

Abstract
Xanthine oxidoreductase is a molybdenum-containing enzyme which catalyzes the hydroxylation reaction on sp2-hybridized carbon centers of a variety of substrates including purines, aldehydes and other heterocyclic compounds. The complex of arsenite-inhibited xanthine oxidase has been characterized previously by UV-visible, EPR and X-ray absorption spectroscopy, and the catalytically essential sulfido ligand of the square-pyrimidal molybdenum center suggested to be involved in arsenite binding through either μ-sulfido, μ-oxo double-bridge or a single μ-sulfido bridge. However, this is contrary to the crystallographically observed single μ-oxo bridge between molybdenum and arsenic in the desulfo-form of aldehyde oxidoreductase from Desulfovibrio gigas (an enzyme closely related to xanthine oxidase) whose molybdenum center has an oxo ligand replacing the catalytically essential sulfur as seen in the functional form of xanthine oxidase. Here, we use X-ray crystallography to characterize the molybdenum center of arsenite-inhibited xanthine oxidase and solve the structures of oxidized and reduced inhibition complexes at 1.82 and 2.11 Å resolution, respectively. We observe μ-sulfido, μ-oxo double-bridge between molybdenum and arsenic in the active sites of both complexes. Arsenic exhibits four-coordinate with a distorted trigonal-prismatic geometry in the oxidized complex and three-coordinate with a distorted trigonal-planar geometry in the reduced complex. The doubly-bridged binding mode is in agreement with previous XAS data that the catalytically essential sulfur is also essential for high affinity of reduced xanthine oxidoreductase for arsenite.
Xanthine oxidoreductase catalyzes the final two steps in purine metabolism in mammals, the oxidation of hypoxanthine to xanthine, and xanthine to uric acid. The the enzyme’s active site is a square-pyramidal LMo(VI)OS(OH) center, where L represents a pyranopterin cofactor coordinated to the metal via a bidentate enedithiolate side chain (Chart 1, depicting the molybdenum coordination sphere and the structure of the pyranopterin moiety).1 The sulfido ligand is essential for enzyme activity toward both xanthine and aldehyde substrates, and if replaced by a second oxo ligand via cyanolysis yields an inactive desulfo form.2
Chart 1.
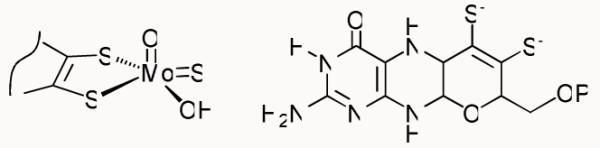
Structure of the molybdenum center of xanthine oxidase.
Arsenite is known to inhibit xanthine oxidase, binding particularly tightly to the reduced form of the enzyme.3a,b EPR studies of arsenite-complexed enzyme have demonstrated strong anisotropic hyperfine and quadrupole coupling to arsenic (I = 3/2) to the electronic spin of Mo(V) (S = 1/2, d1 system), and also that the inactive desulfo enzyme, as well as the native form, can bind arsenite.3a-d X-ray absorption spectroscopy (XAS) studies have indicated a Mo-S-As core with a bond angle of 80 ± 4° and the following distances in the reduced enzyme: Mo-S, 2.39 Å; Mo-As, 3.00 Å; and As-S, 2.27 Å.3e Two alternate binding modes for arsenite have been proposed, with either single μ-sulfido bridge or a μ-sulfido μ-oxo double bridge (Chart 2).3
Chart 2.
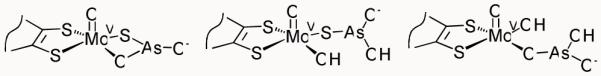
Previously proposed structures for the arsenite-inhibited forms of xanthine oxidase, based on EXAFS data (left and center), and aldehyde oxidoreductase, based on crystallographica data (right).
Recently, X-ray crystallographic studies on the arsenite-inhibited desulfo form of aldehyde oxidoreductase (AOR) from Desulfovibrio gigas (which is closely related to xanthine oxidase, but which has been reported to be active in a MoO2 form lacking the Mo=S required by xanthine oxidase for activity4c) showed a single μ-oxo bridge between As and Mo in the arsenite-inhibited complex (Chart 2).4a,b The observed binding mode for arsenite was distinctly different than proposed for the functional form of xanthine oxidase, and a crystal structure of arsenite-inhibited xanthine oxidase is thus highly desirable to elucidate the mechanism of arsenite inhibition.
Xanthine oxidase was purified and crystals grown as described in the Supporting Information. Arsenite was introduced to enzyme crystals through soaking at a concentration of 10 mM. For the arsenite–inhibited enzyme in the reduced form, dithionite was used to reduce the crystals subsequent to addition to arsenite. A single X-ray data set was collected at the LRL-CAT beamline of Argonne National Laboratory to 1.82 Å resolution for the complex of arsenite with oxidized enzyme and 2.11 Å resolution for the complex with reduced enzyme. The protein structures were solved using the MOLREP program of the CCP4 package6a, using the reported structure of xanthine oxidase solved by Enroth et al.1b (PDB code: 1FIQ) as the search model and then further improved by running rigid-body and restrained refinement using REFMAC.6b For the reduced form of the enzyme complex, non-crystallographic symmetry constraints were set as “tight” between the corresponding peptide chains during refinement. In the somewhat lower-resolution data for the reduced complex, bond distance restraints based on EXAFS-derived values3e were incorporated during refinement. In this way, uncertainties in metal-ligand distances owing to Fourier truncation artifacts in the immediate vicinity of the metal were minimized. Refinemen statistics are summarized in the online Supporting Information (Table S1).
The overall protein structure of arsenite-inhibited xanthine oxidase in both the oxidized and reduced forms as determined here is very similar to that previously reported for the oxidized enzyme, with all differences limited to the active site. Figure 1 shows the molybdenum centers overlaid with the Fo-Fc omit map (the map omitted the molybdenum cofactor and arsenite). In both the oxidized and reduced structures, molybdenum has a square-pyrimidal coordination geometry with the same ligand set and similar metal-ligand distances as seen in the free enzyme (Table S2). As shown in Figure 2, the oxidized structure has a bridging oxygen between molybdenum and arsenic similar to the arsenite-inhibited (desulfo) aldehyde oxidoreductase from D. gigas.4a The reduced structure also suggests a similar bridging oxygen, although its electron density is not as strong as in the oxidized structure. In contrast to the aldehyde oxidoreductase structure, however, in both the oxidized and reduced xanthine oxidoreductase we see clear and strong electron density connecting the equatorial sulfido ligand to the arsenic atom., with an As-S bond of 2.53 Å in the oxidized structure and 2.08 Å in the reduced structure (Figure 1, Table S2); the latter distance is generally consistent with the As-S distance determined for the reduced structure by XAS (2.27 ± 0.03 Å).3e These distances are significantly shorter than the corresponding As-O distances for the non-bridging equatorial oxo ligand of Mo as observed in the X-ray crystal structures of the oxidized (3.07 Å) and reduced (3.24 Å) desulfo D. gigas enzyme (PDB files 1SIJ and 3L4P, respectively).4a,b In addition, the Mo-As distance of 3.19 Å seen here with reduced xanthine oxidoreductase is similar to that seen previously by XAS (3.00 Å)3e and too long for a direct Mo-As bond (Table S2). The Mo-S-As angle of 100° with reduced xanthine oxidoreductase which is somewhat larger than that seen by XAS (80°).3e However, the corresponding Mo-S-As angle with oxidized xanthine oxidoreductase is 86°.
Figure 1.

Stereo images of the active sites of xanthine oxidase complexed with arsenite. (A) and (B) for oxidized enzyme; (C) and (D) for reduced enzyme. (B) and (D) are rotated approximately 90o about the horizontal axis from the perspective shown in (A) and (C) respectively. All the electron density maps were Fo - Fc omit maps calculated before the introduction of the arsenite-inhibited molybdenum co-factor for the refinement of the model and contoured at 3.0 σ within 2 Å of all atoms shown. Molybdenum is in teal, carbon in white, nitrogen in blue, oxygen in red, sulfur in yellow, phosphorus in orange and arsenic in purple. Figures were rendered with PyMOL (Ref. 6c). Only one of the two active sites of xanthine oxidase in each complex is shown although both sites in the asymmetric unit showed similar structures.
Figure 2.
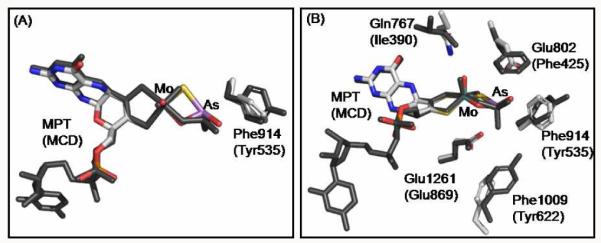
Superposition of the active sites of arsenite-inhibited xanthine oxidase and aldehyde oxidoreductase. (B) is rotated approximately 90° about the horizontal axis from the perspective shown in (A). Desulfo-aldehyde oxidoreductase complex (Protein Data Bank Code: 1SIJ, Ref. 4a) is shown in dark gray. Current xanthine oxidase complex is color-coded as in Figure 1. Amino acids are labeled for both structures with those for aldehyde oxidoreductase in parenthesis. The superposition was performed using COOT (Ref. 6d) to maximize the overlap between the molybdenum atoms and enedithiolate ligands of the two structures. One of the two active sites of xanthine oxidase was used for the superposition although both yielded similar results. Figures were rendered with PyMOL (Ref. 6c).
The current structures of arsenite-complexed xanthine oxidoreductase (with functional, sulfurated enzyme) contrast with that seen previously with the desulfo form of the D. gigas enzyme in clearly demonstrating a double bridge between the molybdenum and arsenic, with μ-sulfido bonding in addition to μ-oxo bonding (Figure 1). In the structure of the complex with oxidized xanthine oxidoreductase, arsenic is four-coordinate with a distorted trigonal pyramidal geometry consisting of the bridging S and two non-bridging O in the base plane and the bridging O to molybdenum occupying the apical position. Its bonding suggests that arsenite has been oxidized to arsenate in the synchrotron beam. (Xanthine oxidase is not known to possess arsenite oxidase activity, and extensive efforts on our part were unable to identify any here.) Chamberlain has previously reported that certain elements of inorganic compounds were subject to reduction or oxidation due to X-ray radiation based on different stability of alternative electron configurations, where the reduction of iodic acid and of potassium permanganate and oxidation of sulfide and sulfite to sulfurous acid were found.7 The bridging oxygen occupies a position in the molybdenum coordination sphere equivalent to the catalytically labile oxygen of the native enzyme.1 In the complex with reduced enzyme, we find arsenic to be three-coordinate with a distorted trigonal-planar geometry consisting of the bridging S and O and one non-bridging O (although the non-bridging O shows strong electron density in only one active site of the dimer, its density is considerably weaker in the other). In the strongly reducing conditions under which the sample was prepared, the arsenic would have remained in the As(III) state. In the aldehyde oxidoreductase structure, arsenic is bound only to three oxygens (one bridging and two non-bridging) in a distorted trigonal-planar geometry (Figure 2). In the oxidized xanthine oxidase structure, the MoOSAs unit is essentially planar with a butterfly angle of approximately 44° relative to the dithiolene-Mo plane, and is orthogonal to the trigonal base plane of the arsenic coordination sphere; similar butterfly angles are seen in the complex with reduced enzyme.
Figure 2 shows a superposition of the active sites of the current structures for the arsenite complex of oxidized xanthine oxidase and D. gigas AOR.4a The alignment was performed using COOT, maximizing overlap of the corresponding enedithiolate ligands and the molybdenum atoms. The arsenic atom occupies similar positions in the two structures, with an RMSD of ~0.6 Å. In the following discussion, the amino acid residue numbers are given first for the bovine enzyme, followed by that of D. gigas enzyme. Glu1261/869, universally conserved in the xanthine oxidase family, occupies almost precisely the same position in the two structures and is within hydrogen bonding distance to one of the two non-bridging oxygen atoms of the bound arsenite (2.52 and 2.71 Å for XO and AOR respectively). Glu802 (which corresponds to the non-conserved Phe425 in the D. gigas proteinis within hydrogen bonding distance (2.52 Å) to the other non-bridging oxygen of arsenite; Phe425 itself lies quite further away at 3.76 Å.
Phe1009/Tyr622 occupy similar, but not identical, positions. The phenolic oxygen of Tyr622 is 3.52 Å from one of the non-bridging oxygens of arsenite, but Phe1009 is 5.37 Å away from the corresponding oxygen of arsenite. Still, the key difference is the bond between arsenic and the cyanolyzable sulfur of the molybdenum coordination sphere in the xanthine oxidoreductase structure, absent in the structure of the D. gigas enzyme because of the desulfo nature of the latter. The sulfur content of the bridging sulfido position of the molybdenum center of xanthine oxidase used for current crystal structures was in excess of 80% based on the percentage functionality determined by standard enzymatic assays of AFR (activity to flavin ratio) for xanthine oxidase (as described in Supporting Information). An examination of the electron densities around the bridging sulfide group and oxo group at different σ levels in the reduced complex clearly indicated that the bridging sulfido group showed a comparable electron density to the two sulfur ligands from the enediolate moiety of the pterin cofactor whose identities were known, while the density for the bridging oxo group was much weaker indicating a less electron-rich ligand (Figure S1 in the Supporting Information).
In summary, the current crystal structures of arsenite-inhibited xanthine oxidase in both the oxidized and reduced state clearly demonstrate a double-bridge between Mo and As that involves the catalytically essential sulfur of this enzyme, consistent with earlier XAS work.3e This bidentate binding appears to be essential for the high affinity of reduced xanthine oxidoreductase for arsenite.3a,b Our structure provides a structural basis for understanding enzyme inhibition by arsenite, which simultaneously blocks the equatorial Mo-OH ligand that initiates nucleophilic attack on substrate at the outset of catalysis and the Mo=S that serves as hydride acceptor in the course of the reaction.1,5c
Supplementary Material
ACKNOWLEDGMENT
We thank the National Institutes of Health for financial support (GM 075036 and ES 012658 to R.H.). The Lilly Research Laboratory Collaborative Access Team (LRL-CAT) beamline at Sector 31 of the Advanced Photon Source is operated by Eli Lilly & Company. We thank Dr. Takeshi Nishino for advice in use of the folate column procedure and for helpful discussions. Use of the Advanced Photon Source at Argonne National Laboratory was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357.
Footnotes
ASSOCIATED CONTENT
Supporting Information. Details of crystallization and structure refinement. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).(a) Hille R. Chem. Rev. 1996;96:2757–2816. doi: 10.1021/cr950061t. [DOI] [PubMed] [Google Scholar]; (b) Enroth C, Eger BT, Okamoto K, Nishino T, Pai EF. Proc. Natl. Acad. Sci. U.S.A. 2000;97:10723–10728. doi: 10.1073/pnas.97.20.10723. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Okamoto K, Matsumoto K, Hille R, Eger BT, Pai EF, Nishino T. Proc. Natl. Acad. Sci. U.S.A. 2004;101:7931–7936. doi: 10.1073/pnas.0400973101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Doonan CJ, Stockert A, Hille R, George GN. J. Am. Chem. Soc. 2005;127:4518–4522. doi: 10.1021/ja042500o. [DOI] [PubMed] [Google Scholar]
- (2).Massey V, Edmondson D. J. Biol. Chem. 1970;245:6595–6598. [PubMed] [Google Scholar]
- (3).(a) Stewart RC, Hille R, Massey V. J. Biol. Chem. 1984;259:14426–14436. [PubMed] [Google Scholar]; (b) Hille R, Stewart RC, Fee JA, Massey V. J. Biol. Chem. 1983;258:4849–2856. [PubMed] [Google Scholar]; (c) Barber MJ, Siegel LM. Biochemistry. 1983;22:618–624. doi: 10.1021/bi00272a014. [DOI] [PubMed] [Google Scholar]; (d) George GN, Bray RC. Biochemistry. 1983;22:1013–1021. doi: 10.1021/bi00274a003. [DOI] [PubMed] [Google Scholar]; (e) Cramer SP, Hille R. J. Am. Chem. Soc. 1985;107:8164–8169. [Google Scholar]
- (4).(a) Boer DR, Thapper A, Brondino CD, Romao MJ, Moura JJG. J. Am. Chem. Soc. 2004;126:8614–8615. doi: 10.1021/ja0490222. [DOI] [PubMed] [Google Scholar]; (b) Thapper A, Boer DR, Brondino CD, Moura JJG, Romao MJ. J. Biol. Inorg. Chem. 2007;12:353–366. doi: 10.1007/s00775-006-0191-9. [DOI] [PubMed] [Google Scholar]; (c) Santos-Silva T, Ferroni F, Thapper A, Marangon J, Gonzalez PJ, Rizzi AC, Moura I, Moura JJG, Romao MJ, Brondino CD. J. Am. Chem. Soc. 2009;131:7990–7998. doi: 10.1021/ja809448r. [DOI] [PubMed] [Google Scholar]
- (5).(a) Massey V, Brumby PE, Komai H, Palmer G. J. Biol. Chem. 1969;244:1682–1691. [PubMed] [Google Scholar]; (b) Nishino T, Nishino T, Tsushima K. FEBS Lett. 1981;131:369–372. doi: 10.1016/0014-5793(81)80406-1. [DOI] [PubMed] [Google Scholar]; (c) Pauff JM, Cao H, Hille R. J. Biol. Chem. 2009;284:8760–8767. doi: 10.1074/jbc.M804517200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).(a) Collaborative Computational Project, Number 4. Acta. Cryst. Sect. D Biol. Crystallogr. 1994;50:760–763. [Google Scholar]; (b) Murshudov G, Vagin A, Dodson EJ. Acta. Cryst. Sect. D Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]; (c) DeLano WL. The PyMOL Molecular Graphics System. DeLano Scientific LLC; San Carlos, CA: [Google Scholar]; (d) Emsley P, Cowtan K. Acta. Cryst. Sect. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- (7).Chamberlain K. Phys. Rev. 1925;26:525–536. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.