

Abstract
Liver regeneration, following partial hepatectomy (PHx) occurs through precisely controlled and synchronized cell proliferation, in which quiescent hepatocytes undergo 1–2 rounds of replication, with restoration of liver mass and function. We previously demonstrated that loss of the Smad3/4 adaptor protein β-2 spectrin (β2SP) is associated with faster entry into S phase, and hepatocellular cancer formation. These observations led us to further pursue the role of β2SP in cell cycle progression in vivo. Liver regeneration studies with partial hepatectomy (PHx) in β2SP+/− mice reveal a surprising and significant decrease in Liver/body weight ratio at 48 hours after PHx in β2SP+/− mice in comparison to wild type mice. At 48 hours after PHx, we also observe decreased levels of cyclin E (2.4-fold, P <0.05), Cdk1 (7.2-fold, P <0.05), cyclin A, pRb (Ser249/Thr252), PCNA, cyclin D1 with elevated levels of pCdk1 (Thr14) (3.6-fold, P <0.05). Strikingly, at 24 hours elevated levels of p53 (4-fold, P <0.05), phospho-p53 (ser15 and ser20) and p21 (200-fold, P <0.05) persisting to 48 hours after PHx further correlated with raised expression of the DNA damage markers pChk2 (Thr68) and γH2AX (S139). However, compromised cell cycle progression with loss of β2SP is not rescued by inhibiting p53 function, and that G2/M phase arrest observed is independent and upstream of p53.
Conclusions
β2SP deficiency results in dysfunctional hepatocyte cell cycle progression and delayed liver regeneration at 48 hours after PHx, which is p53 independent. β2SP loss may increase susceptibility to DNA damage, impair cell cycle progression, and ultimately lead to Hepatocellular Cancer.
Keywords: β2SP, p53, liver regeneration, DNA damage
Introduction
Liver regeneration represents an example of precisely controlled and synchronized cell proliferation in vivo. Following two-thirds partial hepatectomy (PHx), 95% of normally quiescent hepatocytes exit G0, rapidly re-enter the cell cycle, and undergo one or two rounds of replication, with restoration of liver mass and function (1). Cell cycle progression proceeds in a synchronized pattern following PHx. In mid to late G1, phosphorylation of the retinoblastoma protein (Rb) by Cdk4/6-cyclin D complexes initiates the cell cycle and mediates the G1/S-phase transition (2). Cdk2 then successively associates with cyclins E and A, completes phosphorylation of Rb, promotes activation of the DNA replication machinery, and regulates centrosome duplication, completing the transition into S phase. Cdk1, in association with cyclins A and B, is then essential for entry and exit from mitosis. Cyclin D1 has been demonstrated to be activated by 6 hours, and maximal levels of Cdk4 are present at 24 hours after PHx in rats (3). Cdk1 is sharply induced between 18 and 24 hours, followed by a transient decrease, before another increase at 30 hours post-PHx in rats (4). In most mouse strains, it takes 28–34 hours for quiescent (G0) hepatocytes to enter the cell cycle (G1 phase) and DNA synthesis (S phase) peaks at 40–44 hours post-PHx. Restoration of liver mass is nearly complete by 5–7 days in rodents and by 3–4 months in humans (5).
However, little is known about the mechanisms that inhibit proliferation and return hepatocytes to quiescence after regeneration is complete. Cyclin-dependent kinase-inhibitory proteins (CKIs) such as p21 have been demonstrated to be induced during G1 and peak during the post-replicative phase (48–72 hours) after PHx, whereas p27 is expressed in quiescent liver and is only minimally induced during the regenerative process (6). Similarly, TGF-β signaling has been demonstrated to reversibly inhibit the proliferative response following partial hepatectomy (7). TGF-β1 synthesis is up-regulated at 4 hours with peak expression at 72 hours following PHx, and expression of downstream Smad proteins phospho-Smad2, Smad2, and Smad4 are significantly elevated (5, 8, 9). TGF-β type II receptor (TBRII)-conditional knockout mice demonstrate accelerated proliferation and an increased liver-mass to body weight ratio following PHx (10).
We have previously demonstrated the role of a non-pleckstrin homology (PH) domain β-general-spectrin, β2SP (also known as embryonic liver fodrin, ELF, or spectrin β, non-erythrocytic 1 isoform 2), as a Smad3/4 adaptor protein, which regulates TGF-β signaling. We have also demonstrated that β2SP is a key suppressor of tumorigenesis in hepatocellular carcinoma (11, 12). Reduced β2SP expression is observed in human hepatocellular carcinoma specimens, and nearly 40% of β2SP+/− mice spontaneously develop hepatocellular carcinoma by 15 months of age (12, 13). Recently, we have demonstrated that loss of β2SP is associated with an expansion of hepatic progenitor cells in the portal tracts of β2SP+/− mice during liver regeneration (14).
These results led us to further evaluate the role of β2SP in liver regeneration and specifically hepatocyte cell cycle progression. Given its role as a Smad3/4 adaptor protein in TGF-β signaling, we hypothesized that loss of β2SP would result in accelerated proliferation, following partial hepatectomy. Our analysis, however, demonstrated that loss of β2SP results in a delay in liver regeneration. This defect appears to be mediated by dysfunctional expression of cell cycle proteins and by increased DNA damage. This study suggests a unique role for β2SP in liver regeneration, coordinated DNA synthesis, and cell cycle progression and provides further insight into its potential tumor-suppressive function in hepatocellular cancer.
Methods
Animals and Experimental Protocols
Wild-type and β2SP+/− 129 SvEv mice 8–12 weeks of age were subjected to two-thirds partial hepatectomy (PHx) (15). All mice were maintained as described previously (16). All mice underwent PHx between 0900 and 1200 hour (17) and were then sacrificed at 0, 24, 48, 72, and 168 hours following PHx. Liver tissues were collected for immunohistochemical, protein, and RNA analyses.
Protein Extraction and Western Blots
Whole-cell lysates were prepared from pooled livers (n ≥ 3) from each experimental group. Western blot analysis was performed as described previously (16) (Supplementary table 1).
Immunohistochemical Studies
Sections from mouse liver following partial hepatectomy were prepared and processed for immunohistochemistry as described previously (16) (Supplementary table 1).
Cell Cycle Analysis
For cell cycle analysis, wild-type and β2SP−/− MEFs were plated overnight in serum-containing media. The following day, MEFs were transduced with p53shRNA (Supplementary table 1) and further cultured for 48 hours. Control MEFs were transduced with copGFP control lentiviral particles (Supplementary table 1) for analysis of transfection efficiency. After 48 hours, cells were collected and RNA extracted using the RNeasy kit (Qiagen). RT-PCR was then performed to evaluate the knockdown efficiency. We performed FACS analysis as described previously (16). Primary hepatocytes were isolated using a two-step perfusion method (18) and were treated in similar manner as MEFs for cell cycle analysis. To knockdown β2SP in mouse hepatocytes, the hepatocytes were transfected with spectrin β II siRNA (m) (Supplementary table 1).
Microarray Analysis
Custom-designed 44K mouse 60-mer oligo microarrays (Agilent Technologies) were used for the array experiments. Total RNA was extracted from mouse liver using the RNeasy kit (Qiagen).
Statistical Analysis
Results are expressed as the means ± SD or ± SEM. Student’s t-test was used for comparison between groups. P values <0.05 were considered statistically significant.
Results
β2SP+/− Mice Demonstrate Decreased Liver Mass to Body Weight Ratio and Persistent Mitotic Figures 1 Week after Partial Hepatectomy
Fig. 1.

Gross morphologic and histological features of wild-type and β2SP+/− mouse livers following PHx. (A) No gross morphologic differences were noted between wild-type and β2SP+/−mouse livers at any time point. (B) The liver mass to body weight ratio was then calculated at each time point. (C) Hematoxylin and eosin staining showed prominent mitotic figures (denoted by arrows) in β2SP+/− mice at 168 hours post-PHx.
Upon initial examination after hepatectomy, no gross morphologic differences were noted between wild-type and β2SP+/− mouse livers at any time point (Fig. 1A). At baseline, liver mass in β2SP+/− mice is greater, however, the liver mass to body weight ratio is almost the same in β2SP+/− mice in comparison to wild-type mice (4%) (Fig. 1B). However, this ratio in β2SP+/− mice, was significantly lower in comparison to wild-type mice at 48 hours post-PHx, (P < 0.05). Although β2SP+/− mice had yet to return to pre-PHx levels (Fig. 1B), the ratio was nearly identical between wild-type and β2SP+/− mice by 72 hours and at 168 hours. Continued presence of mitotic figures only in mutant mouse livers at 168 hours post-PHx suggests that there is a potential defect in termination of liver regeneration in β2SP+/− mice compared with wild-type mice (Fig. 1C).
β2SP+/− Mouse Livers Demonstrate Dysregulated Cell Cycle and G2/M Phase Arrest Following Partial Hepatectomy
Immunohistochemical and protein expression analysis of pRb (Ser249/Thr252) demonstrated positive labeling beginning at 24 hours in wild-type and mutant mice, with persistently intense labeling in mutant mice through 72 hours post-PHx. Peak labeling in wild-type mice, however, was at 48 hours (Fig. 2A, 2B). Reduction in level of pRb (Ser249/Thr252) in β2SP+/− mouse at 48 hours after PHx in comparison to wild type mice, supporting the alteration of G1/S checkpoint regulation. There was no significant difference in pRb (Ser249/Thr252), PCNA, and pH3 (Ser10) staining between the untreated wild-type mice and β2SP+/− mice (Supplementary Figure 3). Further analysis of cyclin D1 expression found significantly elevated levels in β2SP+/−mouse livers at baseline and 24, 72, and 168 hours post-hepatectomy and the absence of a 48 hours peak as seen in wild-type mice (Fig. 2B). These results do suggest, that while quiescent hepatocytes in mutant mice respond to the mitogenic stimulus of hepatectomy, exit G0, and proceed through G1 to S phase of the cell cycle, this transition is not synchronized as it is in wild-type mice.
Fig. 2.

Analysis of cell cycle phase-specific proteins in wild-type and β2SP+/− mouse livers following partial hepatectomy. (A) Immunohistochemical analysis of pRb (Ser249/Thr252), PCNA, and p-histone (Ser10) demonstrated increased pRb and PCNA labeling in β2SP+/− mice at 24 hours; however, p-histone labeling was absent in β2SP+/− mice at 48 hours after hepatectomy. (B) Western blot analysis demonstrated significantly elevated pRb and cyclin D1 expressions in mutant mice at 24 hours and diminished expressions of cyclin E, cyclin A, cyclinB1 and Cdk1, especially at 48 hours. (*P<0.05)
We demonstrated significant impairment in the expression of PCNA, cyclin A and cyclin E in β2SP+/− mouse livers in comparison to wild type at different time points (Fig. 2B, Supplementary table 2). These results suggest β2SP+/− mice seemed to demonstrate accelerated DNA synthesis beginning at 24 hours, with dysregulated levels of pRb (Ser249/Thr252), cyclin D1, and cyclin A post-PHx. Wild-type mice on the other hand, underwent synchronized G1/S-phase transition and DNA synthesis at about 48 hours.
Further analysis of cell cycle progression through G2/M phase by p-histone (Ser10) demonstrated deregulation in labelling in β2SP+/− mouse liver sections in comparison to wild-type at different time points (Fig. 2A, Supplementary table 2). Analysis of cyclin B1 and Cdk1 expression by western blot confirmed mitotic delay. These results clearly demonstrate dysregulated cell cycle progression in the hepatocytes of β2SP+/− mice and suggest that, whereas mutant hepatocytes seem to have an accelerated G1/S phase, there is a definite delay in progression through the G2 and M phases.
β2SP+/− Mouse Livers Demonstrate Elevated Expression of the Cell Cycle Inhibitors p53 and p21 Following Partial Hepatectomy
Given evidence of dysregulated cell cycle in β2SP+/− mice following partial hepatectomy, we then assessed the expression of the checkpoint proteins p53 and p21 in wild-type and mutant mouse livers. Analysis of p53 and p21 expression by western blot demonstrated a significant impairment in their expressions at different time points post-PHx in β2SP+/− mouse livers in comparison to wild type mouse livers (Fig. 3, Supplementary table 2). Moreover, the peak in p53 and p21 expression in mutant mice correlated with diminished levels of cyclin A and Cdk1 and delayed expression of p-histone (Ser10) (Fig. 2), suggesting that p53 and p21 may activate the G2/M-phase cell cycle checkpoint following PH.
Fig. 3.

Analysis of cell cycle checkpoint proteins in wild-type and β2SP+/− mouse livers following partial hepatectomy. (A) Western blot analysis and densitometric analysis (B) of p53 and p21 expression demonstrated significant elevation in mutant mice beginning at 24 hours post-PHx. Mutant mice also demonstrated increased pCdk1 (Thr14) expression and delayed peak in pSTAT3 (Tyr705), consistent with growth arrest. (* P<0.05)
To evaluate whether elevated p53 expression in mutant mice was secondary to impaired p53 regulation, we assessed MDM2 and PTEN expression by western blot. PTEN may protect p53 from MDM2-mediated degradation and enhance p53 stability, whereas p53 can enhance the transcription of PTEN (19). We demonstrated identical levels of PTEN and significantly lower expression of MDM2 in mutant mice at 24 and 48 hours post-PHx (Supplementary Figure 1), suggesting enhanced p53 expression secondary to cellular or genotoxic stress and the DNA-damage response. G2/M-phase delay was further confirmed by analysis of phosphorylated Cdk1 (Thr14) (Fig. 2B, Supplementary table 2) and pSTAT3 (Tyr705) (Fig. 3, Supplementary table 2) expression. We then performed the TUNEL assay to determine whether p53 may mediate increased apoptosis leading to impaired regeneration. Cleaved caspase expression was virtually absent or significantly reduced in mutant mice compared with wild-type mice, particularly at 48 hours post-PHx. There was no evidence of correlating cleaved caspase-3 expression with either elevated p53 or p21 in mutant mice (Fig. 3). Similarly, results for the TUNEL assay were also negative at 48 hours (Supplementary Figure 2). These results further confirm a temporary p53-p21–mediated G2/M-phase arrest leading to delayed liver regeneration in β2SP+/− mice.
β2SP+/− Mouse Livers Demonstrate Increased DNA Damage and Activation of p53-mediated Cell Cycle Checkpoint Following Partial Hepatectomy
Activation of p53 in response to DNA damage is mediated by phosphorylation on serine 15 and 20 by the kinases ATM/ATR and Chk2. Phosphorylation of p53 then results in an activated protein with numerous transcriptional targets necessary for cell cycle arrest, DNA repair, or apoptosis. Initial gene expression microarray analysis of wild-type and β2SP+/− mouse livers collected at time zero and 48 hours post-PHx found significant elevations (greater than threefold) in mRNA of several DNA damage-related genes, including Tp53, Gadd45, Cdc6, and Chk1 at 48 hours (Supplementary table 3). Further analysis by immunohistochemistry demonstrated elevated expression of pChk2, γH2AX (S139), phosphorylated p53 (on serine 15 and 20) in β2SP+/− mice at different time points in comparison to wild type mice (Fig. 4A, 4B, Supplementary table 2). Moreover, peak phosphorylated p53 expression correlated with elevated p21 expression, suggesting activation of the DNA damage response and downstream target gene regulation by p53, leading to growth arrest and delayed liver regeneration.
Fig. 4.

DNA damage in β2SP+/− mice following partial hepatectomy. (A) Immunohistochemical analysis of wild-type and β2SP+/− mouse livers for DNA damage markers p-Chk2 (Thr68) and γH2AX (S139) at time points following PHx were performed. Arrows denote examples of positive labeling. (B) Elevated levels of phospho-p53 (ser15 and ser20) in β2SP+/− mice by western blot are in consistent with activation of the DNA damage pathway.
β2SP−/− MEFs Demonstrate Dysregulated Cell Cycle in a p53-independent Manner
Given the elevated levels of p53 in β2SP+/− mouse livers following PHx, we further investigated whether down-regulation of p53 influences cell cycle progression of β2SP+/− MEFs and primary hepatocytes in which β2SP levels were reduced by shRNA-mediated knockdown. Under serum-starved conditions, β2SP−/− mutant (MT) MEFs showed a marked reduction of the G0/G1 population in comparison to wild-type control MEFs (Fig. 5), consistent with in vivo data obtained from β2SP+/− PHx mouse livers. There was no significant difference between wild-type MEFs transfected with p53shRNA and untransfected wild-type MEFs. These results suggest that whereas p53 induction is associated with reduced β2SP levels in vivo following PHx, p53 is not the exclusive mediator of aberrant cell cycle in β2SP+/− hepatocytes. It is likely that factors other than p53 also contribute to the defects in cell cycle progression observed in regenerating β2SP+/− hepatocytes.
Fig. 5.
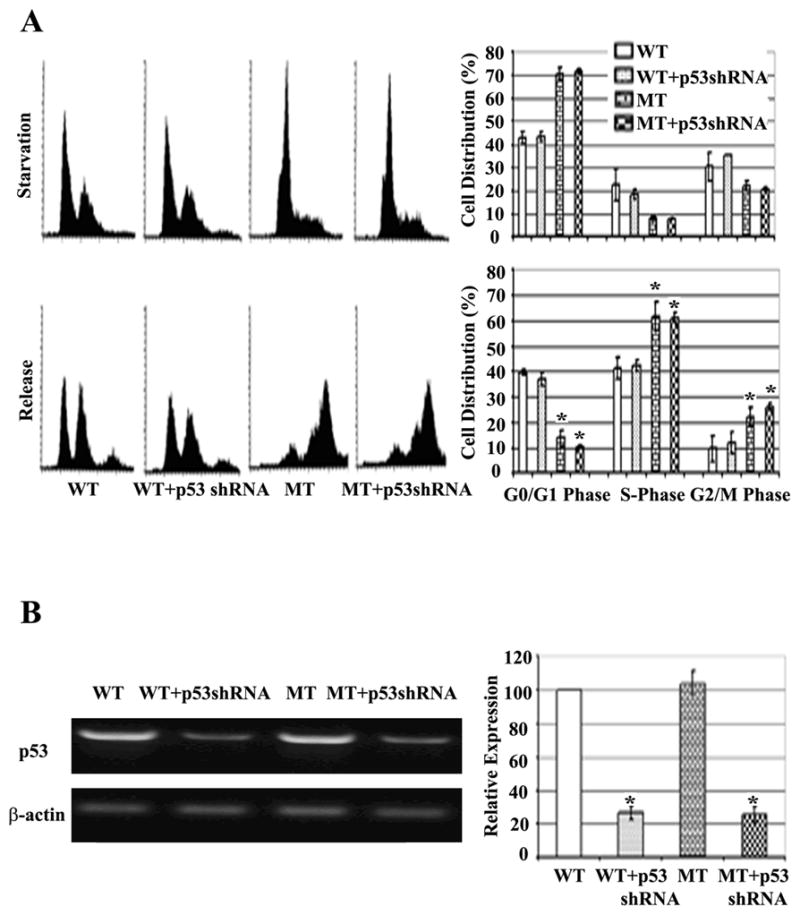
Loss of β2SP alters the cell cycle in a p53-independent manner. (A) Representative histograms of FACS analysis of synchronized and released wild-type (WT), wild-type with p53 shRNA (WT+p53shRNA), and β2SP−/− (MT), β2SP −/− with p53shRNA (MT+p53shRNA) MEFs. (* represents P<0.05). (B) Semi-quantitative RT-PCR of p53 in wild-type (WT), wild-type with p53 shRNA (WT+p53shRNA), and β2SP−/− (MT), β2SP −/− with p53shRNA (MT+p53shRNA) MEFs after 48 hours of shRNA delivery. (*P<0.05)
Discussion
β2SP has been previously demonstrated to be a key adaptor protein in TGF-β signaling and a key tumor-suppressor protein in foregut cancers: nearly 40% of β2SP+/− mice develop hepatocellular carcinoma by 15 months of age and nearly 90% of β2SP+/−/Smad4+/− double heterozygote mutants develop gastric tumors (13, 20). Moreover, FACS analysis of β2SP−/−MEFs following serum starvation and release demonstrated reduction of the G1 population and faster entry into S phase in mutant MEFs compared with wild-type (16). These observations suggest that β2SP plays a key role in cell cycle progression and tumorigenesis. To further investigate this role in vivo, we examined liver regeneration following PHx. Interestingly; our results demonstrated that diminished expression of β2SP is associated with increased DNA damage, activation of the DNA damage response proteins p53 and p21, and G2/M-phase arrest following PHx.
TGF-β, which is capable of inducing an anti-proliferative gene response at any point in the cell cycle, is most effective at inhibiting cell cycle progression during G1 via inhibition of cyclins D, E, and A or via Cdk4 synthesis (21–24). Disruption of TGF-β signaling in liver regeneration, via conditional knockout of the TGF-β type II receptor (TBRII), has similarly demonstrated a nearly two-fold increase and acceleration of S-phase entry following hepatectomy (10, 25). Likewise, our results demonstrated that diminished levels of the Smad3/4 adaptor protein β2SP are associated with elevated levels of pRb (Ser249/Thr252) cyclin D1 and accelerated DNA synthesis, with increased PCNA labeling at 24 hours following PHx. Diminished levels of Cdk1, elevated levels of pCdk1 (Thr14), and delayed expression of p-histone suggest impairment in the entry of mutant hepatocytes into mitosis.
In particular, p53 has been well described as acting as a hub for incoming stress signals, which are then transduced to growth arrest, DNA repair, or apoptosis (26). Our results demonstrate a significant induction of p53 in mutant mice beginning at 24 hours post-PHx, correlating with initial expression of PCNA and elevated levels of pRb (Ser249/Thr252) and cyclin D1, suggesting evidence of cellular or genomic stress during DNA synthesis. This is further supported by increased expression of pChk2 (Thr68) and elevated levels of phospho-p53 (ser15 and 20). In response to DNA damage, ATM/ATR-kinases activate Chk2 (an evolutionarily conserved and well-described kinase) by phosphorylating Thr68 (27). This in turn mediates a chain of phosphorylation events, including phosphorylation of p53 on ser20 and disruption of p53-MDM2 binding, stabilized p53 (26), and recruitment of the transcription factors p300, CBP, and P/CAF, which stimulate transcription from p53-responsive promoters (28). In response to DNA damage, p53 is also modified by ATM-mediated ser15 phosphorylation, resulting in increased stability and biochemical activation (29).
Elevated p53 levels remained through 48 hours post-PHx in mutant mice, correlating with persistent expression of pChk2 and increased expression of phosphorylated histone protein H2AX (ser139) or γH2AX, a marker representative of double-strand DNA breaks. Elevated p53 in mutant mice also correlates with regulation of several downstream target genes like p21, GADD45, and MDM2 (30). We found a similar induction of p21 in correlation with elevated p53 and hepatocyte DNA synthesis in mutant mice. Previous studies demonstrated the key role of p21 in G1/S phase arrest (6) and prolongation of G2/M-phase arrest via phosphorylation of Cdk1 at the Thr14 and Tyr15 inhibitory sites (31). Mitosis occurred by 72 hours after PHx in the mutant mice, correlating with elevated p21 expression, increased Cdk1, and diminished phosphorylation of Cdk1. p53-p21–mediated growth arrest in β2SP mutant mice is also demonstrated with diminished expression and phosphorylation of STAT3, a key mitogen necessary for liver regeneration (32). Further microarray analysis demonstrated significant induction of several p53 high-affinity DNA repair genes, including GADD45 and Cdc6. Interestingly, previous work has also demonstrated that the p53-p21 checkpoint pathway induces accumulation of the growth suppressive, hypophosphorylated, form of pRb (Ser249/Thr252) (33).
Induction of p53 is not the only factor mediating aberrant cell cycle progression in regenerating β2SP+/− hepatocytes. Surprisingly, even with greater than 70% depletion of p53, there was no obvious change in cell cycle progression of wild-type cells, nor was there rescue of cell cycle defects caused by β2SP loss or depletion in MEFs or primary cultured hepatocytes. Further studies with better p53 down-regulation in vitro as well as analysis of PHx in β2SP+/− mice that also lack p53 will be required to resolve these possibilities. Nevertheless, our results clearly show that reduction of β2SP+/− levels in vivo and in vitro (Supplementary Figure 4) leads to clear cell cycle progression defects and are associated with elevated DNA damage and p53 induction following partial hepatectomy.
Growth arrest in β2SP mutant mice seems temporary, as mitosis proceeded by 72 hours and significant liver mass was recovered by 1 week post-PHx. Temporary growth arrest is reflective of successful repair of damaged DNA in otherwise normal hepatocytes that are only missing β2SP. Loss of β2SP and cellular/genomic stress during DNA synthesis, also suggest an additional mechanism for its tumor-suppressive role. Repeated DNA damage in actively proliferating cells likely results in the accumulation of mutations and increased susceptibility to tumorigenesis over time. The precise mechanism for loss of β2SP resulting in DNA damage remains to be elucidated. Our current study, however, conclusively demonstrates that diminished β2SP is associated with dysregulated cell cycle progression, increased DNA damage, and activation of the p53-p21 cell cycle checkpoint leading to G2/M-phase arrest and delay in liver regeneration following PHx (Fig. 6), and that this process is independent of p53. The results of this work and its future implications will certainly contribute to a greater understanding of the DNA damage response, cell cycle synchrony, and ultimately tumorigenesis, particularly in such difficult-to-treat cancers as hepatocellular carcinoma.
Fig. 6.

Proposed model for the mechanism by which loss of β2SP leads to delayed liver regeneration following partial hepatectomy. (A) Cell cycle promoters are in green font, whereas inhibitors are in red. β2SP+/− mice appear to have slightly accelerated DNA synthesis but temporary G2/M-phase arrest leading to delayed liver regeneration. (B) Loss of β2SP increases susceptibility to cellular/DNA damage in proliferating hepatocytes and subsequent induction/activation of p53. p53, then induces expression of the cell cycle checkpoint protein p21, diminishes expression and phosphorylation of the key liver mitogen STAT3, and represses the mitosis-promoting factor Cdk1. These signals then lead to G2/M phase arrest until DNA can be adequately repaired and a subsequent delay in liver regeneration.
Supplementary Material
Supplementary Figure 1. Regulation of p53 expression. Western blot analysis of the p53 regulatory proteins at 24 and 48 hours post-PHx demonstrates significant depression of MDM2 in β2SP+/− mice at 48 hours and no change in PTEN expression.
Supplementary Figure 2. TUNEL assay for regenerating liver tissue. No evidence of TUNEL-positive apoptotic cells was noted. Representative samples from wild-type and β2SP+/− mouse livers are demonstrated from 48 hours post-PHx.
Supplementary Figure 3. Immunostaining of pRb (Ser249/Thr252), PCNA, and pH3 (Ser10) of untreated wild-type and β2SP+/− mice.
Supplementary Figure 4. FACS assay showed normal cell cycle profile in wild type (WT) hepatocytes, while β2SPshRNA (Mut)-transfected hepatocytes showed G2/M arrest at 0 hour and 24 hours after extraction.
Supplementary table 1. List of Antibodies (Abs), shRNA and siRNA.
Supplementary table 2. Expression of cell cycle proteins at different time points after PHx in wild type and β2SP+/− mouse livers.
Supplementary table 3. Microarray data at 48 hrs after PHx in wild type and β2SP+/− mouse livers.
Acknowledgments
This work was supported by NIH Grants RO1CA042857 (L.M.), RO1CA106614 (L.M.), RC2-AA019392 (L.M.), PO1CA130821 (L.M.), Ben Orr Award (L.M.), VA Merit grant and Georgetown Department of Surgery Huffnagel Resident Research fund (A.T.). We thank Dr. Shumei Song, Dr. Hailong Piao and Dr. Nileshkumar Dubey for thoughtful comments and suggestions.
Abbreviations
- β2SP
β-2 spectrin
- PHx
partial hepatectomy
- MAPK
mitogen-activated protein kinase
- CKIs
cyclin-dependent-kinase-inhibitory proteins
- ELF
embryonic liver fodrin
- MT
β2SP mutant
- MEFs
mouse embryonic fibroblasts
References
- 1.Michalopoulos GK, DeFrances MC. Liver regeneration. Science. 1997;276:60–66. doi: 10.1126/science.276.5309.60. [DOI] [PubMed] [Google Scholar]
- 2.Morgan DO. Cyclin-dependent kinases: engines, clocks, and microprocessors. Annu Rev Cell Dev Biol. 1997;13:261–291. doi: 10.1146/annurev.cellbio.13.1.261. [DOI] [PubMed] [Google Scholar]
- 3.Jaumot M, Estanyol JM, Serratosa J, Agell N, Bachs O. Activation of cdk4 and cdk2 during rat liver regeneration is associated with intranuclear rearrangements of cyclin-cdk complexes. Hepatology. 1999;29:385–395. doi: 10.1002/hep.510290226. [DOI] [PubMed] [Google Scholar]
- 4.Garnier D, Loyer P, Ribault C, Guguen-Guillouzo C, Corlu A. Cyclin-dependent kinase 1 plays a critical role in DNA replication control during rat liver regeneration. Hepatology. 2009;50:1946–1956. doi: 10.1002/hep.23225. [DOI] [PubMed] [Google Scholar]
- 5.Taub R. Liver regeneration: from myth to mechanism. Nat Rev Mol Cell Biol. 2004;5:836–847. doi: 10.1038/nrm1489. [DOI] [PubMed] [Google Scholar]
- 6.Albrecht JH, Poon RY, Ahonen CL, Rieland BM, Deng C, Crary GS. Involvement of p21 and p27 in the regulation of CDK activity and cell cycle progression in the regenerating liver. Oncogene. 1998;16:2141–2150. doi: 10.1038/sj.onc.1201728. [DOI] [PubMed] [Google Scholar]
- 7.Russell WE, Coffey RJ, Jr, Ouellette AJ, Moses HL. Type beta transforming growth factor reversibly inhibits the early proliferative response to partial hepatectomy in the rat. Proc Natl Acad Sci U S A. 1988;85:5126–5130. doi: 10.1073/pnas.85.14.5126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Braun L, Mead JE, Panzica M, Mikumo R, Bell GI, Fausto N. Transforming growth factor beta mRNA increases during liver regeneration: a possible paracrine mechanism of growth regulation. Proc Natl Acad Sci U S A. 1988;85:1539–1543. doi: 10.1073/pnas.85.5.1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Macias-Silva M, Li W, Leu JI, Crissey MA, Taub R. Up-regulated transcriptional repressors SnoN and Ski bind Smad proteins to antagonize transforming growth factor-beta signals during liver regeneration. J Biol Chem. 2002;277:28483–28490. doi: 10.1074/jbc.M202403200. [DOI] [PubMed] [Google Scholar]
- 10.Romero-Gallo J, Sozmen EG, Chytil A, Russell WE, Whitehead R, Parks WT, Holdren MS, et al. Inactivation of TGF-beta signaling in hepatocytes results in an increased proliferative response after partial hepatectomy. Oncogene. 2005;24:3028–3041. doi: 10.1038/sj.onc.1208475. [DOI] [PubMed] [Google Scholar]
- 11.Mishra L, Derynck R, Mishra B. Transforming growth factor-beta signaling in stem cells and cancer. Science. 2005;310:68–71. doi: 10.1126/science.1118389. [DOI] [PubMed] [Google Scholar]
- 12.Kitisin K, Ganesan N, Tang Y, Jogunoori W, Volpe EA, Kim SS, Katuri V, et al. isruption of transforming growth factor-beta signaling through beta-spectrin ELF leads to hepatocellular cancer through cyclin D1 activation. Oncogene. 2007;26:7103–7110. doi: 10.1038/sj.onc.1210513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tang Y, Katuri V, Dillner A, Mishra B, Deng CX, Mishra L. Disruption of transforming growth factor-beta signaling in ELF beta-spectrin-deficient mice. Science. 2003;299:574–577. doi: 10.1126/science.1075994. [DOI] [PubMed] [Google Scholar]
- 14.Thenappan A, Li Y, Kitisin K, Rashid A, Shetty K, Johnson L, Mishra L. Role of transforming growth factor beta signaling and expansion of progenitor cells in regenerating liver. Hepatology. 2010;51:1373–1382. doi: 10.1002/hep.23449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mitchell C, Willenbring H. A reproducible and well-tolerated method for 2/3 partial hepatectomy in mice. Nat Protoc. 2008;3:1167–1170. doi: 10.1038/nprot.2008.80. [DOI] [PubMed] [Google Scholar]
- 16.Kim SS, Shetty K, Katuri V, Kitisin K, Baek HJ, Tang Y, Marshall B, et al. TGF-beta signaling pathway inactivation and cell cycle deregulation in the development of gastric cancer: role of the beta-spectrin, ELF. Biochem Biophys Res Commun. 2006;344:1216–1223. doi: 10.1016/j.bbrc.2006.03.236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bucher NL. Regeneration of Mammalian Liver. Int Rev Cytol. 1963;15:245–300. doi: 10.1016/s0074-7696(08)61119-5. [DOI] [PubMed] [Google Scholar]
- 18.Lu L, Li Y, Kim SM, Bossuyt W, Liu P, Qiu Q, Wang Y, et al. Hippo signaling is a potent in vivo growth and tumor suppressor pathway in the mammalian liver. Proc Natl Acad Sci U S A. 107:1437–1442. doi: 10.1073/pnas.0911427107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stambolic V, MacPherson D, Sas D, Lin Y, Snow B, Jang Y, Benchimol S, et al. Regulation of PTEN transcription by p53. Mol Cell. 2001;8:317–325. doi: 10.1016/s1097-2765(01)00323-9. [DOI] [PubMed] [Google Scholar]
- 20.Katuri V, Tang Y, Marshall B, Rashid A, Jogunoori W, Volpe EA, Sidawy AN, et al. Inactivation of ELF/TGF-beta signaling in human gastrointestinal cancer. Oncogene. 2005;24:8012–8024. doi: 10.1038/sj.onc.1208946. [DOI] [PubMed] [Google Scholar]
- 21.Ko TC, Sheng HM, Reisman D, Thompson EA, Beauchamp RD. Transforming growth factor-beta 1 inhibits cyclin D1 expression in intestinal epithelial cells. Oncogene. 1995;10:177–184. [PubMed] [Google Scholar]
- 22.Slingerland JM, Hengst L, Pan CH, Alexander D, Stampfer MR, Reed SI. A novel inhibitor of cyclin-Cdk activity detected in transforming growth factor beta-arrested epithelial cells. Mol Cell Biol. 1994;14:3683–3694. doi: 10.1128/mcb.14.6.3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Geng Y, Weinberg RA. Transforming growth factor beta effects on expression of G1 cyclins and cyclin-dependent protein kinases. Proc Natl Acad Sci U S A. 1993;90:10315–10319. doi: 10.1073/pnas.90.21.10315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ewen ME, Sluss HK, Whitehouse LL, Livingston DM. TGF beta inhibition of Cdk4 synthesis is linked to cell cycle arrest. Cell. 1993;74:1009–1020. doi: 10.1016/0092-8674(93)90723-4. [DOI] [PubMed] [Google Scholar]
- 25.Oe S, Lemmer ER, Conner EA, Factor VM, Leveen P, Larsson J, Karlsson S, et al. Intact signaling by transforming growth factor beta is not required for termination of liver regeneration in mice. Hepatology. 2004;40:1098–1105. doi: 10.1002/hep.20426. [DOI] [PubMed] [Google Scholar]
- 26.Meek DW. The p53 response to DNA damage. DNA Repair (Amst) 2004;3:1049–1056. doi: 10.1016/j.dnarep.2004.03.027. [DOI] [PubMed] [Google Scholar]
- 27.Lee CH, Chung JH. The hCds1 (Chk2)-FHA domain is essential for a chain of phosphorylation events on hCds1 that is induced by ionizing radiation. J Biol Chem. 2001;276:30537–30541. doi: 10.1074/jbc.M104414200. [DOI] [PubMed] [Google Scholar]
- 28.Li W, Liang X, Kellendonk C, Poli V, Taub R. STAT3 contributes to the mitogenic response of hepatocytes during liver regeneration. J Biol Chem. 2002;277:28411–28417. doi: 10.1074/jbc.M202807200. [DOI] [PubMed] [Google Scholar]
- 29.Appella E. Modulation of p53 function in cellular regulation. Eur J Biochem. 2001;268:2763. doi: 10.1046/j.1432-1327.2001.02224.x. [DOI] [PubMed] [Google Scholar]
- 30.Chen X, Ko LJ, Jayaraman L, Prives C. p53 levels, functional domains, and DNA damage determine the extent of the apoptotic response of tumor cells. Genes Dev. 1996;10:2438–2451. doi: 10.1101/gad.10.19.2438. [DOI] [PubMed] [Google Scholar]
- 31.Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, et al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497–1501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- 32.Cressman DE, Greenbaum LE, DeAngelis RA, Ciliberto G, Furth EE, Poli V, Taub R. Liver failure and defective hepatocyte regeneration in interleukin-6-deficient mice. Science. 1996;274:1379–1383. doi: 10.1126/science.274.5291.1379. [DOI] [PubMed] [Google Scholar]
- 33.Brugarolas J, Bronson RT, Jacks T. p21 is a critical CDK2 regulator essential for proliferation control in Rb-deficient cells. J Cell Biol. 1998;141:503–514. doi: 10.1083/jcb.141.2.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Regulation of p53 expression. Western blot analysis of the p53 regulatory proteins at 24 and 48 hours post-PHx demonstrates significant depression of MDM2 in β2SP+/− mice at 48 hours and no change in PTEN expression.
Supplementary Figure 2. TUNEL assay for regenerating liver tissue. No evidence of TUNEL-positive apoptotic cells was noted. Representative samples from wild-type and β2SP+/− mouse livers are demonstrated from 48 hours post-PHx.
Supplementary Figure 3. Immunostaining of pRb (Ser249/Thr252), PCNA, and pH3 (Ser10) of untreated wild-type and β2SP+/− mice.
Supplementary Figure 4. FACS assay showed normal cell cycle profile in wild type (WT) hepatocytes, while β2SPshRNA (Mut)-transfected hepatocytes showed G2/M arrest at 0 hour and 24 hours after extraction.
Supplementary table 1. List of Antibodies (Abs), shRNA and siRNA.
Supplementary table 2. Expression of cell cycle proteins at different time points after PHx in wild type and β2SP+/− mouse livers.
Supplementary table 3. Microarray data at 48 hrs after PHx in wild type and β2SP+/− mouse livers.