

Abstract
Interferon (IFN)-α, a type-I IFN, is widely used to treat chronic hepatitis C virus (HCV) infection, but the broad expression of IFN-α receptors often leads to adverse reactions in many organs. Here, we examine IFN-λ, a type-III IFN, as a therapeutic alternative to IFN-α. Like IFN-α, IFN-λ also induces antiviral activity in hepatocytes, but may induce fewer adverse reactions because its receptor is largely restricted to cells of epithelial origin. We also discuss the recent discovery of single nucleotide polymorphisms (SNPs) near the human IFN-λ3 gene, IL28B, that correlate strongly with the ability to achieve a sustained virologic response (SVR) to therapy with pegylated IFN-α plus ribavirin in patients with chronic hepatitis C.
The interferon subtypes
Interferons (IFNs) are characterized in large by their ability to induce antiviral activity in receptor-bearing target cells. IFNs are divided into type-I, type-II and type-III subtypes, based in part on the differential use of unique receptors through which they mediate signal transduction. The human genome contains thirteen distinct but highly related IFN-α genes and single genes that encode IFN-p, -β, -ε, -κ and -ω. Together these comprise the type-I IFNs, which signal through IFN-alpha receptor (IFNAR) complexes composed of the IFNAR1 and IFNAR2 polypeptide chains [1]. The essential role of the type-I IFNs in the induction of antiviral activity was demonstrated using type-I IFN receptor knockout mice [2,3]. These animals are highly susceptible to infection by many types of viruses. IFN-γ, which signals through the IFN-γ receptor (IFNGR) complex, is the only type-II IFN. Because it is not commonly used as a treatment for HCV infection, it will not be a focus of this review.
The type-III subset of the IFN family was co-discovered by two independent research groups in 2003 [4,5]. It includes three members: IFN-λ1, -λ2 and -λ3 [4], also known as IL-29, IL-28A and IL-28B, respectively [5]. Here, when referring to type-III IFNs, we use the gene symbols IL29, IL28A and IL28B, as recommended by the Human Genome Organization (HUGO) Gene Nomenclature Committee, but the IFN-λ, designations when referring to the corresponding proteins to emphasize their antiviral functions. The type-III IFNs signal through a distinct heterodimeric receptor complex composed of the ligand-binding chain, IFN-λR1 (also known as IL-28R), and the accessory chain, IL-10R2 [4,5,6]. Like type-I IFNs (IFN-α/β), the type-III IFNs induce antiviral activity in cells that express IFN-λ receptors [4,5].
The potent antiviral activity IFN-α has been exploited clinically, and there are several FDA-approved IFN-α drug products that are widely used to treat chronic viral infections, including hepatitis B virus (HBV) and hepatitis C virus (HCV) infections [7]. IFNARs are broadly expressed on most cell types and tissues. As a consequence, IFN-α binds to many cell types throughout the body, and elicits biological responses in many organs besides the primary target tissue. For example, in addition to liver cells, hematopoietic cell types such as neutrophils and lymphocytes express IFN-α receptors, and IFN-α therapy often induces neutropenia and lymphopenia in many patients [7]. In contrast, expression of IFN-λ, receptors is largely restricted to cells of epithelial origin such as keratinocytes, bronchial epithelial cells and hepatocytes, suggesting that clinical administration of recombinant IFN-λ will be less likely to induce the hematopoietic and neurologic adverse reactions associated with IFN-α therapy. Recombinant human IFN-λ is now being tested clinically as a potential novel therapeutic agent for the treatment of chronic HCV infection [8-10]. The results derived from the initial Phase-1 clinical trials support the hypothesis that IFN-λ can induce antiviral activity comparable to that induced by IFN-α but without many of the undesirable side effects that are typically associated with IFN-α therapy [9].
Several recent articles have reviewed the biology of the IFN-λ genes, their corresponding proteins and the signal transduction pathway by which they induce their biological functions [6,11]. In contrast, this review will focus primarily on the preclinical studies that provided the rationale for the current clinical trials that are now evaluating the use of recombinant human IFN-λ as a novel therapeutic agent for treating chronic HCV infection. We also review the very interesting genetic associations between specific single nucleotide polymorphisms (SNPs) in the IL28B (IFN-λ3) gene region and differential responsiveness to treatment with recombinant human IFN-α in patients with chronic HCV infection.
Organization and transcriptional regulation of the IFN-λ genes
IFNs are part of a larger family of structurally related class-2 proteins that also includes six IL-10-related cytokines: IL-10, IL-19, IL-20, IL-22, IL-24 and IL-26 [12,13]. This subset of cytokines can be grouped in the same family because they all signal via receptors that share certain common motifs in their extracellular domains [12-14]. These receptors comprise the class 2 cytokine receptor family. The IFN-λ proteins share common structural features with the IL-10-related cytokines, particularly IL-22 [15,16]; however, unlike IFN-λ, IL-22 does not induce antiviral activity in IL-22 receptor-positive target cells. This supports the functional classification of the IFN-λ proteins as a unique group of IFNs instead of as a novel group of interleukins. Phylogenetically, IL28A, IL28B and IL29 reside somewhere between the type-I IFN and IL-10 gene families [17]. Amino acid sequence comparisons have shown that the type-III IFNs share only weak sequence similarity (∼5-18% identity) with either type-I IFNs or the IL-10-related cytokines.
As shown in Figure 1, IL28A, IL28B and IL29 are clustered together on chromosome 19 (19q13.13 region). The coding region for each of these genes is divided into five exons. The exon-intron structure of the IFN-λ, genes is similar to the organization of the genes encoding the IL-10-related cytokines [6,17]. Although the intron lengths vary significantly, the exon sizes and positions and frames of the exon/intron junctions are highly conserved within the genes for the type-III IFNs and the IL-10-related cytokines (IL19, IL20, IL22, IL24 and IL26). In contrast, the type-I IFN genes (e.g., IFNA1, IFNB1, IFNE, IFNK) do not contain introns.
Figure 1.
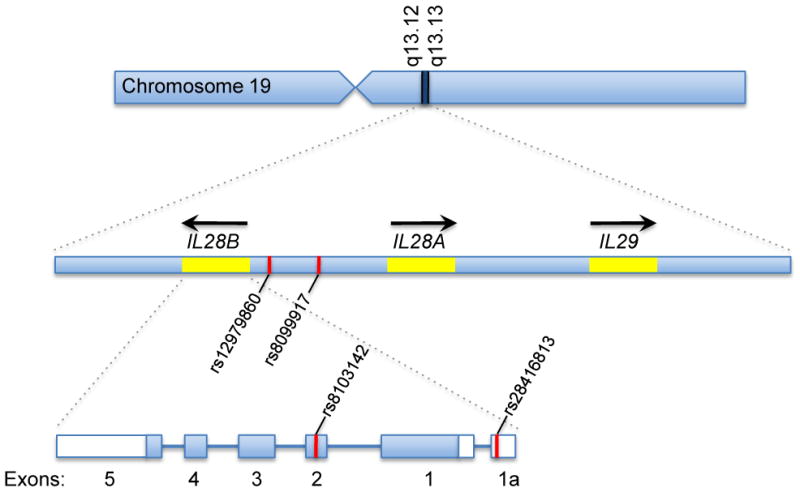
Genomic organization of the IFN-λ genes (IL28B, IL28A and IL29) and locations of key single nucleotide polymorphisms (SNPs). These genes are located on chromosome 19 in the region between q13.12 and q13.13. IL28A and IL28B lie adjacent to each other, but are transcribed in opposite directions. Several of the SNPs identified in the original GWAS studies, including rs12979860 and rs8099917, are located in the intergenic region between IL28A and IL28B [54-56]. Other SNPs such as rs8103142 and rs28416813 are located within the IL28B gene itself. SNP rs 8103142 is located in the coding region of exon 2 and results in a single amino acid change from lysine to arginine at position 70 (K70R); however, this mutation does not appear to induce any significant changes in the potency or bioactivity of the corresponding protein.
The high degree of sequence similarity between the IFN-λ, genes suggests that these genes evolved from a common predecessor relatively recently. It appears that, after the divergence of the IL29 and IL28A genes, a more recent duplication event occurred in which a fragment containing the IL29 and IL28A genes was copied and integrated back into the genome in the “head-to-head” orientation with the IL29-IL28A segment. Modest divergence within this region generated IL28B, which is almost identical to IL28A not only in the coding region but also in the upstream and downstream flanking sequences [4,5,17]. The sequence of IL29 is less similar to IL28A and IL28B than these genes are to each other. In addition, the segment that contained the duplicated IL29 was mutated significantly, which resulted in formation of a non-functional pseudogene, referred to as IFN-λ4ψ (or IFNL4) [6,17].
Not surprisingly, the promoters for the IL28A and IL28B genes are very similar, and share several transcriptional regulatory elements in common with the IL29 promoter, suggesting that all three genes are co-regulated in a similar manner [18-20]. As depicted in Figure 2, binding sites for several key transcription factors, including NF-κB and various IRF proteins, are present in the promoters of both the IFN-β gene (IFNB) and the IFN-λ genes [18-20]. The IFNB promoter contains several IRF-binding elements (IBE) (also known as “virus response elements” (VRE) or “positive regulatory domains” (PRD)) that provide binding sites for phosphorylated IRF3 and/or IRF7 [21,22]. Similar binding sites are also present in the promoters of the IFN-λ genes [18-20]. Therefore, it appears that the same set of transcription factors that regulate IFNB transcription also control expression of the IFN-λ, genes. Furthermore, expression of the type-III IFN genes is inducible by many of the same stimuli that activate expression of the type-I IFN genes [23,24].
Figure 2.
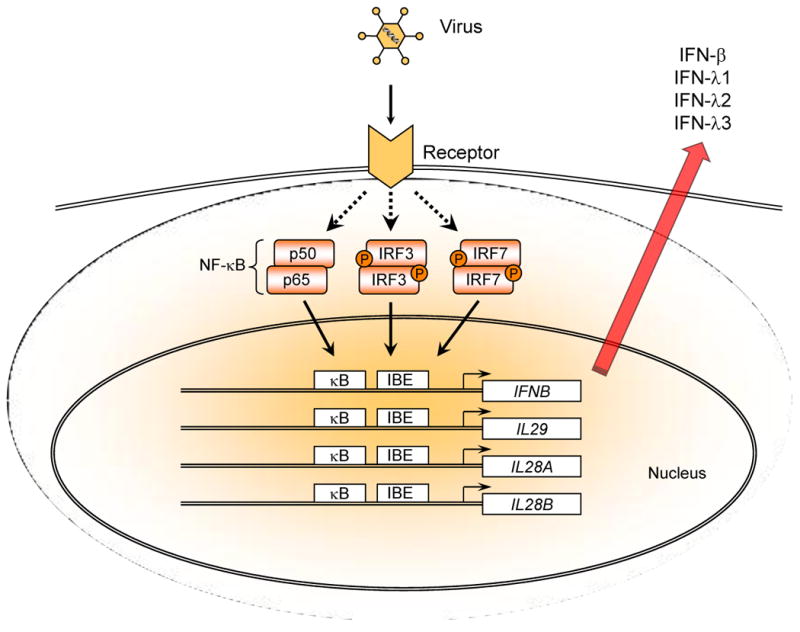
Viral infection induces co-expression of type-I (IFN-α/β) and type-III (IFN-λ) IFNs. Viruses, including HCV, induce co-expression of type-I and type-III IFNs by triggering activation of the Toll-like receptor-3 (TLR3) and/or RIG-I-like receptor (RLR) signaling pathways. This in turn results in the activation of several key transcription factors, including IRF3, IRF7 and NF-κB. IRF3 and IRF7 are activated by serine phosphorylation, and form homodimers or heterodimers which then translocate to the nucleus where they bind to specific DNA sequences known as “IRF-binding elements” (IBE) in the promoters of a number of genes, including the IFN-α, -β and -λ genes. Viral infection also induces simultaneous activation and nuclear translocation of NF-κB. The coordinated binding of NF-κB to κB elements together with the binding of IRF3 and/or IRF7 to cognate IBEs catalyzes transcription of the IFN-α, IFN-β and IFN-λ, (IL28A, IL28B and IL29) genes.
Consistent with their antiviral activity, the IFN-λs are usually co-expressed together with type-I IFNs by virus-infected cells [4,5]. Virtually any cell type can express IFN-λ following viral infection. The original IFN-λ, reports showed that four distinct viruses (Sindbis virus, Dengue virus, vesicular stomatitis virus and encephalomyocarditis virus) induce co-expression of IFN-λ1, -λ2 and -λ3 together with IFN-α and -β in various human cell types, including HeLa (cervical epithelial carcinoma), HT-29 (colorectal carcinoma), Huh7 (hepatoma) and primary human PBMCs [4,5]. It has also been shown that infection of human epithelial cells by respiratory syncytial virus (RSV) induces co-expression of type-I and type-III IFNs [25]. Other viruses, such as influenza virus and Sendai virus also induce expression of IFN-λ in human monocyte-derived dendritic cells (mDC) [23,24,26]. Furthermore, recent reports have shown that HCV infection also induces expression of IFN-λ, [27,28].
IFN-α amplifies induction of IFN-λ expression by influenza or Sendai virus [26,29]. This amplification of IFN-λ production by IFN-α is due, at least in part, to the ability of IFN-α to up-regulate TLR and IRF7 gene expression [29,30]. Viral infection or treatment with certain synthetic TLR agonists induces differential expression of the IFN-α, -β, and -λ genes in plasmacytoid dendritic cells (pDC) and myeloid DCs [23,30,31]. pDCs, in particular, can produce large amounts of IFN-α and -λ in response to viral infection. Influenza virus infection of pDCs or mDCs induces co-expression of all of the IFN-α subtypes as well as IFN-β and IFN-λ [23]. TLR agonists such as CpG DNA, which signals via TLR9, also induces co-expression of IFN-α, -β, and -λ in pDCs. In contrast, other TLR agonists such as lipopolysaccharide and poly(I:C), which signal via TLR4 and TLR3 respectively, preferentially induce co-expression of IFN-β and IFN-λ, but do not induce expression of IFN-α in monocyte-derived DCs [23]. Therefore, infection by live viruses induces co-expression of IFN-α, -β, and -λ, whereas other microbial agents or their structural components such as bacterial DNA, endotoxin and double-stranded RNA induce a more selective expression of the IFN subtypes.
Biological functions induced by IFN-λ
As mentioned above, the IFN-λ proteins bind and signal through a heterodimeric receptor complex composed of the IFN-λR1 and IL-10R2 chains [4,5]. As shown in Figure 3, IFN-λ binds initially to the IFN-λR1 chain, and the binary complex formed by the association of IFN-λ with the IFN-λR1 chain causes a rapid conformational change that facilitates recruitment of the IL-10R2 chain to the complex [15,16]. Once assembly of the ternary complex is complete, the receptor-associated Janus tyrosine kinases, Jak1 and Tyk2, mediate trans-phosphorylation of the receptor chains resulting in the formation of phosphotyrosine-containing peptide motifs on the intracellular domain (ICD) of the IFN-λR1 chain [32]. These phosphorylated peptide motifs provide transient docking sites for the latent cytosolic STAT proteins, STAT1 and STAT2. Signaling through type I (IFN-α/β) or type-III (IFN-λ) IFN receptor complexes results in the formation of a transcription factor complex known as IFN-stimulated gene factor 3 (ISGF3). This complex consists of three proteins: STAT1, STAT2 and IRF-9 (also known as ISGF3γ or p48). After it is fully assembled, ISGF3 translocates to the nucleus where it binds to interferon-stimulated response elements (ISREs) in the promoters of various IFN-stimulated genes (ISGs). These include a number of genes that are classically associated with the antiviral phenotype, including OAS1, MX1, IRF7, and EIF2AK2 (double-stranded RNA-activated protein kinase (PKR)). Recent comparative cDNA microarray analyses by several groups have shown that the repertoire of genes that are induced by IFN-λ is essentially the same as that induced by IFN-α [33,34]. The proteins encoded by these genes mediate the antiviral activity induced by the IFNs. As a result, the downstream biological activities induced by either IFN-α or IFN-λ, are very similar, including induction of anti-viral and anti-proliferative activity in many cell types.
Figure 3.
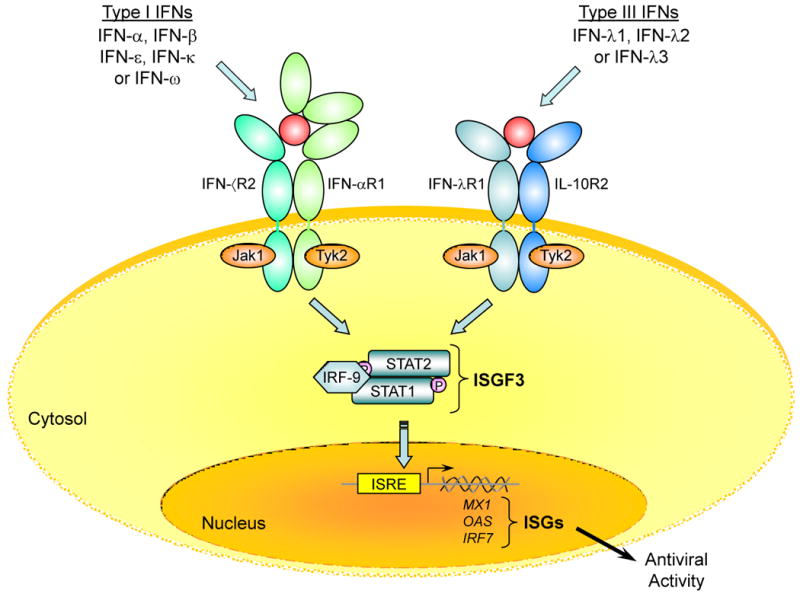
IFN-α (type-I IFN) and IFN-λ (type-III IFN) signal through distinct receptor complexes, but activate the same intracellular signaling pathway. IFN-α binds to a cell surface receptor complex composed two trans-membrane proteins: IFNAR1 and IFNAR2. IFN-λ binds to a distinct receptor complex composed of the IFN-λR1 (IL-28RA) and IL-10R2 chains. The binding of IFN-α or IFN-λ to their cognate receptors induces a common signaling cascade that results in the activation of STAT1 and STAT2 which together with IRF-9 (p48) form ISGF3 transcription factor complexes. The newly formed ISGF3 complexes then translocate from the cytosol to the nucleus where they bind to IFN-stimulated response elements (ISRE) in the promoters of IFN-stimulated genes (ISGs) such as MX1, OAS1 and IRF7. The proteins encoded by these genes in turn mediate the antiviral activity of the type-I and type-III IFNs.
IFN-λ-induced antiviral activity has been demonstrated against many different viruses, including encephalomyocarditis virus (EMCV), vesicular stomatitis virus (VSV) and influenza virus [23,24]. IFN-λ has also been shown to inhibit replication of hepatitis B virus (HBV) in a differentiated murine hepatocyte cell line [35] and to inhibit replication of both subgenomic and full-length HCV replicons in the human hepatoma cell line, Huh7 [33-35]. Although the pattern of gene expression induced by either type-I IFN (IFN-α) or type-III IFN (IFN-λ) is very similar, the magnitude of the response to IFN-α is often greater than that induced by IFN-λ in many cell types. This may reflect a difference in the relative strength of signaling through type-I IFN receptors versus type-III IFN receptors. Alternatively, this may reflect a significant difference in the relative levels of expression of these receptors on the cell membrane.
Recent studies have shown that the IFN-α and IFN-λ ligand-receptor systems can be activated independently in response to certain viruses [36,37]. Furthermore, inhibition of signaling through type-I IFN receptors with neutralizing anti-IFNAR antibodies does not block signaling through type-III IFN (IFN-λ) receptors. Similarly, inhibition of signaling through type-III IFN receptors using neutralizing anti-IL-10R2 antibodies does not block signaling through type-I IFN (IFN-α) receptors. The evolution and conservation of these two independent, antiviral, ligand-receptor systems underscores the critical importance of IFNs in host defense against pathogenic viruses.
Clinical development of IFN-λ as a therapeutic agent for chronic HCV infection
Clinical use of IFN-α as a treatment for chronic HCV infection began in 1986, before the causative virus was identified [38,39], with a small clinical trial in patients with “non-A, non-B hepatitis” [40]. The original IFN-α-based drugs (recombinant human IFN-α2a (Roferon) and IFN-α2b (Intron A)) were not pegylated, and therefore needed to be administered frequently (every other day) to induce significant biological responses in vivo. These early clinical studies showed that recombinant human IFN-α can be highly effective as a treatment for at least a subset of patients with this disease. They also revealed, however, that administration of recombinant IFN-α frequently induces adverse reactions/side effects unrelated to the desired antiviral activity.
The use of recombinant human (rHu) IFN-α as a mono-therapeutic agent to treat chronic HCV infection was approved by the FDA in 1992. A few years later, it was reported that co-treatment with rHu IFN-α plus the anti-viral nucleoside analogue, ribavirin, significantly increased the rate of sustained virologic response (SVR) in patients with chronic HCV infection [41]. As a result, ribavirin received FDA approval in 1998 for use in combination with IFN-α as a treatment for chronic hepatitis C. Co-treatment with rHu IFN-α plus ribavirin resulted in SVR in a significant proportion (∼40%) of patients, however there were still a significant percentage of patients who did not achieve SVR.
The next significant advance occurred when the pegylated versions of IFN-α were developed [42]. The addition of a polyethylene glycol (PEG) moiety to rHu IFN-α generates a more long-lived form of the protein. This modification of the wild-type form of IFN-α allowed for less frequent dosing of the drug: once per week versus three times per week. More importantly, it resulted in higher rates of SVR (45-55%) compared to the rates achieved by treatment with non-pegylated IFN-α [43].
Preclinical studies by several groups showed that recombinant human IFN-λ, inhibits replication of both partial and full-length HCV replicons in human hepatoma cultures [33-35]. IFN-λ receptors are notably absent from most hematopoietic cells [33,44], and pre-clinical toxicology studies conducted by ZymoGenetics, Inc. (Seattle, WA) showed that, unlike pegylated IFN-α, pegylated IFN-λ does not induce dose-dependent inhibition of granulocyte-macrophage (GM) colony formation in a bone marrow stem cell colony formation assay [8]. These findings indicate that pegylated IFN-λ, is not likely to induce the hematopoietic toxicities such as neutropenia that are commonly induced by IFN-α therapy in patients with chronic HCV infection.
Comparative analysis of recombinant human IFN-λ versus IFN-α in cynomolgus monkeys showed that pegylated IFN-λ does not induce expression of interferon-stimulated genes (e.g., protein kinase R (PKR), MX1 or oligoadenylate synthase (OAS)) in peripheral blood leukocytes [8]. In contrast, IFN-α induced expression of all three of these ISGs in monkey leukocytes, consistent with the presence of IFN-α receptors but not IFN-λ receptors on leukocytes. Pharmacokinetic data derived from preclinical modeling in monkeys were used to determine optimal starting doses for the initial Phase I clinical studies in humans.
Pegylated recombinant human IFN-λ (PEG-IFN-λ) has now been evaluated in two Phase 1 clinical trials [9,10]. The initial Phase 1A study was a randomized, blinded, placebo-controlled dose escalation study that evaluated single doses of PEG-IFN-λ administered by subcutaneous injection to healthy volunteers [10]. This study was designed to evaluate the safety, tolerability, pharmacokinetic and pharmacodynamic activity of a single dose of PEG-IFN-λ. A total of 20 subjects were enrolled in this study. Seventeen subjects received active drug, and three received placebo. The doses tested were: 0.5, 1.5, 5.0 and 7.5 μg/kg. PEG-IFN-λ was well tolerated at all dose levels except the 7.5 μg/kg dose. A few study participants developed reversible, dose-related increases in liver transaminases (ALT), and these changes were determined to be the dose-limiting toxicity for this agent. The terminal half-life range was estimated to be: 50 to 80 hours. Single subcutaneous doses of PEG-IFN-λ did not induce fever, fatigue or any overt hematologic changes.
A subsequent Phase 1B study was designed to evaluate the safety and tolerability of repeated doses of PEG-IFN-λ administered either once per week or once every two weeks for a total of four weeks in patients with chronic HCV infection. In this study, PEG-IFN-λ was either administered as a single agent (monotherapy) or in combination with ribavirin [9]. All of the subjects in this study were infected with HCV genotype 1, and many had relapsed following a standard course of treatment with pegylated IFN-α plus ribavirin. Even though the duration of study drug administration was only four weeks, it was obvious that pegylated recombinant human IFN-λ, induced significant decreases in the levels of HCV. At four weeks, six out of six patients who were treated with PEG-IFN-λ at the 1.5 μg/kg dose level exhibited at least a 2-log decrease in HCV RNA levels, and some of the patients who received PEG-IFN-λ in combination with ribavirin exhibited greater than a 4-log reduction in HCV RNA levels. Results from the Phase 1B study as well as an interim analysis of the clinical data from an ongoing Phase 2 study showed that PEG-IFN-λ was well tolerated, and did not induce any significant hematologic toxicities such as neutropenia, thrombocytopenia or anemia [45]. The results from these initial clinical trials of PEG-IFN-λ suggest that a longer course of treatment with this drug might be highly effective in patients with chronic hepatitis C.
IL28B genetic variation
Association of IL28B single nucleotide polymorphisms (SNPs) with response to HCV infection
In 2009, three independent genome-wide association studies (GWAS) identified SNPs in the IL28B gene region as associated with response to co-treatment with pegylated IFN-α plus ribavirin among patients with chronic HCV infection [46-48]. Two groups identified rs8099917 (located ∼8 kb upstream of IL28B) as the variant most strongly associated with SVR [47,48]; another group [46] found the strongest association with rs12979860, which is located ∼3 kb upstream of IL28B. SNPs rs12979860 and rs8099917 are in strong linkage disequilibrium (i.e., highly correlated) in Asian and European populations, but not in those of African ancestry [46,48]. The rs12979860-T allele, which was associated with treatment failure, was much more common in those of African compared to European ancestry [46]. This finding appears to explain, at least in part, previous observations that African American patients with chronic hepatitis C are less responsive to treatment with pegylated IFN-α plus ribavirin than otherwise similar patients of European ancestry [49]. One of the GWAS studies identifying rs8099917 [48] was set among Japanese patients, and showed a very strong association with treatment response. Together, these reports provide strong evidence for an association between IL28B genotype and response to therapy with pegylated IFN-α plus ribavirin in patients of African, European and Asian ancestry. Subsequent reports confirmed these findings, and showed that IL28B genotype was associated with spontaneous HCV clearance as well [50,51].
Functional impact of the IL28B polymorphisms
The identity of the functional variant(s) underlying this association is unknown. Ge et al. [46] identified two polymorphisms (SNPs rs28416813 and rs8103142) in high linkage disequilibrium with rs12979860 as potential functional variants. As shown in Figure 1, rs28416813 is located 37 bp upstream of the transcription start site for IL28B, and, therefore, might potentially regulate gene transcription. SNP rs8103142 is located in the coding region of exon 2 of IL28B, and results in a single amino acid change from lysine to arginine at position 70 (K70R) [46]. However, there were no differences in the relative potencies of IFN-λ3-Arg70 versus IFN-λ3-Lys70 when these variants were evaluated in two different bioassays [52].
Regarding a functional mechanism for the IL28B SNPs, some studies found that alleles associated with treatment failure are also associated with lower expression of IL28A and/or IL28B [47,48] but other studies have not confirmed these findings [46,52,53]. More clearly, there is a link between IL28B genotype and regulation of ISGs that are induced via the Jak-STAT signaling pathway. Prior to the initial reports regarding the IL28B SNP genotypes, several groups had shown that up-regulation of ISGs in the liver of patients with chronic hepatitis C is associated with impaired response to treatment with IFN-α plus ribavirin [54,55]. Recently, several groups have shown that the adverse IL28B variants are also associated with higher constitutive expression of ISGs in HCV-infected livers [52,53,56]. These findings appear to provide a partial functional explanation for the association between IL28B genotype and the variable response to IFN-α/ribavirin therapy in patients with chronic HCV infection.
Potential clinical use of the IL28B SNP genotypes
The Human Genome Project promised to introduce an era of ‘personalized medicine’ in which genetic testing would improve medical practice by predicting the likelihood of response to drugs [57]. That promise may be fulfilled for the treatment of chronic hepatitis C. There is now a commercially available laboratory test for IL28B rs12979860 genotype (see Test No. 480630 at www.labcorp.com); however, the optimal use for this test remains to be determined. Clinical decision making for treatment of HCV-infected patients requires a prediction of the likelihood of achieving SVR that considers not only IL28B genotype, but also other factors that influence treatment response such as racial ancestry, viral genotype, pre-treatment HCV RNA levels and fibrosis stage among others. A prototype model was developed for prediction of SVR among the subjects enrolled in the HALT-C Trial of patients with advanced fibrosis or cirrhosis who had failed prior treatment with IFN-α [58]. This model, which is based on IL28B genotype and four clinical variables, appears to have much better discrimination and predictive ability than can be obtained by using IL28B genotype alone. Efforts are underway to extend this approach to a wider range of patients with chronic hepatitis C in hopes that it can be used clinically.
SVR prediction models may become more important with the advent of direct-acting antiviral (DAA) agents such as Telaprevir and Boceprevir that specifically target viral enzymes that are critical for HCV replication. Data from several clinical trials suggest that adding a protease inhibitor to the standard pegylated-IFN-α plus ribavirin regimen increases the overall SVR rate among treatment-naïve HCV genotype 1-infected patients from approximately 50% to 65-75% [59-61]. While these new treatment regimens promise to increase the likelihood of SVR, they also carry an increased risk of resistance if treatment fails. Patients who fail to achieve SVR on a treatment regimen that includes a protease inhibitor such as Boceprevir or Telaprevir might harbor resistant viruses that could limit future use of such agents in these patients. Initial studies suggest that IL28B genotype may also be predictive of response to treatment with viral protease inhibitors [62]; therefore, prediction models based on IL28B genotype may prove useful for predicting the likelihood of achieving SVR in response to treatment with a combination peginterferon/ribavirin/protease inhibitor regimen. Incorporation of IL28B genotyping into treatment decision-making may increase the number of patients for whom treatment is successful while minimizing those in whom it could be deleterious [63].
IL28B genetic variation and therapeutic use of IFN-λ
It is plausible that therapeutic use of recombinant IFN-λ, might overcome the effects of the unfavorable IL28B genetic variants. Recently, initial results were reported for a Phase 2A clinical study that compared different doses of pegylated IFN-λ to pegylated IFN-α (all given in combination with ribavirin) for treatment of chronic hepatitis C through 12 weeks [64]. Among 19 subjects who received PEG-IFN-λ1 at doses ranging from 120-240 μg, the percentage with undetectable virus at 12 weeks post initiation was significantly higher in patients with the most favorable rs12979860 genotype-CC (86%) than in those with genotype CT or TT (50%). Although based on a very small number of patients and a limited treatment period (12 weeks), these results suggest that the antiviral response of patients to treatment with pegylated IFN-λ plus ribavirin will differ according to IL28B genotype.
Concluding remarks
The current standard of care for treatment of chronic HCV infection is a combination of pegylated IFN-α2a or -α2b plus ribavirin for up to 48 weeks that frequently results in severe adverse reactions. IFN-λ resembles IFN-α functionally in terms of its anti-viral activity, but because IFN-λ, signals via a unique receptor complex, it may elicit fewer side effects and less hematopoietic toxicity than IFN-α. Indeed, the results from early clinical trials of recombinant pegylated-IFN-λ1 in patients with chronic HCV infection support the prediction that IFN-λ will be less toxic than IFN-α as a therapeutic agent. It will be interesting to see if results from the larger Phase 2 and 3 trials confirm the initial clinical findings regarding this novel IFN.
Acknowledgments
This work was supported, in part, by the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Division of Cancer Epidemiology and Genetics. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.de Weerd NA, et al. Type I interferon receptors: biochemistry and biological functions. J Biol Chem. 2007;282(28):20053–20057. doi: 10.1074/jbc.R700006200. [DOI] [PubMed] [Google Scholar]
- 2.Müller U, et al. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264(5167):1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- 3.Hwang SY, et al. A null mutation in the gene encoding a type I interferon receptor component eliminates antiproliferative and antiviral responses to interferons alpha and beta and alters macrophage responses. Proc Natl Acad Sci USA. 1995;92(24):11284–11288. doi: 10.1073/pnas.92.24.11284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kotenko SV, et al. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat Immunol. 2003;4(1):69–77. doi: 10.1038/ni875. [DOI] [PubMed] [Google Scholar]
- 5.Sheppard P, et al. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat Immunol. 2003;4(1):63–68. doi: 10.1038/ni873. [DOI] [PubMed] [Google Scholar]
- 6.Donnelly RP, Kotenko SV. Interferon-lambda: a new addition to an old family. J Interferon Cytokine Res. 2010;30(8):555–564. doi: 10.1089/jir.2010.0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aghemo A, et al. Pegylated interferons alpha2a and alpha2b in the treatment of chronic hepatitis C. Nat Rev Gastroenterol Hepatol. 2010;7(9):485–494. doi: 10.1038/nrgastro.2010.101. [DOI] [PubMed] [Google Scholar]
- 8.Miller DM, et al. Interferon-lambda as a potential new therapeutic for hepatitis C. Ann NY Acad Sci. 2009;1182:80–87. doi: 10.1111/j.1749-6632.2009.05241.x. [DOI] [PubMed] [Google Scholar]
- 9.Muir AJ, et al. Phase 1b study of pegylated interferon-lambda 1 with or without ribavirin in patients with chronic genotype 1 hepatitis C virus infection. Hepatology. 2010;52(3):822–832. doi: 10.1002/hep.23743. [DOI] [PubMed] [Google Scholar]
- 10.Ramos EL. Preclinical and clinical development of pegylated interferon-lambda 1 in chronic hepatitis C. J Interferon Cytokine Res. 2010;30(8):591–595. doi: 10.1089/jir.2010.0066. [DOI] [PubMed] [Google Scholar]
- 11.Li M, et al. Interferon-lambdas: the modulators of antivirus, antitumor, and immune responses. J Leukoc Biol. 2009;86(1):23–32. doi: 10.1189/jlb.1208761. [DOI] [PubMed] [Google Scholar]
- 12.Kotenko SV. The family of IL-10-related cytokines and their receptors: related, but to what extent? Cytokine Growth Factor Rev. 2002;13(3):223–240. doi: 10.1016/s1359-6101(02)00012-6. [DOI] [PubMed] [Google Scholar]
- 13.Pestka S, et al. Interleukin-10 and related cytokines and receptors. Annu Rev Immunol. 2004;22:929–979. doi: 10.1146/annurev.immunol.22.012703.104622. [DOI] [PubMed] [Google Scholar]
- 14.Bazan JF. Structural design and molecular evolution of a cytokine receptor superfamily. Proc Natl Acad Sci USA. 1990;87(18):6934–6938. doi: 10.1073/pnas.87.18.6934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gad HH, et al. Interferon-lambda is functionally an interferon but structurally related to the interleukin-10 family. J Biol Chem. 2009;284(31):20869–20875. doi: 10.1074/jbc.M109.002923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Miknis ZJ, et al. Crystal structure of human interferon-λ1 in complex with its high-affinity receptor interferon-λR1. J Mol Biol. 2010;404(4):650–664. doi: 10.1016/j.jmb.2010.09.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fox BA, et al. The role of genomic data in the discovery, annotation and evolutionary interpretation of the interferon-lambda family. PLoS One. 2009;4(3):e4933. doi: 10.1371/journal.pone.0004933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Onoguchi K, et al. Viral infections activate types I and III interferon genes through a common mechanism. J Biol Chem. 2007;282(10):7576–7581. doi: 10.1074/jbc.M608618200. [DOI] [PubMed] [Google Scholar]
- 19.Osterlund PI, et al. IFN regulatory factor family members differentially regulate the expression of type III IFN (IFN-lambda) genes. J Immunol. 2007;179(6):3434–3442. doi: 10.4049/jimmunol.179.6.3434. [DOI] [PubMed] [Google Scholar]
- 20.Thomson SJ, et al. The role of transposable elements in the regulation of IFN-lambda1 gene expression. Proc Natl Acad Sci USA. 2009;106(28):11564–11569. doi: 10.1073/pnas.0904477106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Génin P, et al. The role of differential expression of human interferon-alpha genes in antiviral immunity. Cytokine Growth Factor Rev. 2009;20(4):283–295. doi: 10.1016/j.cytogfr.2009.07.005. [DOI] [PubMed] [Google Scholar]
- 22.Honda K, et al. Type I interferon gene induction by the interferon regulatory factor family of transcription factors. Immunity. 2006;25(3):349–360. doi: 10.1016/j.immuni.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 23.Coccia EM, et al. Viral infection and Toll-like receptor agonists induce a differential expression of type I and lambda interferons in human plasmacytoid and monocyte-derived dendritic cells. Eur J Immunol. 2004;34(3):796–805. doi: 10.1002/eji.200324610. [DOI] [PubMed] [Google Scholar]
- 24.Ank N, et al. Lambda interferon (IFN-lambda), a type III IFN, is induced by viruses and IFNs and displays potent antiviral activity against select virus infections in vivo. J Virol. 2006;80(9):4501–4509. doi: 10.1128/JVI.80.9.4501-4509.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Spann KM, et al. Suppression of the induction of alpha, beta, and lambda interferons by the NS1 and NS2 proteins of human respiratory syncytial virus in human epithelial cells and macrophages. J Virol. 2004;78(8):4363–4369. doi: 10.1128/JVI.78.8.4363-4369.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Osterlund P, et al. Gene expression and antiviral activity of alpha/beta interferons and interleukin-29 in virus-infected human myeloid dendritic cells. J Virol. 2005;79(15):9608–9617. doi: 10.1128/JVI.79.15.9608-9617.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Langhans B, et al. Interferon-lambda serum levels in hepatitis C. J Hepatol. 2011;54(5):859–865. doi: 10.1016/j.jhep.2010.08.020. [DOI] [PubMed] [Google Scholar]
- 28.Diegelmann J, et al. Comparative analysis of the lambda-interferons IL-28A and IL-29 regarding their transcriptome and their antiviral properties against hepatitis C virus. PLoS One. 2010;5(12):e15200. doi: 10.1371/journal.pone.0015200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sirén J, et al. IFN-alpha regulates TLR-dependent gene expression of IFN-alpha, IFN-beta, IL-28, and IL-29. J Immunol. 2005;174(4):1932–1937. doi: 10.4049/jimmunol.174.4.1932. [DOI] [PubMed] [Google Scholar]
- 30.Dai J, et al. Regulation of IFN regulatory factor-7 and IFN-alpha production by enveloped virus and lipopolysaccharide in human plasmacytoid dendritic cells. J Immunol. 2004;173(3):1535–1548. doi: 10.4049/jimmunol.173.3.1535. [DOI] [PubMed] [Google Scholar]
- 31.Lauterbach H, et al. Mouse CD8-alpha+ DCs and human BDCA3+ DCs are major producers of IFN-lambda in response to poly IC. J Exp Med. 2010;207(12):2703–2717. doi: 10.1084/jem.20092720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dumoutier L, et al. Role of the interleukin (IL)-28 receptor tyrosine residues for antiviral and anti-proliferative activity of IL-29/interferon-lambda 1: similarities with type I interferon signaling. J Biol Chem. 2004;279(31):32269–32274. doi: 10.1074/jbc.M404789200. [DOI] [PubMed] [Google Scholar]
- 33.Doyle SE, et al. Interleukin-29 uses a type 1 interferon-like program to promote antiviral responses in human hepatocytes. Hepatology. 2006;44(4):896–906. doi: 10.1002/hep.21312. [DOI] [PubMed] [Google Scholar]
- 34.Marcello T, et al. Interferons alpha and lambda inhibit hepatitis C virus replication with distinct signal transduction and gene regulation kinetics. Gastroenterology. 2006;131(6):1887–1898. doi: 10.1053/j.gastro.2006.09.052. [DOI] [PubMed] [Google Scholar]
- 35.Robek MD, et al. Lambda interferon inhibits hepatitis B and C virus replication. J Virol. 2005;79(6):3851–3854. doi: 10.1128/JVI.79.6.3851-3854.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang X, et al. Ku70 is a novel cytosolic DNA sensor that induces type III rather than type I IFN. J Immunol. 2011;186(8):4541–4545. doi: 10.4049/jimmunol.1003389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mordstein M, et al. Lambda interferon renders epithelial cells of the respiratory and gastrointestinal tracts resistant to viral infections. J Virol. 2010;84(11):5670–5677. doi: 10.1128/JVI.00272-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Choo QL, et al. Isolation of a cDNA clone derived from a blood-borne non-A, non-B viral hepatitis genome. Science. 1989;244(4902):359–362. doi: 10.1126/science.2523562. [DOI] [PubMed] [Google Scholar]
- 39.Kuo G, et al. An assay for circulating antibodies to a major etiologic virus of human non-A, non-B hepatitis. Science. 1989;244(4902):362–364. doi: 10.1126/science.2496467. [DOI] [PubMed] [Google Scholar]
- 40.Hoofnagle JH, et al. Treatment of chronic non-A, non-B hepatitis with recombinant human alpha interferon: a preliminary report. N Engl J Med. 1986;315(25):1575–1578. doi: 10.1056/NEJM198612183152503. [DOI] [PubMed] [Google Scholar]
- 41.McHutchison JG, et al. Interferon alfa-2b alone or in combination with ribavirin as initial treatment for chronic hepatitis C. N Engl J Med. 1998;339(21):1485–1492. doi: 10.1056/NEJM199811193392101. [DOI] [PubMed] [Google Scholar]
- 42.Harris JM, Chess RB. Effect of pegylation on pharmaceuticals. Nat Rev Drug Discov. 2003;2(3):214–221. doi: 10.1038/nrd1033. [DOI] [PubMed] [Google Scholar]
- 43.Fried MW, et al. Peginterferon alfa-2a plus ribavirin for chronic hepatitis C virus infection. N Engl J Med. 2002;347(13):975–982. doi: 10.1056/NEJMoa020047. [DOI] [PubMed] [Google Scholar]
- 44.Sommereyns C, et al. IFN-lambda is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLoS Pathog. 2008;4(3):e1000017. doi: 10.1371/journal.ppat.1000017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zeuzem S, et al. Pegylated interferon-lambda (PEG-IFN-λ,) shows superior viral response with improved safety and tolerability versus PEG-IFN-α-2A in HCV patients (G1/2/3/4): EMERGE Phase IIB through week 12. J Hepatol. 2011;54:S538–S539. [Google Scholar]
- 46.Ge D, et al. Genetic variation in IL28B predicts hepatitis C treatment-induced viral clearance. Nature. 2009;461(7262):399–401. doi: 10.1038/nature08309. [DOI] [PubMed] [Google Scholar]
- 47.Suppiah V, et al. IL28B is associated with response to chronic hepatitis C interferon-alpha and ribavirin therapy. Nat Genet. 2009;41(10):1100–1104. doi: 10.1038/ng.447. [DOI] [PubMed] [Google Scholar]
- 48.Tanaka Y, et al. Genome-wide association of IL28B with response to pegylated interferon-alpha and ribavirin therapy for chronic hepatitis C. Nat Genet. 2009;41(10):1105–1109. doi: 10.1038/ng.449. [DOI] [PubMed] [Google Scholar]
- 49.Conjeevaram HS, et al. Peginterferon and ribavirin treatment in African American and Caucasian American patients with hepatitis C genotype 1. Gastroenterology. 2006;131(2):470–477. doi: 10.1053/j.gastro.2006.06.008. [DOI] [PubMed] [Google Scholar]
- 50.Thomas DL, et al. Genetic variation in IL28B and spontaneous clearance of hepatitis C virus. Nature. 2009;461(7265):798–801. doi: 10.1038/nature08463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rauch A, et al. Genetic variation in IL28B is associated with chronic hepatitis C and treatment failure: a genome-wide association study. Gastroenterology. 2010;138(4):1338–1345. doi: 10.1053/j.gastro.2009.12.056. [DOI] [PubMed] [Google Scholar]
- 52.Urban TJ, et al. IL28B genotype is associated with differential expression of intrahepatic interferon-stimulated genes in patients with chronic hepatitis C. Hepatology. 2010;52(6):1888–1896. doi: 10.1002/hep.23912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Honda M, et al. Hepatic ISG expression is associated with genetic variation in IL28B and the outcome of IFN therapy for chronic hepatitis C. Gastroenterology. 2010;139(2):499–509. doi: 10.1053/j.gastro.2010.04.049. [DOI] [PubMed] [Google Scholar]
- 54.Chen L, et al. Hepatic gene expression discriminates responders and non-responders in treatment of chronic hepatitis C viral infection. Gastroenterology. 2005;128(5):1437–1444. doi: 10.1053/j.gastro.2005.01.059. [DOI] [PubMed] [Google Scholar]
- 55.Sarasin-Filipowicz M, et al. Interferon signaling and treatment outcome in chronic hepatitis C. Proc Natl Acad Sci USA. 2008;105(19):7034–7039. doi: 10.1073/pnas.0707882105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dill MT, et al. Interferon-induced gene expression is a stronger predictor of treatment response than IL28B genotype in patients with hepatitis C. Gastroenterology. 2011;140(3):1021–31. doi: 10.1053/j.gastro.2010.11.039. [DOI] [PubMed] [Google Scholar]
- 57.Feero WG, et al. Genomic medicine - an updated primer. N Engl J Med. 2010;362(21):2001–2011. doi: 10.1056/NEJMra0907175. [DOI] [PubMed] [Google Scholar]
- 58.O'Brien TR, et al. An IL28B genotype-based clinical prediction model for treatment of chronic hepatitis C. PLoS One. 2011 doi: 10.1371/journal.pone.0020904. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.McHutchison JG, et al. Telaprevir with peginterferon and ribavirin for chronic HCV genotype 1 infection. N Engl J Med. 2009;360(18):1827–1838. doi: 10.1056/NEJMoa0806104. [DOI] [PubMed] [Google Scholar]
- 60.Hézode C, et al. Telaprevir and peginterferon with or without ribavirin for chronic HCV infection. N Engl J Med. 2009;360(18):1839–1850. doi: 10.1056/NEJMoa0807650. [DOI] [PubMed] [Google Scholar]
- 61.Kwo PY, et al. Efficacy of boceprevir, an NS3 protease inhibitor, in combination with peginterferon alfa-2b and ribavirin in treatment-naive patients with genotype 1 hepatitis C infection (SPRINT-1): an open-label, randomised, multicentre phase 2 trial. Lancet. 2010;376(9742):705–716. doi: 10.1016/S0140-6736(10)60934-8. [DOI] [PubMed] [Google Scholar]
- 62.Akuta N, et al. Amino acid substitution in hepatitis C virus core region and genetic variation near the IL28B gene predict viral response to telaprevir with peginterferon and ribavirin. Hepatology. 2010;52(2):421–429. doi: 10.1002/hep.23690. [DOI] [PubMed] [Google Scholar]
- 63.Morgan TR, O'Brien TR. IL28B-genotype testing now and in the era of direct-acting antiviral agents. Clin Gastroenterol Hepatol. 2011;9(4):293–294. doi: 10.1016/j.cgh.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Muir AJ, et al. Pegylated interferon-lambda (PEG-IFN-λ) phase 2, dose-ranging, active-controlled study in combination with ribavirin (RBV) for treatment-naïve HCV patients (Genotypes 1, 2, 3 or 4): Safety, viral response, and impact of IL28B host genotype through week 12. Hepatology. 2010;52(4):715A–716A. [Google Scholar]