

Abstract
The switch to an angiogenic phenotype is a fundamental determinant of neoplastic growth and tumor progression. We demonstrate that homozygous deletion of the p53 tumor suppressor gene via homologous recombination in a human cancer cell line promotes the neovascularization and growth of tumor xenografts in nude mice. We find that p53 promotes Mdm2-mediated ubiquitination and proteasomal degradation of the HIF-1α subunit of hypoxia-inducible factor 1 (HIF-1), a heterodimeric transcription factor that regulates cellular energy metabolism and angiogenesis in response to oxygen deprivation. Loss of p53 in tumor cells enhances HIF-1α levels and augments HIF-1-dependent transcriptional activation of the vascular endothelial growth factor (VEGF) gene in response to hypoxia. Forced expression of HIF-1α in p53-expressing tumor cells increases hypoxia-induced VEGF expression and augments neovascularization and growth of tumor xenografts. These results indicate that amplification of normal HIF-1-dependent responses to hypoxia via loss of p53 function contributes to the angiogenic switch during tumorigenesis.
Keywords: p53, hypoxia-inducible factor-1 (HIF-1), angiogenesis, vascular endothelial growth factor (VEGF), hypoxia, cancer
Regions of vascular deficiency or defective microcirculation in growing tumors are deprived of O2, glucose, and other nutrients. Apoptosis induced by nutrient deficiency counterbalances cell proliferation and limits tumor growth (Holmgren et al. 1995; O'Reilly et al. 1996; Parangi et al. 1996). Clonal evolution of tumor cells in this hypoxic microenvironment results from selection of subpopulations that not only resist apoptosis (Graeber et al. 1996) but also promote the formation of new blood vessels (for review, see Hanahan and Folkman 1996; Folkman 1997). In addition to promoting further growth of the primary tumor, cellular adaptation to hypoxia and tumor neovascularization strongly correlate with the risk of invasion and metastasis (Brown and Giaccia 1998; Dang and Semenza 1999; for review, see Folkman 1997). The switch to an angiogenic phenotype is considered to be a fundamental determinant of neoplastic progression (Gimbrone et al. 1972; Folkman et al. 1989; Bergers et al. 1999). This realization has, in turn, fueled an intense search for the molecular mechanisms by which the angiogenic switch is activated during tumorigenesis.
Hypoxia-inducible factor 1 (HIF-1) is a heterodimeric transcription factor that regulates O2 homeostasis and physiologic responses to O2 deprivation (for review, see Guillemin and Krasnow 1997; Semenza 1999). HIF-1 consists of two subunits, HIF-1α and HIF-1β, that belong to a subfamily of basic helix-loop-helix (bHLH) transcription factors containing a PAS (Per-ARNT-Sim) motif (Wang et al. 1995). A decrease in cellular O2 tension leads to elevation of HIF-1 activity via stabilization of the HIF-1α protein; conversely, ubiquitin-mediated proteolysis of HIF-1α on reexposure to a normoxic environment results in rapid decay of HIF-1 activity (Semenza and Wang 1992; Wang et al. 1995; Salceda and Caro 1997; Huang et al. 1998; Kallio et al. 1999). The binding of HIF-1α, in conjunction with its dimerization partner HIF-1β, to DNA (consensus binding sequence, 5′-RCGTG-3′) leads to the transcriptional activation of genes that mediate anaerobic metabolism (glucose transporters and glycolytic enzymes), O2-carrying capacity (erythropoietin, transferrin), and vasodilatation (inducible nitric oxide synthase and heme oxygenase-1) (for review, see Guillemin and Krasnow 1997; Semenza 1999). HIF-1 also binds to the 5′ flanking sequence of the vascular endothelial growth factor (VEGF) gene and is required for transactivation of VEGF in response to hypoxia (Forsythe et al. 1996; Carmeliet et al. 1998; Iyer et al. 1998; Ryan et al. 1998). The binding of VEGF to the receptor tyrosine kinases flk1/KDR, flt-1, and flt-4 (VEGFR-1–VEGFR-3) on vascular endothelial cells promotes their proliferation and leads to vessel formation (for review, see Ferrara 1993; Risau and Flamme 1995; Brown et al. 1996). In contrast to wild-type cells, VEGF gene expression is not induced by hypoxia in HIF-1α-deficient embryonic stem cells, and dramatic vascular regression occurs in HIF-1α-null mouse embryos (Iyer et al. 1998; Kotch et al. 1999). Therefore, HIF-1 is a key transcriptional mediator of metabolic adaptation and VEGF-mediated angiogenesis in response to hypoxia. Although these responses serve to maintain O2 homeostasis in normal tissues, they are also co-opted by tumors to facilitate neovascularization and growth. Akin to their role in vascular development and remodeling in normal tissues, HIF-1α (Maxwell et al. 1997; Carmeliet et al. 1998; Ryan et al. 1998) and VEGF (Plate et al. 1992; Shweiki et al. 1992; Kim et al. 1993; Millauer et al. 1994) facilitate tumor angiogenesis, and both HIF-1α (Zhong et al. 1999) and VEGF (for review, see Folkman 1997) are overexpressed in a wide variety of human cancers.
The genetic alterations that are responsible for oncogenesis and tumor progression may also underlie the ability of tumors to switch to an angiogenic phenotype. The human p53 tumor suppressor gene encodes a multifunctional transcription factor that mediates cellular responses to diverse stimuli, including DNA damage and hypoxia (for review, see Giaccia and Kastan 1998). In addition to being an integral component of the surveillance mechanisms that arrest cell cycle progression under adverse conditions, p53 is also involved in mediating hypoxia-induced apoptosis (Graeber et al. 1996) and inducing inhibitors of angiogenesis such as thrombospondin-1 (Dameron et al. 1994; Van Meir et al. 1994). Evidence also suggests that p53 negatively regulates VEGF expression (Mukhopadhyay et al. 1995; Bouvet et al. 1998; Fontanini et al. 1998). Somatic mutations of the p53 gene represent one of the most common genetic alterations in human cancers, and the acquisition of such defects is strongly associated with tumor progression and metastasis (for review, see Levine 1997).
In this study, we demonstrate that genetic inactivation of p53 in cancer cells provides a potent stimulus for tumor angiogenesis and identify a novel mechanism by which loss ofp53 function contributes to activation of the angiogenic switch in tumors. We find that homozygous deletion of p53 via homologous recombination in human colon cancer cells promotes the neovascularization and growth of tumor xenografts in nude mice. We show that p53 inhibits HIF-1 activity by targeting the HIF-1α subunit for Mdm2-mediated ubiquitination and proteasomal degradation. Conversely, the loss of p53 enhances hypoxia-induced HIF-1α levels and augments HIF-1-dependent expression of VEGF in tumor cells. We further demonstrate that forced expression of HIF-1α in p53-expressing tumor cells promotes VEGF expression and neovascularization of tumor xenografts. These findings indicate that inactivation of p53 in tumor cells contributes to activation of the angiogenic switch via amplification of normal HIF-1-dependent responses to hypoxia.
Results
Inhibition of tumor angiogenesis and growth by p53
The effect of p53 on tumor cell growth and angiogenesis was examined by comparing an isogenic set of human colon adenocarcinoma cell lines differing only in their p53 status (Bunz et al. 1998). The parental HCT116 line, containing wild-type p53 (p53+/+), and a p53-deficient derivative (p53−/−), generated by homologous recombination, demonstrated equivalent growth kinetics in tissue culture, with doubling times of 29 and 32 hr, respectively (Fig. 1A). However, xenografts (2.5 × 104–2.5 × 105 cells) of p53−/− HCT116 cells in athymic BALB/c (nu/nu) mice exhibited a significantly shorter latency and marked increase in tumor growth kinetics compared with their p53+/+ counterparts (Fig. 1B,C). Whereas 12/12 animals inoculated with 2.5 × 104 p53−/− cells developed tumors within 3 weeks, only 1/12 mice receiving the same number of p53+/+ cells was able to establish a tumor during the entire 8-week observation period. To examine whether the observed differences in growth kinetics in vivo were associated with variation in tumor vascularity, tumors established from p53+/+ and p53−/− cells were subjected to histologic analysis and nuclear magnetic resonance (NMR) imaging. Immunohistochemical analyses of tumor sections using an antibody against von Willebrand Factor (vWF) demonstrated significantly increased blood vessel density in p53−/− tumors compared with their p53+/+ counterparts (Fig. 1D,E). Analyses of neovascularization by NMR imaging showed that compared with p53+/+ tumors, p53−/− tumors had a higher vascular volume (14 ± 2.6 μl/g vs. 8.4 ± 2.4 μl/g in highly permeable regions), as well as a threefold greater vascular permeability (0.4 ± 0.18 μl/g/min vs. 0.13 ± 0.04 μl/g/min in highly vascular zones) (Fig. 1F). Thus, loss of p53 function has a profound effect on the neovascularization and growth of human colorectal cancer xenografts in nude mice.
Figure 1.
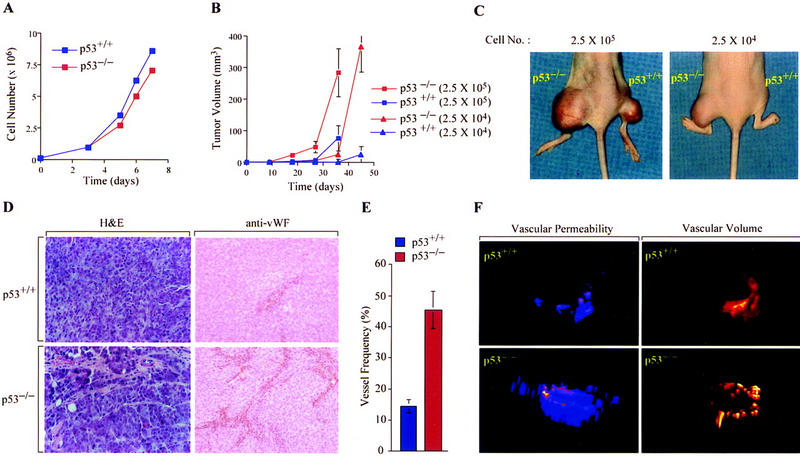
Effect of p53 genotype on tumor growth and angiogenesis. (A) Growth of p53+/+ (blue) and p53−/− (red) HCT116 cells cultured in DMEM supplemented with 10% fetal calf serum at 37°C and 95%air/5%CO2. (B, C) Growth of p53+/+ (blue) and p53−/− (red) HCT116 xenografts [2.5 × 104 (▴) or 2.5 × 105 (█) cells] injected subcutaneously into right (p53+/+) or left (p53−/−) hind legs of athymic BALB/c (nu/nu) mice. Values expressed represent mean ± s.e. of 12 xenografts of each cell type. (D) Histologic analysis of blood vessels in p53+/+ and p53−/− HCT116 xenograft tumors by staining with H&E or immunoperoxidase detection of endothelial cells using an anti-vWF antibody (×25). (E) Quantification of blood vessel density in p53+/+ (blue) and p53−/− (red) xenografts. The data represent the mean ± s.e. of the frequency of vessel hits among 300 random sampling points from each of three tumors of either genotype. (F) Representative NMR analysis of in vivo vascular volume (right) and permeability (left) of p53+/+ and p53−/− (bottom) HCT116 xenografts.
Effect of p53 genotype on hypoxia-induced VEGF expression and HIF-1 activity
Hypoxia-induced, HIF-1-mediated expression of VEGF stimulates angiogenesis and vascular permeability in neoplastic tissues (Plate et al. 1992; Shweiki et al. 1992; Forsythe et al. 1996; Maxwell et al. 1997; Carmeliet et al. 1998). p53+/+ and p53−/− HCT116 cells were analyzed for expression of VEGF mRNA and protein under tissue culture conditions simulating the hypoxic tumor microenvironment. Following exposure to 1% O2, p53−/− cells exhibited a greater induction of VEGF mRNA and protein compared with their p53+/+ counterparts (Fig. 2A,B). Transcriptional activation of the VEGF gene in response to hypoxia is mediated by binding of HIF-1 to a 47-bp hypoxia-response element in the 5′ flanking region, and a reporter plasmid containing this sequence (VEGF-p11w) is transactivated by cotransfection of an expression vector encoding HIF-1α (pCEP4/HIF-1α) (Forsythe et al. 1996). To examine whether p53 influences HIF-1-mediated transcriptional activation of VEGF, p53+/+ and p53−/− cells were cotransfected with the VEGF-p11w reporter and CMVβgal [encoding β-galactosidase (β-gal)]. Analyses of luciferase and β-gal activity in response to hypoxia (1% O2) revealed a fourfold greater increase in VEGF-p11w transcription (relative to β-gal) in p53−/− cells compared with p53+/+ cells (Fig. 2C). These differences were not seen when the reporter contained a 3-bp substitution in the hypoxia response element that eliminated HIF-1 binding (VEGF-p11m), suggesting that HIF-1 was a target for p53-mediated inhibition. Coexpression of pCEP4/HIF-1α in p53+/+ cells increased hypoxia-induced activation of VEGF-p11w to levels that approached the reporter activity exhibited by hypoxic p53−/− cells in the absence of exogenous HIF-1α (Fig. 2C). Conversely, cotransfection of an expression vector encoding wild-type human p53 into p53−/− cells completely repressed hypoxia-induced VEGF-p11w expression (Fig. 2C). Electrophoretic mobility shift assays demonstrated that hypoxia-induced HIF-1 DNA-binding activity was reduced in p53+/+ cells compared with p53−/− cells (Fig. 2D). The specificity of binding of HIF-1 to DNA was confirmed by competing hypoxia-induced DNA–protein complexes with excess unlabeled wild-type probe but not with an unlabeled mutant probe containing the same 3-bp substitution in the HIF-1 binding site as in reporter VEGF-p11m. Thus, p53 inhibits HIF-1 activity and VEGF expression in response to hypoxia.
Figure 2.
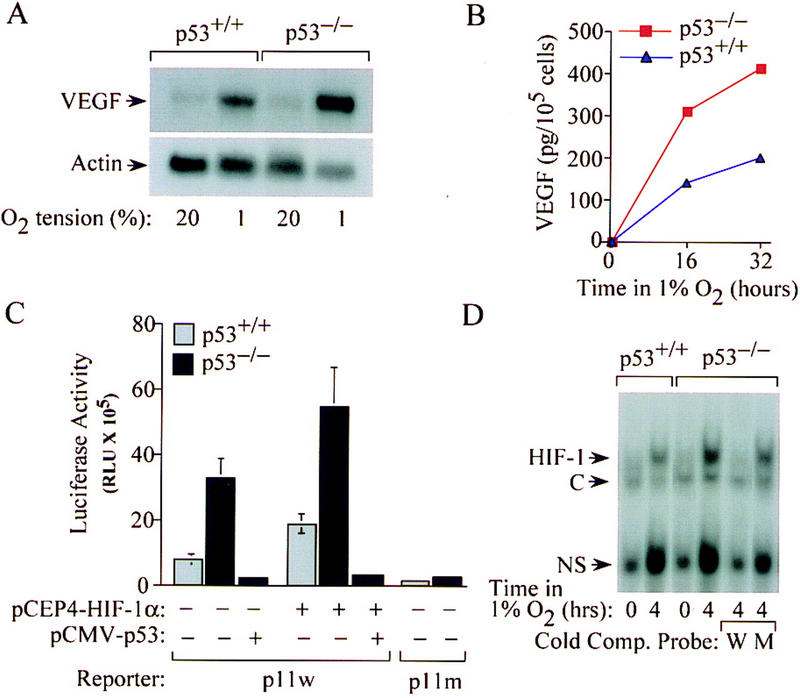
Effect of p53 genotype on hypoxia-induced VEGF expression and HIF-1 activity. (A) Northern blot analysis of VEGF mRNA expression in p53+/+ and p53−/− HCT116 cells incubated for 16 hr in either 20%or 1%O2. (B) ELISA of VEGF protein concentration in supernatant medium of p53+/+ (blue ▴) or p53−/− (red █) HCT116 cells incubated for 16–32 hr in 1% O2. (C) Hypoxia-induced and HIF-1-dependent activation of VEGF-reporter activity in p53+/+ (shaded bars) and p53−/− (solid bars) HCT116 cells. Wild-type (p11w) and mutant (p11m) copies of the hypoxia response element from the VEGF gene were inserted 5′ to a SV40 promoter–luciferase transcription unit. Cells were cotransfected with either VEGF–p11w or VEGF–p11m and CMVβgal, with or without pCEP4/HIF-1α or pCMV–p53, exposed to 1% O2 for 20 hr, and harvested for luciferase assays. The data represent the mean ± s.e. luciferase activity (normalized for β-gal activity) from three independent experiments. (D) Electrophoretic mobility shift assays of HIF-1 DNA-binding activity in nuclear extracts from p53+/+ and p53−/− HCT116 cells exposed to 20% (lanes 1 and 3) or 1% (lanes 2 and 4–6) O2. HIF-1 DNA binding was confirmed by competition assays using either unlabeled wild-type oligonucleotide (W) or a mutant oligonucleotide (M) containing the same 3-bp substitution as in p11m. Complexes containing HIF-1, constitutive (C), and nonspecific (NS) DNA-binding activities (Semenza and Wang 1992) are indicated.
Effect of p53 on oxygen-regulated expression and stability of HIF-1α
Hypoxia-induced HIF-1 DNA-binding and transcriptional activity are dependent on increased levels of HIF-1α protein and its heterodimerization with HIF-1β (Wang and Semenza 1993; Wang et al. 1995; Jiang et al. 1996; Huang et al. 1998). To investigate whether p53 influences HIF-1 activity by altering expression of HIF-1α, the levels of HIF-1α protein and mRNA were assessed in p53+/+ and p53−/− cells exposed to either 20% or 1% O2. In response to hypoxia, p53−/− HCT116 cells or mouse embryonic fibroblasts (MEFs) expressed higher levels of HIF-1α protein compared with their p53+/+ counterparts (Fig. 3A,B). In contrast to HIF-1α protein levels, HIF-1α mRNA was expressed at equivalent levels in hypoxic p53+/+ and p53−/− cells (Fig. 3C), suggesting an effect of p53 on HIF-1α protein expression. To confirm this effect, p53−/− cells were cotransfected with pCEP4-HIF-1α and either pCMV-p53 (encoding wild-type human p53) or empty vector (pCMV0) and exposed to 1% O2 for 8 hr. Immunoblot analysis showed that p53−/− cells cotransfected with pCMV-p53 exhibited reduced levels of HIF-1α compared with cells receiving the control vector (Fig. 3D).
Figure 3.

Effect of p53 on oxygen-regulated expression and stability of HIF-1α. (A) Immunoblot analysis of HIF-1α expression in p53+/+ and p53−/− HCT116 cells cultured for 8 hr in 20%or 1%O2. The blot was analyzed sequentially with monoclonal antibodies against HIF-1α (H1α67), p53 (DO-1), and β-actin. (B) Immunoblot analysis of HIF-1α expression in p53+/+ and p53−/− MEFs cultured for 8 hr in 20% or 1% O2. (C) Northern blot analysis of HIF-1α mRNA expression in p53+/+ and p53−/− HCT116 cells cultured as in A. (D). Immunoblot analysis of HIF-1α protein in p53−/− HCT116 cells cultured in 1% O2 for 8 hr following cotransfection with pCEP4–HIF-1α and either pCMV–p53 or empty vector. The blot was analyzed sequentially with anti-HIF-1α and anti-p53 monoclonal antibodies. (E) Half-life of HIF-1α protein in p53+/+ and p53−/− cells exposed to 100 μm cobalt chloride following addition of 100 μm cycloheximide. Lysates of cells harvested at the indicated time intervals were subject to immunoblot analysis of HIF-1α and p53 expression.
The steady state level of HIF-1α protein is regulated by an oxygen-dependent and iron-sensitive mechanism of ubiquitin-mediated proteasomal degradation (Salceda and Caro 1997; Huang et al. 1998; Kallio et al. 1999). The 20S proteasome is the core catalytic subunit of the 26S proteasome complex that mediates degradation of ubiquitin-tagged proteins (for review, see Hershko and Ciechanover 1998). HIF-1α expression is induced by exposure to hypoxia or treatment with cobalt chloride (Wang et al. 1995). To examine whether p53 influences the stability of HIF-1α protein, HIF-1α expression was analyzed in lysates of cobalt-treated p53+/+ and p53−/− cells at serial time intervals following addition of cycloheximide. HIF-1α protein decayed with a half-life of <20 min in p53+/+ cells, compared with >40 min in p53−/− cells (Fig. 3E).
HPV-E6 augments HIF-1α stability and VEGF expression in response to hypoxia
The human papilloma virus (HPV16) E6 oncoprotein promotes ubiquitin-dependent conjugation and degradation of p53 (Scheffner et al. 1990). To investigate whether E6-induced degradation of endogenous p53 promotes expression of HIF-1α and induction of VEGF, the PA-1 ovarian teratocarcinoma cell line was stably transfected with an expression vector encoding HPV-16 E6 (PA-1 E6) or empty vector (PA-1 Neo) (Ravi et al. 1998). Under hypoxic conditions, PA-1 E6 cells expressed higher levels of HIF-1α protein compared with PA-1 Neo cells (Fig. 4A). Analyses of HIF-1α protein stability in cycloheximide-treated cells showed that HIF-1α protein decayed with a half-life of ∼15 min in PA-1 cells, compared with >30 min in PA-1 E6 cells (Fig. 4B). PA-1 Neo or PA-1 E6 cells were cotransfected with either VEGF-p11w or VEGF-p11m reporter and CMVβgal. Analyses of luciferase and β-gal activity in response to hypoxia (1% O2) revealed a twofold greater increase in VEGF-p11w transcription (relative to β-gal) in PA-1 E6 cells compared with PA-1 Neo cells (Fig. 4C). Neither cell line exhibited significant transcription of the VEGF-p11m reporter. Consistent with the promotion of HIF-1-dependent VEGF transcription by E6 expression, exposure to 1% O2 resulted in greater induction of VEGF protein expression in PA-1 E6 cells compared with PA-1 Neo cells (Fig. 4D).
Figure 4.

HPV E6 increases expression of HIF-1α and VEGF in response to hypoxia. (A) Immunoblot analysis of HIF-1α expression in PA-1 Neo or PA-1 E6 cells cultured for 8 hr in 20% or 1% O2. (B) Half-life of HIF-1α protein in PA-1 Neo or PA-1 E6 cells exposed to 100 μm cobalt chloride following addition of 100 μm cycloheximide. Lysates of cells harvested at the indicated time intervals were subject to immunoblot analysis of HIF-1α expression. (C) Hypoxia-induced and HIF-1-dependent activation of VEGF-reporter activity in PA-1 Neo (open bars) and PA-1 E6 (solid bars) cells. Cells were cotransfected with either VEGF–p11w or VEGF–p11m and CMVβgal, exposed to 1% O2 for 20 hr, and harvested for luciferase assays. The data represent the mean luciferase activity (normalized for β-gal activity) from three independent experiments. (D) ELISA of VEGF protein concentration in supernatant medium of PA-1 Neo (open bar) or PA-1 E6 (solid bar) cells incubated for 16 hr in 1% O2.
p53 promotes ubiquitin-dependent of HIF-1α
To determine whether p53 interacts with HIF-1α in HCT116 cells, as previously demonstrated in MCF-7 cells (An et al. 1998), protein lysates from hypoxic p53+/+ and p53−/− cells were immunoprecipitated with an anti-p53 or isotype control antibody, and the resulting immune complexes were subjected to immunoblot assays using an antibody against HIF-1α. HIF-1α was detected in immunoprecipitates derived from p53+/+ cells but not p53−/− cells or immune complexes precipitated with the control antibody (Fig. 5A).
Figure 5.

Effect of p53 expression on ubiquitin-mediated degradation of HIF-1α. (A) Interaction of p53 with HIF-1α. Lysates of p53+/+ or p53−/− HCT116 cells exposed to 1%O2 for 8 hr were immunoprecipitated with either anti-p53 antibody or isotype control antibody (C) and the resultant immune complexes were subjected to immunoblot analysis with anti-HIF-1α monoclonal antibody. (B) Differential ubiquitination of HIF-1α in hypoxic p53+/+ and p53−/− HCT116 cells. Cells were cotransfected with pCMVβgal and pCEP4/HIF-1α with either MT107/His6-Ub or empty vector (MT107), and cultured in 1%O2 for 4 hr in the presence of 50 μm MG132. Aliquots of whole-cell extract (WCE) or His-tagged proteins purified from whole-cell lysates (His-Ub) were subjected to immunoblot analysis with anti-HIF-1β antibody. (C) Effect of p53 expression on HIF-1α protein levels in hypoxic ts20TGR and H38-5 cells. Cells transfected with pCMV–p53 or pCMVβgal were maintained at either 35°C or 39°C for 8 hr and exposed to 1%O2 for an additional 8 hr at their respective temperatures. Whole-cell lysates were subjected to immunoblot analysis with anti-HIF-1α or anti-p53 antibodies. (D) Effect of p53 on complex formation between HIF-1α and Mdm2. Lysates of p53−/− HCT116 cells transfected with either pCMV–p53 or empty vector and transferred to 1%O2 for 6 hr were immunoprecipitated with anti-Mdm2 or isotype control antibody, and the resulting immune complexes were subjected to immunoblot assays using an antibody against HIF-1α. (E) Effect of wild-type p53, p53ΔI, or p53Gln22,Ser23 on expression of HIF-1α in response to hypoxia. p53−/− HCT116 cells transfected with pCMVβgal and either pCMV–p53, pCB6 + p53ΔI, pCMV–p53Gln22,Ser23, or empty vector were exposed to 1%O2 for 8 hr. Whole-cell lysates were subjected to immunoblot analysis with anti-HIF-1α or anti-Mdm2 antibodies. (F) Effect of dominant–negative (RING finger) mutants of Mdm2 on hypoxia-induced expression of HIF-1α. p53+/+ and p53−/− HCT116 cells transfected with vectors encoding human Mdm2 (1–440) (pCHDM1–440), Mdm2 (464Ala) (pCHDM464Ala), or pCMVβgal were exposed to 1%O2 for 8 hr. Whole-cell lysates were subjected to immunoblot analysis with anti-HIF-1α, anti-p53, or anti-Mdm2 antibodies. (G) Effect of dominant–negative (RING finger deletion mutant) Mdm2 on p53-mediated inhibition of HIF-1α expression in ts20TGR and H38-5 cells. Cells cotransfected with pCMV–p53 and either pCHDM1–440 or empty vector were maintained at 39°C for 12 hr and then exposed to 20%or 1%O2 for an additional 8 hr at 39°C. Whole-cell lysates were subjected to anti-HIF-1α immunoblot analysis.
To determine whether p53 promotes ubiquitination of HIF-1α, p53+/+ and p53−/− cells were cotransfected with an HIF-1α expression vector (pCEP4/HIF-1α) and a vector encoding hexahistidine-tagged ubiquitin (His6-Ub) or the empty control vectors. Transfected cells were exposed to 1% O2 for 4 hr in the presence of MG132, a peptide aldehyde inhibitor of the 20S proteasome. Aliquots of whole-cell extracts or His-tagged proteins isolated by affinity purification from cell lysates were subjected to immunoblot assays using an anti-HIF-1α monoclonal antibody (Fig. 5B). Immunoblot analysis of whole cell extracts of p53+/+ cells detected a 120-kD protein corresponding to the apparent molecular mass of HIF-1α (Wang et al. 1995), as well as an additional series of slower migrating complexes. The higher molecular weight complexes represented polyubiquitinated forms of HIF-1α as they were also detected by immunoblot analysis of His-tagged proteins with an anti-HIF-1α monoclonal antibody. Compared with p53+/+ cells, p53−/− cells transfected with vectors encoding HIF-1α and His6-Ub demonstrated a higher level of unconjugated HIF-1α and a reciprocal reduction in polyubiquitinated HIF-1α (Fig. 5B). Introduction of a p53 expression vector (pCMV–p53) into p53−/− cells increased the proportion of HIF-1α that was ubiquitinated under hypoxic conditions (Fig. 5B).
Conjugation of Ub to proteins destined for degradation involves conversion of Ub to a high-energy thiol ester by the E1 Ub-activating enzyme followed by the transfer of activated Ub to the substrate via the activity of an E2 Ub-conjugating enzyme and an E3 Ub–protein ligase (for review, see Hershko and Ciechanover 1998). To confirm the requirement of the Ub-proteasome system for p53-mediated degradation of HIF-1α, we examined the effect of p53 on hypoxia-induced HIF-1α expression in the BALB/c 3T3-derived ts20TGR cell line, which harbors a thermolabile E1, or a derivative cell line (H38-5), in which the temperature-sensitive defect was corrected by introduction of the human E1 cDNA (Chowdary et al. 1994). ts20TGR and H38-5 cells were transfected with either an expression vector encoding human p53 or a control vector and transferred to hypoxic chambers (1% O2) at either the permissive temperature (35°C) or the restrictive temperature (39°C). Transfection of p53 into ts20TGR cells resulted in reduced HIF-1α levels at 35°C but not at 39°C (Fig. 5C). However, E1-expressing H38-5 cells exhibited p53-mediated reduction of HIF-1α levels at both temperatures. Taken together, the data indicate that p53 limits hypoxia-induced expression of HIF-1α by promoting its ubiquitination and proteasomal degradation.
Whereas a single E1 is responsible for activation of ubiquitin, multiple E3 enzymes are responsible for specific selection of proteins destined for degradation. Because p53 induces the Mdm2 E3 Ub–protein ligase and is itself a target for Ub-mediated degradation via its interaction with Mdm2 (Momand et al. 1992; Barak et al. 1993; Wu et al. 1993; Haupt et al. 1997; Honda et al. 1997; Kubbutat et al. 1997), this raised the possibility that HIF-1α is recruited to Mdm2 via its interaction with p53. To test this hypothesis, protein lysates of p53−/− HCT116 cells that were transfected with either pCMV–p53 or empty vector and transferred to 1%O2 for 6 hr were immunoprecipitated with anti-Mdm2 or isotype control antibody, and the resulting immune complexes were subjected to immunoblot assays using an antibody against HIF-1α. Anti-Mdm2 immunoprecipitates derived from cells transfected with p53 displayed significantly higher levels of coprecipitated HIF-1α protein compared to immune complexes derived from p53−/− HCT116 cells with the empty vector (Fig. 5D).
Amino acid residues Phe-19, Leu-22, and Trp-23 in the amino-terminal transactivation domain of p53 are critical for its interaction with Mdm2 (Lin et al. 1994). A p53 double mutant at residues 22 and 23 (p53 Gln22, Ser23) fails to interact with Mdm2 and is also transactivation deficient (Lin et al. 1994). A p53 mutant carrying a deletion of residues 13–19 (p53ΔI) is also unable to bind to Mdm2 but retains its transactivation function (Marston et al. 1995). To investigate whether p53 requires interaction with Mdm2 to mediate degradation of HIF-1α, p53−/− HCT116 cells were transfected with encoding either wt p53, p53 22-23, p53ΔI, or control vector and analyzed for HIF-1α expression under hypoxic conditions. In contrast to wild-type p53, the p53 mutants (p53ΔI or p53 Gln22, Ser23) or the control vector were unable to reduce the levels of HIF-1α (Fig. 5E).
The Ub–protein ligase function of Mdm2 is dependent on a RING finger domain (residues 434–490) at the carboxyl terminus (Honda et al. 1997). Mdm2 mutants with a deletion of the RING finger domain [Mdm2 (1–440)] or a substitution of a cysteine residue at position 464 to alanine [Mdm2 (464Ala)] are deficient in Ub–protein ligase function but retain the ability to bind p53, thereby behaving in a dominant negative manner (Kubbutat et al. 1999). Introduction of Mdm2 (1–440) or Mdm2 (464Ala) augmented hypoxia-induced HIF-1α levels in p53+/+ HCT116 cells but did not significantly influence HIF-1α expression in hypoxic p53−/− HCT116 cells (Fig. 5F). To determine whether Mdm2 functions as an E3-ligase that mediates p53-induced degradation of HIF-1α, ts20TGR and H38-5 cells were cotransfected with expression vectors encoding wild-type p53 and either Mdm2 (1–440) or empty control vector and transferred to hypoxic chambers (1%O2) at 39°C. Cotransfection of Mdm2 (1–440) increased hypoxia-induced HIF-1α expression in E1-proficient H38-5 cells coexpressing p53 to levels observed in E1-deficient ts20TGR cells (Fig. 5G). Together, the data in Figure 5 are consistent with a model in which p53 acts as a molecular chaperone that facilitates recognition and recruitment of HIF-1α for ubiquitination by Mdm2.
Enhancement of tumor angiogenesis in p53+/+ cells by forced expression of HIF-1α
To determine whether p53-mediated degradation of HIF-1α contributes to the suppression of tumor angiogenesis and growth, p53+/+ HCT116 cells were stably transfected with pCEP4/HIF-1α (HCT116–HIF-1α) (Fig. 6A). Under hypoxic conditions, stable transfectants overexpressing HIF-1α demonstrated significantly increased VEGF mRNA levels compared with the parental p53+/+ cells (Fig. 6B). When inoculated into athymic nude mice, HCT116–HIF-1α cells established tumors with a shorter latency and exhibited a significant increase in tumor growth kinetics compared with the parental cells (Fig. 6C). Histologic evaluation and analyses of NMR maps, as described earlier, revealed a significant increase in blood vessel density, vascular volume (17.4 μl/g) and , (0.8 μl/g/min) in xenografts established from HCT116–HIF-1α cells compared with those derived from the parental p53+/+ HCT116 cells (Fig. 6D).
Figure 6.
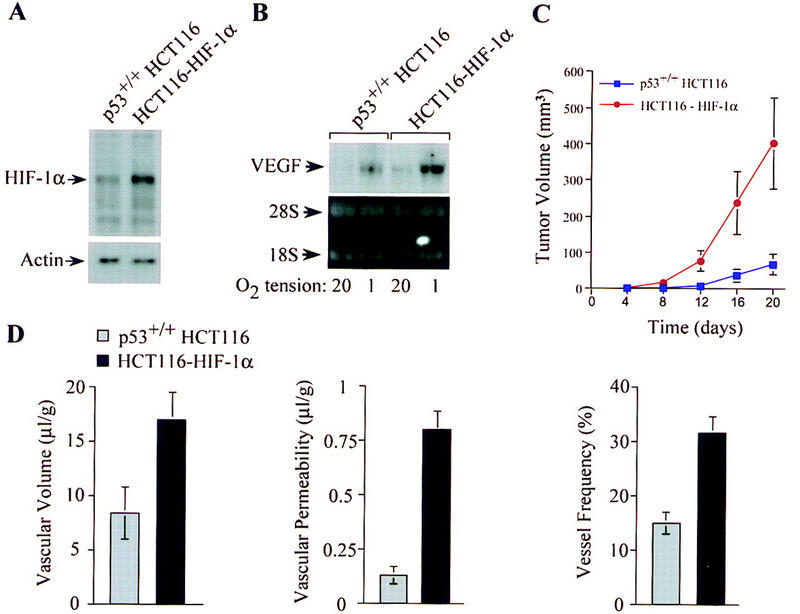
Increased tumor angiogenesis and growth in p53+/+ cells with forced overexpression of HIF-1α. (A) Immunoblot analyses of HIF-1α protein levels in p53+/+ HCT116 cells and p53+/+ HCT116 cells stably transfected with a HIF-1α expression vector (HCT116–HIF-1α following exposure to 1%O2 for 8 hr. (B) Northern blot analysis of VEGF mRNA levels in p53+/+ HCT116 and HCT116–HIF-1α cells cultured for 16 hr in 20%or 1%O2. (C) Growth of p53+/+ HCT116 (blue █) and HCT116–HIF-1α (red ▴) cells (2.5 × 106) injected subcutaneously into the flanks of athymic BALB/c nude mice. Values expressed represent mean ± s.e. of 12 xenografts of each cell type. (D) Quantification of vascular volume, permeability, and blood vessel density in p53+/+ HCT116 (shaded bars) and HCT116–HIF-1α (solid bars) xenograft tumors. In vivo vascular volume and permeability of the tumors were determined by NMR analyses, and blood vessel frequency in stained sections of excised tumors was analyzed as described in Fig. 1.
Discussion
Recognition of the importance of angiogenesis for the growth and metastasis of cancers has raised fundamental questions regarding the molecular mechanisms of the angiogenic switch during tumor progression. The genetic alterations involved in tumorigenesis are also responsible for the phenotypic characteristics of cancer cells. The p53 tumor suppressor gene is one of the most frequently mutated genes in human cancers (for review, see Levine 1997). In addition to p53 mutations, which occur in ∼50%of all cancers (involving >50 tissue types), p53 is also inactivated by viral oncoproteins such the E6 protein of cervical cancer-associated HPV 16 and 18, adenovirus E1A, and SV40 large T antigen (for review, see Levine 1997). Our observations indicate that loss of p53 function, via somatic mutations or expression of viral oncoproteins, contributes to activation of the angiogenic switch during tumorigenesis.
In addition to identifying the loss of p53 as a discrete and potentially rate-limiting event in tumor angiogenesis, we define a novel mechanism by which p53 regulates the angiogenic switch. Our observations indicate that p53 inhibits hypoxia-induced expression of HIF-1α by facilitating its ubiquitination and subsequent degradation. This mechanism is distinct from the proposal that p53 inhibits HIF-1-mediated transactivation by competing for the p300 coactivator (Blagosklonny et al. 1998) and is analogous to the proposed role of the von Hippel-Lindau (VHL) tumor suppressor (Maxwell et al. 1999). As in the case of VHL (Maxwell et al. 1999), we demonstrate that p53 interacts with HIF-1α in vivo, as reported previously (An et al. 1998). In addition, we demonstrate for the first time that a tumor suppressor (p53) promotes the ubiquitin-mediated degradation of HIF-1α via recruitment of an E3 ubiquitin–protein ligase (Mdm2). Although ubiquitination is assumed to be the mechanism by which VHL affects HIF-1α degradation, our data provide the first direct evidence for this mechanism of tumor suppressor action. The constitutive stabilization of HIF-1α and the related HIF-2α protein) resulting from VHL loss of function may underlie the predisposition to highly angiogenic tumors in VHL disease, a rare hereditary cancer syndrome. Our findings indicate that deregulation of HIF-1α expression, leading to overexpression of VEGF, may contribute to the angiogenic switch conferred by inactivation of p53 in a broad array of human cancers. In accordance with this hypothesis, HIF-1α is frequently overexpressed in common human cancers and there is a statistically significant correlation between the presence of mutant p53 and HIF-1α overexpression (Zhong et al. 1999). Our findings suggest that increased HIF-1 activity resulting from loss of p53 function may contribute to the overexpression of VEGF that is observed in a wide variety of human cancers (for review, see Brown et al. 1996; Folkman 1997).
The angiogenic switch is regulated by changes in the relative balance between inducers and inhibitors of endothelial cell proliferation and migration (for review, see Hanahan and Folkman 1996). The switch can be activated by increasing the levels of inducers, such as VEGF, and/or by reducing the concentration of inhibitors, such as thrombospondin-1 (TSP-1). The p53-mediated inhibition of VEGF expression demonstrated in this study, together with the previously reported ability of p53 to upregulate TSP-1 (Dameron et al. 1994), indicates that p53 provides dual functions that regulate angiogenesis. Thus, the loss of p53 function during tumorigenesis deregulates both arms of the balance, providing a potent stimulus for neovascularization and tumor progression.
In addition to loss-of-function mutations in tumor suppressor genes such as p53 or VHL, oncogene activation is also capable of stimulating HIF-1 activity. Expression of the v-Src oncogene induces expression of HIF-1α protein, HIF-1 DNA-binding activity, and transcriptional activation of VEGF and enolase 1 (Jiang et al. 1997). A phosphatidylinositol 3-kinase/Akt pathway of HIF-1 activation may induce VEGF expression in Ha-ras-transformed cells (Mazure et al. 1997). Therefore, increased HIF-1 expression is associated with multiple genetic alterations that promote tumor angiogenesis. Because HIF-1 is also a key transcriptional activator of genes encoding glucose transporters and glycolytic enzymes (Iyer et al. 1998), these genetic alterations also contribute to the metabolic adaptation and enhanced survival of tumor cells in hypoxic microenvironments.
As p53 is an important mediator of DNA damage-induced apoptosis, the angiogenic phenotype conferred by inactivation of p53 in human cancers is frequently associated with resistance to conventional genotoxic anticancer agents (for review, see Lowe 1995). Because p53-deficient tumors remain dependent on angiogenesis for growth and metastasis, inhibition of angiogenesis may represent an effective therapeutic intervention (Boehm et al. 1997; Bergers et al. 1999). Recent studies indicate that inhibition of tumor-derived VEGF expression restricts angiogenesis and promotes vascular regression in experimental tumor models (Kim et al. 1993; Millauer et al. 1994, 1996; Warren et al. 1995; Goldman et al. 1998). Loss of HIF-1 activity is also associated with decreased angiogenesis and growth of tumor xenografts in nude mice (Jiang et al. 1997; Maxwell et al. 1997; Carmeliet et al. 1998; Ryan et al. 1998). By demonstrating that deregulation of HIF-1 underlies the increased expression of VEGF in p53-deficient cancers, our data provide further support for the hypothesis that inhibition of HIF-1 may abrogate the ability of such tumors to establish an adequate vascular supply and adapt their cellular metabolism to hypoxia, thereby curtailing their growth and metastasis.
Materials and methods
Cell lines and culture
The parental HCT116 human colon adenocarcinoma cell line, containing wild-type p53 (p53+/+), and a p53-deficient derivative (p53−/−) created by homozygous deletion via homologous recombination (Bunz et al. 1998), were a gift from Bert Vogelstein. p53+/+ HCT116 cells were transfected with pCEP4/HIF-1α and a pool of stable transformants overexpressing HIF-1α (HCT116–HIF-1α) was selected in the presence of hygromycin (200 μg/ml). p53+/+ or p53−/− HCT116 and HCT116–HIF-1α cells were maintained in McCoy's modified medium (Life Technologies, Inc.) supplemented with 10%fetal calf serum (FCS), penicillin (100 U/ml), and streptomycin (100 μg/ml) at 37°C and 5%CO2. PA-1 ovarian teratocarcinoma cells stably transfected with pCMV–HPV16 E6 or pCMV–Neo, generated as described (Ravi et al. 1998), were maintained in Basal Eagle medium supplemented with 0.5 mg/ml G418, 10%FCS, and antibiotics (as described above) at 37°C and 5%CO2. p53+/+ and p53−/− MEFs (gift from Tyler Jacks, Massachusetts Institute of Technology, Cambridge, MA), were cultured in Dulbecco's modified Eagle medium (DMEM) containing 10%FCS and antibiotics. The BALB/c 3T3-derived ts20TGR or H38-5 cell lines (Chowdary et al. 1994) (gift from Harvey L. Ozer) were maintained at 35°C in DMEM supplemented with 10%fetal bovine serum and antibiotics. The permissive and nonpermissive temperatures for the ts20TGR E1-mutant cell line are 35°C and 39°C, respectively. Cells were plated on 100-mm petri dishes and allowed to approach confluence. For hypoxic conditions, cells were placed in a modular incubator chamber and flushed with a gas mixture containing 1%O2, 5%CO2, and balance N2 (Semenza and Wang 1992).
Growth of tumor xenografts in nude mice
HCT116 cells (2.5 × 104, 2.5 × 105, or 2.5 × 106) suspended in 0.1 ml of PBS were injected subcutaneously into the right (p53+/+) or left (p53−/−) hind legs or flanks of athymic BALB/c (nu/nu) mice. Tumor volumes were determined by external measurement in three dimensions using the equation V = [L × W × H] × /6, where V = volume, L = length, W = width, and H = height. Care of experimental animals was in accordance with institutional animal care and use committee guidelines.
NMR analyses of in vivo vascular volume and permeability of tumor xenografts
Multislice maps of relaxation rates (T1−1) were obtained by a saturation recovery T1 SNAPSHOT-FLASH imaging method (flip angle of 5°, echo time of 2 msec). Images of four slices (slice thickness of 1 mm) acquired with an in-plane spatial resolution of 125 μm (128 × 128 matrix, 16-mm field of view, NS = 8) were obtained for three relaxation delays (100 msec, 500 msec, and 1 sec) for each of the slices; 128 × 128 × 4 T1 maps were acquired within 7 min. An Mo map with a recovery delay of 7 sec was acquired once at the beginning of the experiment. Images were obtained before intravenous administration of 0.2 ml of 60 mg/ml albumin–GdDTPA in saline (dose of 500 mg/kg) and repeated starting after the injection up to 32 min. Relaxation maps were reconstructed from data sets for three different relaxation times and the Mo data set on a pixel by pixel basis. At the end of the imaging studies, the animal was sacrificed, and 0.5 ml of blood was withdrawn from the inferior vena cava. Vascular volume and permeability product surface area (PS) maps were generated from the ratio of Δ(1/T1) values in the images to that of blood. The slope of Δ(1/T1) ratios versus time in each pixel was used to compute PS, whereas the intercept of the line at zero time was used to compute vascular volume. Thus, vascular volumes were corrected for permeability of the vessels. Volume and permeability values (mean ± s.e.) were computed for tumor xenografts established with HCT116 p53+/+ (n = 4), HCT116 p53−/− (n = 5), and HCT116–HIF-1α (n = 2) cells.
Histologic analyses of blood vessel density in tumor xenografts
Five-micrometer sections prepared from paraffin-embedded tissue were stained with hemotoxin & eosin (H & E) and subjected to immunoperoxidase detection of endothelial cells using an anti-vWF antibody. A circular matrix of 25 random sampling points (per unit area) was superimposed on defined fields, and the points overlying a vessel were scored as a percentage of the total points.
Plasmids
Plasmids encoding human wild-type full-length p53 (pC53-SN; gift from Bert Vogelstein, Johns Hopkins Oncology Center, Baltimore, MD), mutant p53 (pCB6+ p53ΔI) (Marston et al. 1994), p53 double mutants (pCMV–p53Gln22, Ser23) (Lin et al. 1994), human mutant Mdm2 (pCHDM1-440 and pCHD464Ala; provided by Karen Vousden) (Kubbutat et al. 1999), His6–Ubiquitin (MT107–His6-Ub; provided by Dirk Bohmann) (Musti et al. 1997), pCMV–HPV16 E6 (provided by Kathy Cho, Johns Hopkins University School of Medicine, Baltimore, MD), and HIF-1α (pCEP4–HIF-1α) (Forsythe et al. 1996; Jiang et al. 1996) have been described previously.
Analysis of VEGF reporter activity
Wild-type (p11w) and mutant (p11m) copies of the hypoxia response element from the VEGF gene cloned 5′ to a SV40-promoter–luciferase transcription unit were described previously (Forsythe et al. 1996). p53+/+ or p53−/− HCT116 cells and PA-1 Neo or PA-1 E6 cells were cotransfected (using Lipofectin) with either VEGF–p11w or VEGF–p11m and CMVβgal, with or without pCEP4/HIF-1α. Transfected cells were exposed to hypoxia (1%O2) for 20 hr and harvested for β-gal and luciferase assays (Promega) in fixed protein aliquots. Luciferase activity was normalized for β-gal activity. The data represent the mean ± s.e. from three independent experiments.
Electrophoretic mobility shift assays of HIF-1 DNA-binding activity
Nuclear extracts (5 μg) prepared from p53+/+ and p53−/− HCT116 cells exposed to either 20%or 1%O2 were incubated with 32P-labeled double-stranded oligonucleotide probe containing a wild-type HIF-1 binding site and DNA/protein complexes were analyzed by polyacrylamide gel electrophoresis as described previously (Semenza and Wang 1992; Jiang et al. 1996). HIF-1 binding to the probe was confirmed by competition assays using 50 ng of either unlabeled wild-type oligonucleotide or a mutant oligonucleotide containing the same 3-bp substitution as in p11m (Semenza and Wang 1992; Forsythe et al. 1996).
Northern blot
VEGF and HIF-1α mRNA was assessed by Northern blot analyses of total RNA prepared from p53+/+ or p53−/− HCT116 and HCT116–HIF-1α cells cultured for 16 hr in either 20%or 1%O2. Total RNA (20-μg aliquots) was fractionated by 1.2%agarose–formaldehyde gel electrophoresis and transferred to nylon membranes. The blots were hybridized to probes for VEGF, HIF-1α, and β-actin mRNA using random primer labeling (Boehringer Mannheim) (Jiang et al. 1997).
ELISA
Quantikine (R & D Systems) was used to measure VEGF protein in supernatant medium of p53+/+ or p53−/− HCT116 cells and PA-1 Neo or PA-1 E6 cells cultured as described above for 16–32 hr.
Immunoblot analyses and immunoprecipation
Nuclear extracts or whole-cell lysates were prepared, fractionated by SDS-PAGE, transferred to PVDF membranes (Millipore, Bedford, MA), and immunoblotted with monoclonal antibodies against HIF-1α (H1α67; Novus Biologicals, Inc.) (Zhong et al. 1999), p53 (DO-1, Ab-6; Oncogene Research Products), Mdm2 (Ab-1; Oncogene Research Products), or β-actin ( Santa Cruz Biotechnology, Inc.). Immunoreactive proteins were detected using enhanced chemiluminescence (Amersham). For analysis of protein interactions, whole-cell lysates were immunoprecipitated with antibodies against either p53 or Mdm2 or isotype control antibody, and the resultant immune complexes were subjected to immunoblot analysis with anti-HIF-1α monoclonal antibody H1α67.
Analysis of HIF-1α protein half-life and ubiquitin-dependent degradation
Cells exposed to 100 μm cobalt chloride for 4 hr were treated with 100 μm cycloheximide. Whole-cell extracts were prepared at intervals of 15–40 min and subjected to immunoblot analyses with anti-HIF-1α antibody. To assess ubiquitination of HIF-1α in hypoxic conditions, p53+/+ and p53−/− HCT116 cells were cotransfected (Lipofectin, Life Technologies, Inc.; 100-mm dishes) with 2 μg of pCMVβgal and 6μg each of pCEP4/HIF-1α and either MT107/His6-Ub or empty vector (MT107) (Musti et al. 1997), and cultured in 1%O2 for 4 hr in the presence of 50 μm MG132 (Peptides International, Inc.). Cells were lysed in buffer supplemented with 5 mm N-ethylmaleimide and 50 mm imidazole, as described (Ravi et al. 1998). Whole-cell extracts or His-tagged proteins [purified from 500 μg of whole cell–protein lysates (normalized to β-gal activity) using Talon Metal Affinity Resin (Clontech)] were subjected to SDS-PAGE and immunoblot analysis with anti-HIF-1α antibody. To analyze whether the effect of p53 on HIF-1α protein levels was dependent on ubiquitination, ts20TGR and H38-5 cells were transfected (using Lipofectin) with pCMV–p53 and pCMVβgal, maintained at either 35°C or 39°C for 8 hr, and then exposed to 1%O2 for an additional 8 hr at their respective temperatures. Whole-cell lysates were subjected to immunoblot analysis with anti-HIF-1α or anti-p53 antibodies.
Acknowledgments
This work was funded in part by grants from the National Institutes of Health (CA71660-01A1 to A.B., RO1-HL55338 to G.L.S., and RO1-CA73850 to Z.M.B.), U.S. Army Medical Research and Material Command Department of Defense (DAMD 17-99-1-9230 to A.B.), and the American Cancer Society. A.B. is a recipient of a Passano Physician Scientist award and a Scholar award from the Valvano Foundation for Cancer Research. We thank Dr. Bert Vogelstein for his gift of p53+/+ and p53−/− HCT116 cell lines, and the vector encoding wild type p53; Dr. Tyler Jacks for his gift of p53+/+ and p53−/− MEFs; Drs. Karen H. Vousden and Arnold J. Levine for vectors encoding mutant p53 and mutant Mdm2; Dr. Harvey L. Ozer for his gift of the ts20TGR and H38-5 cell lines; Dr. Kathy Cho for her gift of the vector encoding HPV-16 E6; and Dr. Dirk Bohmann for providing plasmids MT107 and MT123.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL gsemenza@jhmi.edu; FAX (410) 955-0484.
References
- An WG, Kanekal M, Simon MC, Maltepe E, Blagosklonny MV, Neckers LM. Stabilization of wild-type p53 by hypoxia-inducible factor 1α. Nature. 1998;392:405–408. doi: 10.1038/32925. [DOI] [PubMed] [Google Scholar]
- Barak Y, Juven T, Haffner R, Oren M. Mdm-2 expression is induced by wild type p53 activity. EMBO J. 1993;12:461–468. doi: 10.1002/j.1460-2075.1993.tb05678.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergers G, Javaherian K, Lo KN, Folkman J, Hanahan D. Effects of angiogenesis inhibitors on multistage carcinogenesis in mice. Science. 1999;284:808–812. doi: 10.1126/science.284.5415.808. [DOI] [PubMed] [Google Scholar]
- Blagosklonny MV, An WG, Romanova LY, Trepel J, Fojo T, Neckers L. p53 inhibits hypoxia-inducible factor-stimulated transcription. J Biol Chem. 1998;273:11995–11998. doi: 10.1074/jbc.273.20.11995. [DOI] [PubMed] [Google Scholar]
- Boehm T, Folkman J, Browder T, O'Reilly MS. Antiangiogenic therapy of experimental cancer does not induce acquired drug resistance. Nature. 1997;390:404–407. doi: 10.1038/37126. [DOI] [PubMed] [Google Scholar]
- Bouvet M, Ellis LM, Nishizaki M, Fujiwara T, Liu W, Bucana CD, Fang B, Lee JJ, Roth JA. Adenovirus-mediated wild-type p53 gene transfer down-regulates vascular endothelial growth factor expression and inhibits angiogenesis in human colon cancer. Cancer Res. 1998;58:2288–2292. [PubMed] [Google Scholar]
- Brown JM, Giaccia AJ. The unique physiology of solid tumors: Opportunities (and problems) for cancer therapy. Cancer Res. 1998;58:1408–1416. [PubMed] [Google Scholar]
- Brown LF, Detmar M, Claffey K, Nagy JA, Feng D, Dvorak AM, Dvorak HF. Vascular permeability factor/vascular endothelial growth factor: A multifunctional angiogenic cytokine. In: Goldberg ID, Rosen E, editors. Control of angiogenesis. Berlin, Germany: Birkhauser Verlag; 1996. [Google Scholar]
- Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science. 1998;282:1497–1501. doi: 10.1126/science.282.5393.1497. [DOI] [PubMed] [Google Scholar]
- Carmeliet P, Dor Y, Herbert JM, Fukumura D, Brusselmans K, Dewerchin M, Neeman M, Bono F, Abramovitch R, Maxwell P, et al. Role of HIF-1α in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature. 1998;394:485–490. doi: 10.1038/28867. [DOI] [PubMed] [Google Scholar]
- Chowdary DR, Dermody JJ, Jha KK, Ozer HL. Accumulation of p53 in a mutant cell line defective in the ubiquitin pathway. Mol Cell Biol. 1994;14:1997–2003. doi: 10.1128/mcb.14.3.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dameron KM, Volpert OV, Tainsky MA, Bouck N. Control of angiogenesis in fibroblasts by p53 regulation of thrombospondin-1. Science. 1994;265:1582–1584. doi: 10.1126/science.7521539. [DOI] [PubMed] [Google Scholar]
- Dang CV, Semenza GL. Oncogenic alterations of metabolism. Trends Biochem Sci. 1999;24:68–72. doi: 10.1016/s0968-0004(98)01344-9. [DOI] [PubMed] [Google Scholar]
- Ferrara N. Vascular endothelial growth factor. Trends Cardiovasc Med. 1993;3:244–250. doi: 10.1016/1050-1738(93)90046-9. [DOI] [PubMed] [Google Scholar]
- Folkman J. Tumor angiogenesis. In: Holland JF, Bast RC Jr, Morton DL, Frei III E, Kufe DW, Weichselbaum RR, editors. Cancer Medicine. Baltimore, MD: Williams & Wilkins; 1997. pp. 181–204. [Google Scholar]
- Folkman J, Watson K, Ingber D, Hanahan D. Induction of angiogenesis during the transition from hyperplasia to neoplasia. Nature. 1989;339:58–61. doi: 10.1038/339058a0. [DOI] [PubMed] [Google Scholar]
- Fontanini G, Boldrini L, Vignati S, Chine S, Basolo F, Silvestri V, Lucchi M, Mussi A, Angeletti CA, Bevilacqua G. Bcl2 and p53 regulate vascular endothelial growth factor (VEGF)-mediated angiogenesis in non-small cell lung carcinoma. Eur J Cancer. 1998;34:718–723. doi: 10.1016/s0959-8049(97)10145-9. [DOI] [PubMed] [Google Scholar]
- Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, Semenza GL. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor1. Mol Cell Biol. 1996;16:4604–4613. doi: 10.1128/mcb.16.9.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaccia AJ, Kastan MB. The complexity of p53 modulation: Emerging patterns from divergent signals. Genes & Dev. 1998;12:2973–2983. doi: 10.1101/gad.12.19.2973. [DOI] [PubMed] [Google Scholar]
- Gimbrone MAJ, Leapman SB, Cotran RS, Folkman J. Tumor dormancy in vivo by prevention of neovascularization. J Exp Med. 1972;136:261–276. doi: 10.1084/jem.136.2.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman CK, Kendall RL, Cabrera G, Soroceanu L, Heike Y, Gillespie GY, Siegal GP, Mao X, Bett AJ, Huckle WR, et al. Paracrine expression of a native soluble vascular endothelial growth factor receptor inhibits tumor growth, metastasis, and mortality rate. Proc Natl Acad Sci. 1998;95:8795–8800. doi: 10.1073/pnas.95.15.8795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graeber TG, Osmanian C, Jacks T, Housman DE, Koch CJ, Lowe SW, Giaccia AJ. Hypoxia-mediated selection of cells with diminished apoptotic potential in solid tumours. Nature. 1996;379:88–91. doi: 10.1038/379088a0. [DOI] [PubMed] [Google Scholar]
- Guillemin K, Krasnow MA. The hypoxic response: Huffing and HIFing. Cell. 1997;89:9–12. doi: 10.1016/s0092-8674(00)80176-2. [DOI] [PubMed] [Google Scholar]
- Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell. 1996;86:353–364. doi: 10.1016/s0092-8674(00)80108-7. [DOI] [PubMed] [Google Scholar]
- Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature. 1997;387:299–303. doi: 10.1038/387296a0. [DOI] [PubMed] [Google Scholar]
- Hershko A, Ciechanover A. The ubiquitin system. Annu Rev Biochem. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- Holmgren L, O'Reilly MS, Folkman J. Dormancy of micrometastases: Balanced proliferation and apoptosis in the presence of angiogenesis suppression. Nature Med. 1995;1:149–153. doi: 10.1038/nm0295-149. [DOI] [PubMed] [Google Scholar]
- Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett. 1997;420:25–27. doi: 10.1016/s0014-5793(97)01480-4. [DOI] [PubMed] [Google Scholar]
- Huang LE, Gu J, Schau M, Bunn HF. Regulation of hypoxia-inducible factor 1α is mediated by an oxygen-dependent domain via the ubiquitin-proteasome pathway. Proc Natl Acad Sci. 1998;95:7987–7992. doi: 10.1073/pnas.95.14.7987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, Gassmann M, Gearhart JD, Lawler AM, Yu AY, et al. Cellular and developmental control of O2 homeostasis by hypoxia inducible factor1α. Genes & Dev. 1998;12:149–162. doi: 10.1101/gad.12.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang B-H, Rue E, Wang GL, Roe R, Semenza GL. Dimerization, DNA binding, and transactivation properties of hypoxia-inducible factor 1. J Biol Chem. 1996;271:17771–17778. doi: 10.1074/jbc.271.30.17771. [DOI] [PubMed] [Google Scholar]
- Jiang B-H, Agani F, Passaniti A, Semenza GL. V-SRC induces expression of hypoxia-inducible factor1 (HIF-1) and transcription of genes encoding vascular endothelial growth factor and enolase 1: Involvement of HIF-1 in tumor progression. Cancer Res. 1997;57:5328–5335. [PubMed] [Google Scholar]
- Kallio PJ, Wilson WJ, O'Brien S, Makino Y, Poellinger L. Regulation of the hypoxia-inducible transcription factor 1α by the ubiquitin-proteasome pathway. J Biol Chem. 1999;274:6519–6525. doi: 10.1074/jbc.274.10.6519. [DOI] [PubMed] [Google Scholar]
- Kim KJ, Li B, Winer J, Armanini M, Gillett N, Phillips HS, Ferrara N. Inhibition of vascular endothelial growth factor-induced angiogenesis suppresses tumour growth in vivo. Nature. 1993;362:841–844. doi: 10.1038/362841a0. [DOI] [PubMed] [Google Scholar]
- Kotch LE, Iyer NV, Laughner E, Semenza GL. Defective vascularization of HIF-1α-null embryos is not associated with VEGF deficiency but with mesenchymal cell death. Dev Biol. 1999;209:254–267. doi: 10.1006/dbio.1999.9253. [DOI] [PubMed] [Google Scholar]
- Kubbutat HMG, Jones SN, Vousden KH. Regulation of p53 stability by Mdm2. Nature. 1997;387:299–303. doi: 10.1038/387299a0. [DOI] [PubMed] [Google Scholar]
- Kubbutat MHG, Ludwig RL, Levine AJ, Vousden KH. Analysis of the degradation function of Mdm2. Cell Growth & Diff. 1999;10:87–92. [PubMed] [Google Scholar]
- Levine AJ. p53, the cellular gatekeeper for growth and division. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- Lin J, Chen J, Elenbaas B, Levine AJ. Several hydrophobic amino acids in the p53 amino-terminal domain are required for transcriptional activation, binding to mdm-2 and the adenovirus 5 E1B 55-kD protein. Genes & Dev. 1994;8:1235–1246. doi: 10.1101/gad.8.10.1235. [DOI] [PubMed] [Google Scholar]
- Lowe SW. Cancer therapy and p53. Curr Opin Oncol. 1995;7:547–553. doi: 10.1097/00001622-199511000-00013. [DOI] [PubMed] [Google Scholar]
- Marston NJ, Jenkins JR, Vousden KH. Oligomerisation of full-length p53 contributes to the interaction with mdm2 but not HPV E6. Oncogene. 1995;10:1709–1715. [PubMed] [Google Scholar]
- Maxwell PH, Dachs GU, Gleadle JM, Nicholls LG, Harris AL, Stratford IJ, Hankinson O, Pugh CW, Ratcliffe PJ. Hypoxia-inducible factor 1 modulates gene expression in solid tumors and influences both angiogenesis and tumor growth. Proc Natl Acad Sci. 1997;94:8104–8109. doi: 10.1073/pnas.94.15.8104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399:271–275. doi: 10.1038/20459. [DOI] [PubMed] [Google Scholar]
- Mazure NM, Chen EY, Laderoute KR, Giaccia AJ. Induction of vascular endothelial growth factor by hypoxia is modulated by a phosphatidylinositol 3-kinase/Akt signaling pathway in Ha-ras-transformed cells through a hypoxia inducible factor-1 transcriptional element. Blood. 1997;90:3322–3331. [PubMed] [Google Scholar]
- Millauer B, Shawver LK, Plate KH, Risau W, Ullrich A. Glioblastoma growth inhibited in vivo by a dominant-negative Flk-1 mutant. Nature. 1994;367:576–579. doi: 10.1038/367576a0. [DOI] [PubMed] [Google Scholar]
- Millauer B, Longhi MP, Plate KH, Shawver LK, Risau W, Ullrich A, Strawn LM. Dominant-negative inhibition of Flk-1 suppresses the growth of many tumor types in vivo. Cancer Res. 1996;56:1615–1620. [PubMed] [Google Scholar]
- Momand J, Zambetti GP, George DL, Levine AJ. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell. 1992;69:1237–1245. doi: 10.1016/0092-8674(92)90644-r. [DOI] [PubMed] [Google Scholar]
- Mukhopadhyay D, Tsiokas L, Sukhatme VP. Wild-type p53 and v-Src exert opposing influences on human vascular endothelial growth factor gene expression. Cancer Res. 1995;55:6161–6165. [PubMed] [Google Scholar]
- Musti AM, Treier M, Bohmann D. Reduced ubiquitin-dependent degradation of c-Jun after phosphorylation by MAP kinases. Science. 1997;275:400–402. doi: 10.1126/science.275.5298.400. [DOI] [PubMed] [Google Scholar]
- O'Reilly MS, Holmgren L, Chen C, Folkman J. Angiostatin induces and sustains dormancy of human primary tumors in mice. Nature Med. 1996;2:689–692. doi: 10.1038/nm0696-689. [DOI] [PubMed] [Google Scholar]
- Parangi S, O'Reilly MS, Christofori G, Holmgren I, Grosfeld J, Folkman J, Hanahan D. Antiangiogenic therapy of transgenic mice impairs de novo tumor growth. Proc Natl Acad Sci. 1996;93:2002–2007. doi: 10.1073/pnas.93.5.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plate KH, Breier G, Weich HA, Risau W. Vascular endothelial growth factor is a potential tumour angiogenesis factor in human gliomas in vivo. Nature. 1992;359:845–848. doi: 10.1038/359845a0. [DOI] [PubMed] [Google Scholar]
- Ravi R, Mookerjee B, Hensbergen YV, Bedi GC, Giordano A, El-Deiry WS, Fuchs EJ, Bedi A. p53-mediated repression of nuclear factor κ-B RelA via the transcriptional integrator p 300. Cancer Res. 1998;58:4531–4536. [PubMed] [Google Scholar]
- Risau W, Flamme I. Vasculogenesis. Annu Rev Cell Dev Biol. 1995;11:73–91. doi: 10.1146/annurev.cb.11.110195.000445. [DOI] [PubMed] [Google Scholar]
- Ryan HE, Lo J, Johnson RS. HIF-1α is required for solid tumor formation and embryonic vascularization. EMBO J. 1998;17:3005–3015. doi: 10.1093/emboj/17.11.3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salceda S, Caro J. Hypoxia-inducible factor 1α (HIF-1α) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. J Biol Chem. 1997;272:22642–22647. doi: 10.1074/jbc.272.36.22642. [DOI] [PubMed] [Google Scholar]
- Scheffner M, Werness BA, Huibregtse JM, Levine AJ, Howley PM. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell. 1990;63:1129–1136. doi: 10.1016/0092-8674(90)90409-8. [DOI] [PubMed] [Google Scholar]
- Scheffner M, Nuber U, Huibregtse JM. Protein ubiquitination involving an E1-E2-E3 enzyme ubiquitin thioester cascade. Nature. 1995;373:81–83. doi: 10.1038/373081a0. [DOI] [PubMed] [Google Scholar]
- Semenza GL. Regulation of mammalian oxygen homeostasis by hypoxia-inducible factor 1. Annu Rev Cell Dev Biol. 1999;15:551–578. doi: 10.1146/annurev.cellbio.15.1.551. [DOI] [PubMed] [Google Scholar]
- Semenza GL, Wang GL. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol. 1992;12:5447–5454. doi: 10.1128/mcb.12.12.5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shweiki D, Itin A, Soffer D, Keshet E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature. 1992;359:843–845. doi: 10.1038/359843a0. [DOI] [PubMed] [Google Scholar]
- Van Meir EG, Polverini PJ, Chazin VR, Su Huang H-J, de Tribolet N, Cavenee WK. Release of an inhibitor of angiogenesis upon induction of wild-type p53 expression in glioblastoma cells. Nature Genet. 1994;8:171–176. doi: 10.1038/ng1094-171. [DOI] [PubMed] [Google Scholar]
- Wang GL, Semenza GL. Characterization of hypoxia-inducible factor-1 and regulation of DNA binding activity by hypoxia. J Biol Chem. 1993;268:21513–21518. [PubMed] [Google Scholar]
- Wang GL, Jiang B-H, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci. 1995;92:5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren RS, Yuan H, Matli MR, Gillett NA, Ferrara N. Regulation by vascular endothelial growth factor of human colon cancer tumorigenesis in a mouse model of experimental liver metastasis. J Clin Invest. 1995;95:1789–1797. doi: 10.1172/JCI117857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu XW, Bayle JH, Olson D, Levine AJ. The p53 mdm-2 autoregulatory feedback loop. Genes & Dev. 1993;7:1126–1132. doi: 10.1101/gad.7.7a.1126. [DOI] [PubMed] [Google Scholar]
- Zhong H, DeMarzo AM, Laughner E, Lim M, Hilton A, Zagzag D, Buechler P, Isaacs WB, Semenza GL, Simons JW. Overexpression of hypoxia-inducible factor 1α in common human cancers and their metastases. Cancer Res. 1999;59:5830–5835. [PubMed] [Google Scholar]