 50 × 4 cm) column chromatography, using petroleum ether/CH2Cl2/EtOAc gradient mixtures of increasing polarity, yielding 29 fractions (A1–A29). Fraction A23 (CH2Cl2/EtOAc, 70:30; 2.3 g) was chromatographed further by flash silica gel CC (70 g, 30 × 2.5 cm), using a hexane/EtOAc gradient with an increasing amount of EtOAc, yielding 18 fractions (B1–B18). Fraction B11 (hexane/EtOAc, 50:50; 609.0 mg) was fractionated further over Sephadex LH-20 (Pharmacia Biotech, Uppsala, Sweden) (75 × 1.5 cm), using a CH2Cl2/acetone mixture (85:15) as mobile phase, yielding 83.6 mg of compound 5 (915–975 mL elution volume) and 60.5 mg of compound 1 (1100–1160 mL elution volume). Fraction B5 (hexane/EtOAc, 65:35; 68.3 mg) was purified by Sephadex LH-20 CC (40 × 1.0 cm), using a CH2Cl2/acetone mixture (85:15) as mobile phase, affording 43.0 mg of compound 2 (35–42 mL elution volume). Fraction B6 (hexane/EtOAc, 65–35; 175.1 mg) was fractionated by Sephadex LH20 CC (70 × 1.5 cm) using acetone as mobile phase, yielding 12 fractions (C1–C12). Compound 3 (20.16–21.70 min retention time; 12.2 mg) was obtained by separation of fraction C7 (260–275 mL elution volume; 35.8 mg) by semipreparative HPLC (Phenomenex Synergy Max-RP column (10 μm; 250 × 10 mm); 48% acetonitrile/methanol (75:25), 52% water, isocratic; flow 4.00 mL/min; 30 °C). Fraction B7 (hexane/EtOAc, 60–40; 82.1 mg) was purified further by Sephadex LH-20 CC (75 × 1.5 cm) using CH2Cl2/acetone (85:15) as mobile phase, resulting in 24.4 mg of compound 4 (370–475 mL elution volume). Fraction A20 (petroleum ether/CH2Cl2/EtOAc, 30:66.5:3.5 to 10:85.5:4.5; 882.3 mg) was rechromatographed by Sephadex LH-20 CC (70 × 1.5 cm) using acetone as mobile phase, yielding 12 fractions (D1–D12). Fractions D9 and D10 (290–350 mL elution volume; total 297.0 mg) were combined and purified by Sephadex LH-20 CC (75 × 1.5 cm) using CH2Cl2/acetone (85:15) as mobile phase, affording 64.0 mg of compound 6 (885–925 mL elution volume). Fraction A11 (petroleum ether/CH2Cl2/EtOAc, 55:42.7:2.3; 1.3 g) was further separated by Sephadex LH-20 CC (70 × 1.5 cm) using acetone as mobile phase, resulting in 13 fractions (E1–E13). Purification of fraction E6 (240–250 mL elution volume; 339.7 mg) by Sephadex LH-20 CC (75 × 1.5 cm) using a CH2Cl2/acetone mixture (85:15) resulted in 265.6 mg of compound 7 (508–534 mL elution volume). Fraction A10 (petroleum ether/CH2Cl2/EtOAc, 60:38:2; 1.6 g) was rechromatographed by Sephadex LH-20 CC (75 × 1.5 cm) using CH2Cl2/acetone (85:15), affording 17 fractions (F1–F17). Fraction F15 (1165–1260 mL elution volume; 81.6 mg) was recrystallized from acetone/hexane, yielding 33.2 mg of off-white crystals. These were further purified by semipreparative HPLC (Phenomenex Aqua C18 column (5 μm, 250 × 10 mm); 70% acetonitrile, 30% water, isocratic; flow 3.00 mL/min; 40 °C). Purification of the collected peak (15.25–17.91 min retention time, 23.1 mg) by Sephadex LH-20 CC (35 × 1.0 cm), using acetone as mobile phase, yielded 12.0 mg of compound 8 (24–52 mL elution volume). Fraction E10 (305–330 mL elution volume; 294.3 mg) was further separated by Sephadex LH-20 CC (70 × 1.5 cm), using CH2Cl2/acetone (85:15) as mobile phase, resulting in eight fractions (G1–G8). Compound 9 (460–480 mL elution volume; 22.5 mg) was obtained by separation of combined fractions G4 and G5 (250–320 mL elution volume; total 192.3 mg) by HSCCC. Parameters: hexane/EtOAc/MeOH/CH3CN, 10:3:3:5; upper phase used as mobile phase; tail to head mode; coil volume 230 mL; flow 1 mL/min; 800 rpm. Fraction A6 (petroleum ether/CH2Cl2/EtOAc, 75:23.7:1.3; 130.5 mg) was further purified by Sephadex LH-20 CC (75 × 1.5 cm) using a CH2Cl2/acetone mixture (85:15) as mobile phase, yielding 84.7 mg of compound 10 (350–430 mL elution volume). Fraction A9 (petroleum ether/CH2Cl2/EtOAc, 65:33.2:1.8; 227.5 mg) was separated by Sephadex LH-20 CC (70 × 1.5 cm) using acetone as mobile phase, yielding 120.3 mg of compound 11 (255–335 mL elution volume). The identities of the isolated compounds were confirmed by physical and spectroscopic methods (optical rotation, 1D- and 2D-NMR, and LC-MS) and by comparison with published data.7,12−16 The purity of all isolated compounds was ≥96% (determined by HPLC).
Croton Oil-Induced Dermatitis
Male CD-1 mice weighing 28–32 g were supplied by Harlan Laboratories (San Pietro al Natisone, Italy). Topical inflammation was induced on the right ear (surface: about 1 cm2) of anesthetized mice (145 mg/kg ketamine hydrochloride, intraperitoneally) applying 80 μg of croton oil dissolved in 15 μL of acetone. The left ear remained untreated, since preliminary experiments showed that the vehicle (acetone) neither affected the inflammatory response nor induced irritation. Control animals received only the irritant solution, whereas other animals received both the irritant and the test substances dissolved in acetone.(17) At different times after dermatitis induction, animals were sacrificed and a punch (6 mm ⦶) was taken from both ears to evaluate the edematous response. Ten animals were used for each group of treatment. All animal experiments complied with the Italian D.L. n. 116 of January 27, 1992, and associated guidelines in the European Communities Council Directive of November 24, 1986 (86/609 ECC), concerning animal welfare and Appendix A of the European Convention ETS 123.
Evaluation of the Edematous Response
Edema was quantified by the difference in weight between the punches taken from the treated and untreated (opposite) ears. The antiedema activity was expressed as percent inhibition of the edematous response in animals treated with the test substances in comparison to edema of control animals treated with the irritant alone.(17) The overall effect of the test substances on edema development up to 48 h was quantified by calculating the areas under the curves (AUCs) representing the edematous response up to 48 h and, subsequently, by the ratio between the AUCs of these animals and the AUCs of controls.
Evaluation of the Granulocyte Infiltrate
The cellular infiltrate was quantified by measuring myeloperoxidase activity, as an index of the presence of neutrophilic granulocytes, in the same plug of treated ears used to measure edema. Myeloperoxidase was extracted by HTAB, according to the method of Bradley et al.,(21) and the enzyme activity was measured by a colorimetric assay using TMB as chromogen.(22) Each ear plug, suspended in 1 mL of buffered saline (0.1 M sodium acetate buffer at pH 4.2), containing 0.1% HTAB (w/v), was homogenized by Ultra-Turrax (Ika-Werk, Staufen, Germany) for 5 s at 20 000 rpm. The homogenate was centrifuged at 15000g for 5 min, and the supernatant was used for the colorimetric assay, because preliminary experiments revealed that the pellet contained less than 5% of total myeloperoxidase activity. In each well of a 96-well microplate, 25 μL of the supernatant were mixed with 50 μL of the chromogen solution (2.83 mM TMB dissolved in 0.1 M sodium acetate buffer at pH 4.2, containing 0.1% (w/v) HTAB). The enzyme reaction was started by adding 75 μL of 0.7 mM hydrogen peroxide. After 5 min of incubation at 25 °C, the reaction was blocked by 50 μL of 4 M acetic acid, containing 10 nM sodium azide. The absorbance was read at 620 nm using an automated microplate reader (Bio-Tek Instruments, Winooski, VT). Myeloperoxidase activity was expressed as enzyme units in 1 mL of supernatant. One unit of peroxidase activity was defined as the amount of enzyme oxidizing 1 nM of TMB/min. The enzyme activity of each sample was determined in duplicate. The global effect of the tested substances on the whole cellular infiltrate up to 48 h was quantified by calculating the AUCs representing the time course of myeloperoxidase activity up to 48 h and, subsequently, the ratio between AUCs of these animals and AUCs of controls.
Histological Analysis
Ear biopsies, fixed in 10% formalin, were dehydrated in ascending grades of ethanol, cleared in xylene, and embedded in paraffin wax. Sections (10 μm) were stained with hematoxylin-eosin or Giemsa and evaluated using a light microscope (Zeiss Axiophot, with Photometrics Cool Snaps Camera and the RS-image program).
Cell Culture
HEK-293/NFκB-luc cells were maintained at 37 °C and 5% CO2 in DMEM with phenol red supplemented with 2 mM glutamine, 100 U/mL benzylpenicillin, 100 μg/mL streptomycin, and 10% fetal bovine serum. A549 cells were cultured in DMEM/high glucose (4.5 g/L) medium supplemented with heat-inactivated fetal calf serum, 10% v/v, 100 U/mL penicillin, and 100 μg/mL streptomycin at 37 °C and 5% CO2.
NF-κB Transactivation Activity
HEK-293 cells stably transfected with a NF-κB luciferase reporter were seeded in 10 cm dishes and transfected with 5 μg of pEGFP-C1. Six hours later, the cells were seeded in 96-well plates and incubated at 37 °C and 5% CO2 overnight. On the next day, the medium was exchanged with a serum-free DMEM and cells were treated with the respective test compounds dissolved in dimethyl sulfoxide (DMSO). To avoid nonspecific effects of the solvent, the final concentration of DMSO was always adjusted to 0.1%. One hour after the treatment the cells were stimulated with 2 ng/mL human recombinant TNF-α for 6 h, and, after a lyses step, the luminescence of the firefly luciferase and the fluorescence of EGFP were quantified on a GeniosPro plate reader (Tecan; Grödig, Austria). The luciferase signal derived from the NF-κB reporter was normalized by the EGFP-derived fluorescence to account for differences in the cell number or transfection efficiency.
In Vitro Assays for PGE2 Formation via COX-1, COX-2, and LTB4 Formation Inhibitory Activity
Cyclooxygenase-1 (COX-1) and cyclooxygenase-2 (COX-2) inhibition assays were performed in a 96-well-plate format with purified prostaglandin H synthase 1 (PGHS-1) from ram seminal vesicles for COX-1 and purified PGHS-2 from sheep placental cotyledons for COX-2, as previously described.(23) The concentration of prostaglandin E2 (PGE2), the main arachidonic acid metabolite in this reaction, was determined using a competitive PGE2 EIA kit. Indomethacin (IC50 COX-1, 2.1 μM) and NS-398 (IC50 COX-2, 2.1 μM) were used as positive controls. The bioassay for inhibition of 5-LO-mediated leukotriene B4 (LTB4) formation was carried out in a 96-well-plate format with stimulated human neutrophilic granulocytes, as described by Adams et al.(24) with slight modifications.(25) Zileuton (IC50 6.3 μM) was used as positive control. Test samples were dissolved in absolute ethanol.
Preparation of Crude mPGES-1 in Microsomes of A549 Cells and Determination of mPGES-1 Enzymatic Activity
Determination of mPGES-1 activity was performed as described previously.(26) In brief, A549 cells were treated with 1 ng/mL interleukin-1β for 48 h at 37 °C and 5% CO2. After cell harvesting and sonification, the homogenate was subjected to differential centrifugation at 10000g for 10 min and 174000g for 1 h at 4 °C. The obtained pellet (microsomal fraction) was resuspended in 1 mL of homogenization buffer (0.1 M potassium phosphate buffer pH 7.4, 1 mM phenylmethylsulfonyl fluoride, 60 μg/mL soybean trypsin inhibitor, 1 μg/mL leupeptin, 2.5 mM glutathione, and 250 mM sucrose), and the total protein concentration was determined. Microsomal membranes were diluted in potassium phosphate buffer (0.1 M, pH 7.4) containing 2.5 mM glutathione. Test compounds, MK-886 (reference inhibitor), or vehicle was added, and after 15 min at 4 °C, the reaction (100 μL total volume) was initiated by addition of PGH2 (20 μM, final concentration, unless stated otherwise). After 1 min at 4 °C, the reaction was terminated using stop solution (100 μL; 40 mM FeCl2, 80 mM citric acid, and 10 μM 11β-PGE2 as internal standard). PGE2 was separated by solid-phase extraction and analyzed by RP-HPLC as described.(26)
Statistical Analysis
Pharmacological in vivo data were analyzed by one-way analysis of variance, followed by Dunnett’s test for multiple comparisons of unpaired data. The dose giving a 50% inhibition of the edematous response (ID50) was calculated by graphic interpolation of the logarithmic dose–effect curves. To calculate the IC50 values regarding NF-κB inhibition, at least three different concentrations measured in quadruplicate in three independent transfection experiments were used utilizing nonlinear regression with Data Analysis Toolbox software (MDL Information Systems Inc., Nashville, TN). For the determination of IC50 values in the remaining in vitro assays, samples were tested in at least three different concentrations (duplicates; at least three independent experiments). Calculation of IC50 values was performed by nonlinear regression using SigmaPlot 9.0 (Systat Software Inc., San Jose). Probability levels below 0.05 were considered as statistically significant. Results are given as means ± SD.
Acknowledgments
This work was supported by grants from the Austrian Science Fund (FWF) [NFN S10703-B03, S10704-B03, and S10705-B03]. The authors thank E. P. Ellmerer for recording of NMR spectra.
Supporting Information Available
Sections of mouse ears 3 h (Figure S1) and 24 h (Figure S2) after phlogosis induction, AUCs of edematous response and leukocyte infiltrate (Table S1), as well as a description of the in vitro assays used for the measurement of IKK2 activity, GR, PPARα and -γ activation, and free-radical-scavenging activity are available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- Simpson B. B. In Flora Neotropica; New York Botanical Garden: New York, 1989; pp 1–108. [Google Scholar]
- Simpson B. B. Syst. Bot. 1991, 45, 397–409. [Google Scholar]
- Carini M.; Aldini G.; Orioli M.; Facino R. M. Planta Med. 2002, 68, 193–197. [DOI] [PubMed] [Google Scholar]
- ESCOP Monographs Supplement 2009; Georg Thieme Verlag: Stuttgart, 2009; pp 213–216. [Google Scholar]
- Scholz E.; Rimpler H. Planta Med. 1989, 55, 379–384. [DOI] [PubMed] [Google Scholar]
- Stahl E.; Ittel I. Planta Med. 1981, 42, 144–154. [DOI] [PubMed] [Google Scholar]
- Arnone A.; Di Modugno V.; Nasini G.; Venturini I. Gazz. Chim. Ital. 1988, 118, 675–682. [Google Scholar]
- Arnone A.; Di Modugno V.; Nasini G.; De Pava O. V. Gazz. Chim. Ital. 1990, 120, 397–401. [Google Scholar]
- Maffei Facino R.; Carini M.; Aldini G.; De Angelis L. Rapid Commun. Mass Spectrom. 1997, 11, 1303–1308. [Google Scholar]
- Schühly W.; Hüfner A.; Pferschy-Wenzig E. M.; Prettner E.; Adams M.; Bodensieck A.; Kunert O.; Oluwemimo A.; Haslinger E.; Bauer R. Bioorg. Med. Chem. 2009, 17, 4459–4465. [DOI] [PubMed] [Google Scholar]
- Saleem M.; Kim H. J.; Ali M. S.; Lee Y. S. Nat. Prod. Rep. 2005, 22, 696–716. [DOI] [PubMed] [Google Scholar]
- Achenbach H.; Gross J.; Dominguez X. A.; Cano G.; Verde Star J.; Brussolo L. D. C.; Munoz G.; Salgado F.; López L. Phytochemistry 1987, 26, 1159–1166. [Google Scholar]
- Clive D. L. J.; Stoffman E. J. L. Org. Biomol. Chem. 2008, 6, 1831–1842. [DOI] [PubMed] [Google Scholar]
- Moinuddin S. G. A.; Hishiyama S.; Cho M.; Davin L. B.; Lewis N. G. Org. Biomol. Chem. 2003, 1, 2307–2313. [DOI] [PubMed] [Google Scholar]
- Achenbach H.; Utz W.; Lozano B.; Guajardo Touché E. M.; Moreno S. Phytochemistry 1996, 43, 1093–1095. [Google Scholar]
- Achenbach H.; Gross J.; Bauereiss P.; Dominguez X. A.; Sanchez Vega H.; Verde Star J.; Romboldt C. Phytochemistry 1989, 28, 1959–1962. [Google Scholar]
- Tubaro A.; Dri P.; Delbello G.; Zilli C.; Della Loggia R. Agents Actions 1985, 17, 347–349. [DOI] [PubMed] [Google Scholar]
- Noha S.; Atanasov A. G.; Schuster D.; Markt P.; Fakhrudin N.; Heiss E. H.; Schrammel O.; Rollinger J. M.; Stuppner H.; Dirsch V. M.; Wolber G. Bioorg. Med. Chem. Lett. 2011, 21, 577–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fakhrudin N.; Ladurner A.; Atanasov A. G.; Heiss E. H.; Baumgartner L.; Markt P.; Schuster D.; Ellmerer E.-P.; Wolber G.; Rollinger J. M.; Stuppner H.; Dirsch V. M. Mol. Pharmacol. 2010, 77, 559–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raivio T.; Palvimo J. J.; Kannisto S.; Voutilainen R.; Janne O. A. J. Clin. Endocr. Metab. 2002, 87, 3740–3744. [DOI] [PubMed] [Google Scholar]
- Bradley P. P.; Priebat D. A.; Christensen R. D.; Rothstein G. J. Invest. Dermatol. 1982, 78, 206–209. [DOI] [PubMed] [Google Scholar]
- Andrews P. C.; Krinsky N. I. J. Biol. Chem. 1981, 256, 4211–4218. [PubMed] [Google Scholar]
- Reininger E. A.; Bauer R. Phytomedicine 2006, 13, 164–169. [DOI] [PubMed] [Google Scholar]
- Adams M.; Kunert O.; Haslinger E.; Bauer R. Planta Med. 2004, 70, 904–908. [DOI] [PubMed] [Google Scholar]
- Knödler M.; Conrad J.; Wenzig E. M.; Bauer R.; Lacorn M.; Beifuss U.; Carle R.; Schieber A. Phytochemistry 2008, 69, 988–993. [DOI] [PubMed] [Google Scholar]
- Koeberle A.; Siemoneit U.; Buhring U.; Northoff H.; Laufer S.; Albrecht W.; Werz O. J. Pharmacol. Exper. Ther. 2008, 326, 975–982. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.