

Abstract
Bone marrow-derived plasmacytoid dendritic cells (pDCs) from IRAK2-deficient mice produced more interferons than did wild-type pDCs upon stimulation with the TLR9 ligand CpG. Furthermore, in CpG-stimulated IRAK2-deficient pDCs there was increased nuclear translocation of interferon regulatory factor 7 (IRF7), the key transcription factor for interferon gene transcription in these cells. In IRAK2-deficient macrophages, enhanced NFκB activation and increased expression of CpG-induced genes were detected within 2 hours after treatment. However, at later times, NFκB activation was decreased and, in contrast to the results with interferon, there was less secretion of other pro-inflammatory cytokines (such as TNFα) and chemokines in CpG-stimulated IRAK2-deficient pDCs and macrophages. Therefore, although IRAK2 is a negative regulator of TLR9-mediated interferon production through its modulation of the transcriptional activity of IRF7, it is also a positive regulator of TLR9-mediated pro-inflammatory cytokine and chemokine production at some level subsequent to transcription.
Introduction
Toll-like receptors (TLRs) recognize pathogen-associated molecular patterns (PAMPs) from invading microorganisms, and activate inflammatory responses through the induction of pro-inflammatory cytokines and interferons (1-6). While TLR4 recognizes lipopolysaccharide (LPS) from bacterial cell walls (7), TLRs 1, 2, and 6 detect a variety of lipoproteins and peptidoglycans (8-11). TLR5 senses conserved bacterial flagellin (12) and TLRs 3, 7, 8 and 9, located in the intracellular endosomal-lysosomal compartment, detect nucleic acids derived from viruses and bacteria (13, 14). In addition, TLR7 and TLR8 also recognize synthetic imidazoquinoline-like molecules (13, 14).
Upon stimulation with ligands, the adaptor molecule MyD88 is recruited to all the TLRs except TLR3 (15). MyD88 interacts in turn with the IL-1 receptor-associated kinases (IRAKs), including IRAK4 and IRAK1. IRAKs are activated through cross- or auto-phosphorylation in the receptor-proximal complex, which mediates the recruitment of TRAF6 (16). The IRAK1-TRAF6 complex then dissociates from the receptor to interact with and activate the downstream kinases TAK1 and MEKK3 (17), which then activate NFκB and MAPK, resulting in the production of pro-inflammatory cytokines and chemokines (18). Interestingly, some TLR-induced pro-inflammatory cytokines, such as TNFα, in turn activate NFκB through autocrine or paracrine pathways, which further promote the additional production of pro-inflammatory cytokines and chemokines at late times after TLR ligand stimulation (19, 20).
We and others recently reported that IRAK2 is essential for TLR-mediated pro-inflammatory cytokine and chemokine production (21, 22). IRAK2-deficient macrophages showed normal early, but reduced late activation of NFκB in response to the TLR2 ligand Malp-2 and the TLR7 ligand R848, suggesting that IRAK2 is critical for the late transcriptional response to the activation of TLR2 and TLR7 (21, 22). Importantly, a reduction of cytokine and chemokine mRNA stability and translation was observed in IRAK2-deficient bone marrow (BM)-derived macrophages in response to LPS, demonstrating that IRAK2 is also involved in the post-transcriptional control of TLR-mediated cytokine and chemokine production (22).
It is important to note that the activation of TLR-mediated signaling can also lead to the production of type I interferon (IFN), including IFNα and IFNβ (23). Plasmocytoid dendritic cells (pDCs) are the major cell type that produces type I interferon and regulates antiviral immunity in vivo (24). pDCs express high levels of TLR7 and TLR9, which detect microbial-derived single-strand RNA and unmethylated CpG dinucleotides, respectively. Previous studies showed that TLR9 activation in pDCs leads to high levels of type I interferon through the formation of a MyD88-IRAK1-TRAF6 complex (25, 26) and the subsequent activation of interferon regulatory factor 7 (IRF7), a master transcription factor for type I interferon expression (27).
We now unexpectedly find that IRAK2 is actually a negative regulator of CpG-induced IFN production at the transcriptional level. IRAK2-deficient pDCs displayed increased nuclear translocation of IRF7 and interferon production in response to stimulation with TLR9 ligands. On the other hand, IRAK2 is still required for TLR9-mediated post-transcriptional control of pro-inflammatory cytokine and chemokine production. CpG-stimulated IRAK2-deficient pDCs and macrophages both produced smaller amounts of pro-inflammatory cytokines and chemokines than did wild-type cells, which coincided with reduced TNFα secretion and decreased late NFκB activation in IRAK2-deficient macrophages. Taken together, the results demonstrate the dual functions of IRAK2 in TLR9–mediated interferon and pro-inflammatory cytokine production.
Materials and Methods
Reagents
LPS (Escherichia coli 055:B5) was purchased from Sigma-Aldrich, R848 from GLSynthesis, CpG A (ODN 1585) and CpG B (ODN1826) from Invivogen. Antibodies against phospho-ERK, phospho-p38, phospho-IκBα and were purchased from Cell Signaling. Antibodies against IRAK1, IκBα and ß-actin were from Santa Cruz. Antibodies against IRAK2 were from Abcam. APC-conjugated TNFα antibody, Brefeldin A, and permeabilization buffer were from eBioscience. IRAK2 knockout and control mice were generated as previously described (22). TNFα knockout and control mice were purchased from Taconic Farm. IL-1 receptor, IL-18 receptor and IL-33 receptor knockout and control mice were purchased from the Jackson Laboratory.
Bone marrow-derived macrophages and pDCs
Bone marrow cells were obtained from tibias and femurs by flushing with Dulbecco’s minimal essential medium (DMEM). For bone marrow-derived macrophages, the bone marrow cells were cultured in DMEM with 100 U/ml of penicillin, 100 μg/ml of streptomycin, 20% heat-inactivated fetal bovine serum (FBS), and 30% L929 cells-conditioned medium for 5 days for differentiation. For bone marrow-derived pDCs, the bone marrow cells were plated at 1 × 106 cells per ml in RPMI complete medium (10% FBS, 2 mM L-glutamine, 100 U/ml of penicillin, 100 μg/ml of streptomycin, and 50 μM ß-mercaptoethanol) containing 100 ng/ml of Flt3L (PeproTec), and collected for experiments after 6 days.
Quantitative real-time PCR
Total RNA was isolated with the TRIzol reagent (Invitrogen). Real-time PCR was performed as described previously (28). The specific primer sequences used for ß-actin, TNFα, IL-6, mouse KC, CCL4, CCL5, SOCS3, IFNα4, and IFNβ, are as follows: ß-actin: 5’-GGTCATCACTATTGGCAACG-3’ and 5’-ACGGATGTCAACGTCACACT-3’; TNF-α: 5’-CAAAGGGAGAGTGGTCAGGT-3’ and 5’-ATTGCACCTCAGGGAAGAGT-3’; IL-6: 5’-GGACCAAGACCATCCAATTC-3’ and 5’-ACCACAGTGAGGAATGTCCA-3’; KC: 5’-TAGGGTGAGGACATGTGTGG-3’ and 5’-AAATGTCCAAGGGAAGCGT-3’; CCL4: 5’-CTCCCACTTCCTGCTGTTTC-3’ and 5’-GCTCACTGGGGTTAGCACA-3’ CCL5: 5’-CTGCAATGAAGTGCAGGAGT-3’ and 5’-GTGTTGGCTGGCATTAATCTG-3’; SOCS3 : 5’-GCACGGGGAGCCCCTTTGTA-3’ and 5’-CCGGATCCCGGGGAGGTAGT-3’; IFNα4: 5’-GACCTGCCTCACACTTATAA-3’ and ‘-TCACTCCTTCTCCTCACTCAG-3’; IFNβ: 5 ‘-CACAGCCCTCTCCATCAACT-3’ and 5’-TCCCACGTCAATCTTTCCTC-3’
Western analysis
Cells were harvested, washed once with phosphate-buffered saline (PBS), and lysed for 30 min at 4°C in1.0% NP-40, 100 mM Tris hydrochloride, pH 8.0, 20% glycerol, and 0.2 mM EDTA. Cellular debris was removed by centrifugation at 10,000 × g for 5 min. For immunoblotting, cell extracts were fractionated by sodium dodecyl sulfate-PAGE and transferred to Immobilon-P transfer membranes (Millipore), using a wet transfer apparatus (Bio-Rad Laboratories). Immunoblot analysis was performed and the bands were visualized with horseradish peroxidase–coupled goat anti–rabbit, goat anti–mouse, or donkey anti–goat immunoglobulin as appropriate (Rockland), using the ECL chemiluminescence Western blotting detection system (GE Healthcare).
Murine herpesviruses 68 infection
Bone marrow-derived pDCs were plated in 12-well plates at 5 × 105 cells/ml and infected with MHV68 (multiplicity of infection [MOI] = 5) for 24 h. IFN, proinflammatory cytokine and chemokine concentrations in the supernatant media was measured by ELISA. To analyze the proliferation of virus in pDCs, GFP protein was measured by the Western method.
ELISA assay
Bone marrow-derived pDCs were stimulated with MHV68 (MOI=5) or CpG A (1 μg ml), and supernant media were collected 24 h after stimulation. Bone marrow-derived macrophages were stimulated with LPS (1 μg/ml), R848 (1 μg/ml) or CpG B (1 μg/ml) for 24 hours, and supernant media were collected 24 h after stimulation. IL-6, KC, and TNFα concentrations were measured by ELISA kits from R&D Systems. IFNα4 and IFNß concentrations in pDC culture media were measured by using ELISA kits from PBL Biomedical Laboratories. One million cells was used for ELISA assay.
Preparation and fractionation of nuclear extracts
Bone marrow-derived pDCs were stimulated with CpG A (1 μg ml) for the indicated times. Cell pellets were collected and was hed with ice-cold PBS twice, and resuspended in 6 volumes of buffer A (10 mM HEPES, pH 7.9, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM DTT,1 mM PMSF, 0.3 mM Na3VO4, 5 mM NaF) and incubated on ice for 10 min. The cell suspension was transferred to a Dounce homogenizer and the cells were disrupted with 45 strokes. After centrifugation at 500 × g for 1 minat 4°C, the supernatants were collected as cytoplasmic fractions. The nuclear pellets were washed with 1 ml Nuclei EZ Lysis buffer (Sigma Nuclei EZ Prep kit) followed by washing with 1 ml buffer A. Nuclear pellets were collected by centrifugation at 500 × g for 1 min at 4°C and resuspended in 50 μl of buffer B (20 mM HEPES, pH 7.9, 500 mM NaCl, 1.5 mM MgCl2, 0.5 mM EDTA, 0.5 mM DTT, 1 mM PMSF, 0.3 mM Na3VO4, 5 mM NaF) and incubated on ice for 30 min. After centrifugation at 14,000 × g for 10 min at 4°C, the supernatants nuclear fractions were collected.
Illumina Beadchip Micoarray analysis
250 ng of RNA was reverse transcribed into cRNA and labeled with biotin-UTP, using the TotalPrep RNA amplification kit (Ambion). cRNA was quantified using a nanodrop spectrophotometer and the cRNA quality (size distribution) was further analyzed in a 1% agarose gel. cRNA was hybridized to the Illumina MouseRef8 v1.1 Expression BeadChip using standard protocols (provided by Illumina). The microarray data was submitted to the Gene Expression Omnibus at NCBI (http://www.ncbi.nlm.nih.gov/geo/, accession number, GSE24264).
Intracellular staining
Bone marrow-derived macrophages were stimulated with CpG B (1 μg ml) for indicated times and fixed with formaldehyde (2% in PBS) on ice for 20 min. Two hours before fixing, Brefeldin A was added to block protein secretion and enrich intracellular TNFα. The fixed cells were collected by centrifuge at 1,000 × g for 5 min at 4 °C, washed with PBS twice, and blocked with PBS containing 2% serum on ice for 30 min. The cell pellets were collected by centrifuge at 1,000 × g for 5 min, resuspended in permealization buffer containing 1% serum and incubated for 30 min at room temperature. APC-conjugated TNFα antibody (1 μg/ml) was added to the cell suspension, followed by incubation for 45 min. The cell pellets were collected by centrifuge at 1,000 × g for 5 min, washed with 1 × permealization buffer containing 1% serum twice, and resuspended in PBS. The TNFα-stained cells were analyzed by flowcytometry by using a BD FACSCalibur instrument.
Results
IRAK2 is a negative regulator of TLR9-induced interferon production in pDCs
To examine the role of IRAK2 in interferon production, bone marrow-derived wild-type and IRAK2-deficient pDCs were infected with murine herpesvirus 68 (MHV68) and examined for virus-induced interferon production by ELISA. IRAK2-deficient pDCs produced less cytokines (Fig. 1B), but more interferon than did wild-type pDCs in response to infection (Fig. 1A). In addition, virus-mediated GFP expression (an indication of MHV68 proliferation) was decreased in IRAK2-deficient pDCs compared to wild-type cells (Fig. 1B). Since MHV68 activates interferon production through TLR9, we examined the role of IRAK2 in TLR9-mediated interferon production by treating the cells with TLR9 ligands. A group of synthetic TLR9 ligands, termed CpG A, are potent IFN inducers in pDCs (29). CpG A is characterized by phophodiester backbone CpG motifs and a phosphorothioate-modified poly G stretch at the 5’ and 3’ ends, and is capable of forming stable higher-order structures. In contrast, conventional phosphorothioate-modified CpG (CpG B) generally does not form higher-order structures (30, 31). Compared to CpG A, CpG B is a much weaker inducer of IFNs in pDCs, but a much stronger inducer of pro-inflammatory cytokines in macrophages and conventional DCs (32). Multimeric CpG A tends to be retained in early endosomes, whereas monomeric CpG B typically resides in late endosomes. Although it has been suggested that the endosomal localization of CpGs is critical for their functions, the mechanisms for the differential properties of CpG A and CpG B are still not clear (33). Consistent with the results of virus infection, more interferon was detected in IRAK2-deficient pDCs than that in wild-type pDCs upon CpG A stimulation (Fig. 2A). Furthermore, IRAK2-deficient pDCs showed markedly increased interferon mRNA and IRF7 nuclear translocation in response to CpG A stimulation (Fig. 2C, 2D), suggesting that IRAK2 probably inhibits the TLR9-induced transcription of interferon genes. Moreover, TLR9-induced phosphorylation of IκBα, p38, ERK and JNK was also increased in IRAK2-deficient pDCs compared to wild-type cells, demonstrating increased NFκB and MAPK signaling (Fig. 2E). Taken together, these results indicate that IRAK2 is a negative regulator of TLR9-mediated signaling in pDCs, functioning at a receptor-proximal level to inhibit the activation of all branches of downstream signaling.
Figure 1. IRAK2-deficient pDCs are more resistant to the infection by MHV68.
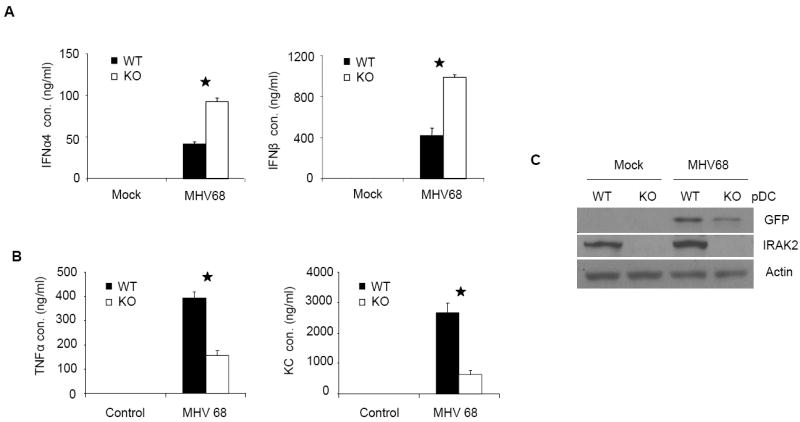
Wild-type and IRAK2-deficient bone marrow-derived pDCs were infected with MHV68 (MOI=5) for 24 h. IFNα and IFNβ (A) TNFα and KC (B) concentrations in culture media were measured by ELISA. Results shown are the means and s. d. of triplicate determinations (* p< 0.05). (C) Cell lysates were analyzed by the Western method with antibodies against GFP, IRAK2 and actin.
Figure 2. Increased responses to CpG A stimulation in IRAK2-deficient pDCs.
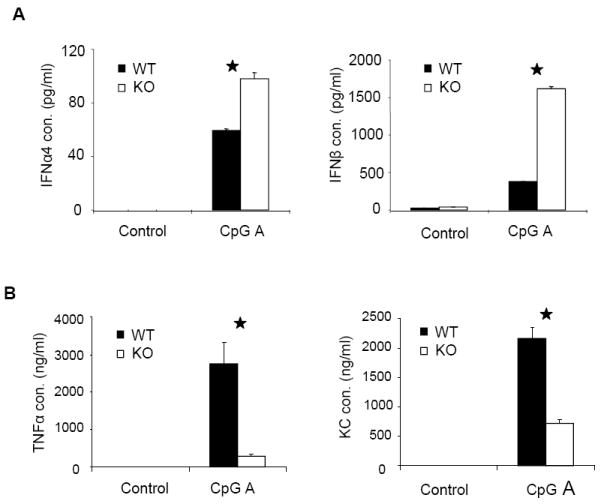

Wild-type and IRAK2-deficient BM-derived pDCs were stimulated with CpG A (1 μg/ml) for 24 h. IFNα and IFNβ (A) TNFα and KC (B) concentrations in culture media were measured by ELISA. Results shown are the means and s. d. of triplicate determinations (* p< 0.05). (C) Wild-type and IRAK2-deficient BM-derived pDCs were treated with CpG A (1 μg/ml) for the indicated times and total RNAs (2 μg) were analyzed by real-time PCR (* p< 0.05). (D) Nuclear and cytoplasmic fractions prepared from wild-type or IRAK2-deficient pDCs, either untreated or treated with CpG A (1 μg/ml), were analyzed by the Western method with antibodies against IRF7, Histone H3 and GAPDH. (E) Wild-type or IRAK2-deficient pDCs were treated with CpG A (1 μg/ml) for the indicated times and cell lysates were analyzed by the Western method with antibodies against IRAK2, phospho-IKBα, IKBα, phospho-ERK, phospho-JNK, phospho-p38, and actin. The protein levels were analyzed by ImageJ 1.43u and normalized to actin.
IRAK2 is a negative regulator of TLR9-mediated signaling in macrophages at the transcriptional level at early times after CpG stimulation
Wild-type and IRAK2-deficient bone marrow-derived macrophages were stimulated with TLR ligands, followed by analyses for the activation of NFκB and MAP kinases. CpG B (instead of CpG A) was utilized because it is a more potent activator of TLR9 signaling in macrophages. As previously reported (32), whereas IRAK2 deficiency did not have much impact on TLR4- or TLR7-mediated phosphorylation of IκBα at early times, TLR4- and TLR7-induced early p38 and ERK activation were reduced in IRAK2-deficient macrophages compared to wild-type cells, within 30 min after LPS and R848 stimulation (Fig. 3A). Interestingly, IRAK2-deficient macrophages showed higher levels of early phosphorylation of IκBα, p38 and ERK than did wild-type macrophages in response to CpG B, within 30 min (Fig. 3A), suggesting that IRAK2 specifically suppresses TLR9-mediated early signaling in macrophages. To further define the differential role of IRAK2 in TLR7- versus TLR9-mediated signaling, we used Illumina microarrays to examine the gene expression profiles of wild-type and IRAK2-deficient macrophages in response to stimulation with R848 and CpG B. Three hundred and thirty four genes were induced more than 2-fold in wild-type macrophages after 2 h of stimulation with CpG B. The majority (213, 63%) of the genes induced by CpG B were expressed at higher levels in IRAK2-deficient macrophages than in wild-type cells. In contrast, the majority of the R848-induced genes (594 out of 716, 83%) were expressed at lower levels in IRAK2-deficient macrophages. A large group of genes was induced by both R848 and CpG B, most of which showed increased expression in response to CpG B, but decreased expression in response to R848 in IRAK2-deficient macrophages (Fig. 3B). The expression patterns of several genes shown in Fig. 3B were confirmed by real time PCR (Fig. 3C).
Figure 3. Increased responses to CpG B stimulation in IRAK2-deficient macrophages.
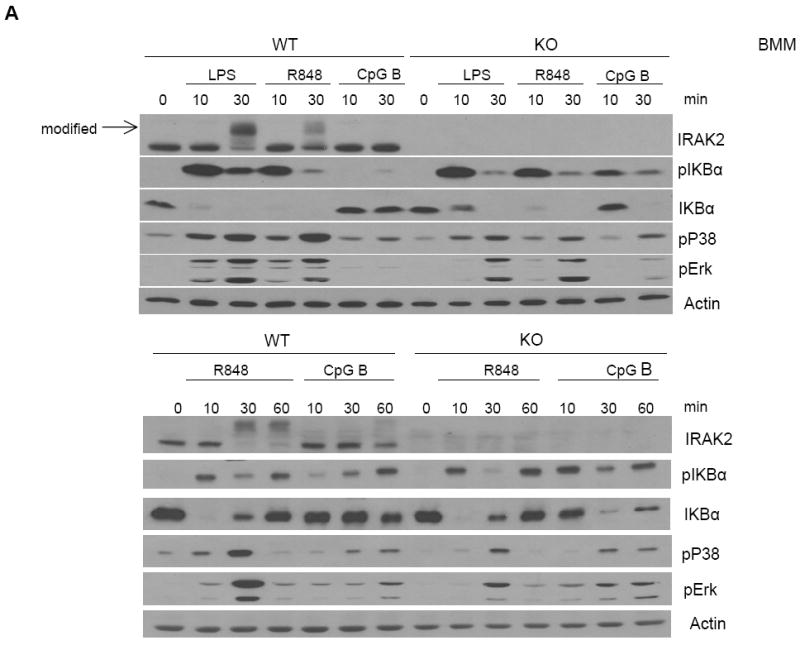

(A) Wild-type and IRAK2-deficient BM-derived macrophages were treated with LPS (1 μg/ml), R848 (1 μg/ml) or CpG B (1 μg/ml) for the indicated times. Cell lysates were analyzed by the Western method with antibodies against IRAK2, phospho-IKBα, IKBα, phospho-ERK, phospho-p38, and actin. (B) Wild-type or IRAK2-deficient BM-derived macrophages were treated with R848 (1 μg/ml) or CpG B (1 μg/ml) for 2 h. A heat map is shown for some genes from Illumina microarray analyses that were induced less by R848 but more by CpG in IRAK2-deficient macrophages. (C) Real-time PCR analysis of selected genes from microarray analysis. Wild-type and IRAK2-deficient BM-derived macrophages were treated with R848 or CpG B for the indicated times or were untreated. (* p< 0.05).
IRAK2 is required for sustained CpG B-initiated NFκB activation at late time
We next examined the overall effect of IRAK2 on TLR9-mediated cytokine and chemokine production. Despite the suppressive effect of IRAK2 in TLR9-mediated gene transcription at early times of CpG B stimulation (Fig. 3B), IRAK2-deficient macrophages still produced much less TNFα, KC and IL-6 than the wild-type macrophages (Fig. 4A). IRAK2 deficient macrophages showed decreased KC and IL6 mRNA expression at late times of LPS stimulation (Fig. 4B), which is consistent with our previous report (22). Interestingly, although IRAK2-deficient macrophages showed increased TNFα and KC mRNA expression at early times after CpG stimulation (0.5, 1 or 2 h), TNFα and KC mRNA expression were dramatically decreased at late times (4 or 8 h, Fig. 4B). Moreover, IRAK2-deficient macrophages also showed decreased phosphorylation of IκBα at late times (4, 8 or 16 h, Fig. 4C), indicating that the activation of NFκB is greatly decreased. Taken together, these results demonstrate that IRAK2 is essential for sustaining CpG B-initiated NFκB activation at late times.
Figure 4. Impaired activation of NFκB at late times after CpG B stimulation in IRAK2-deficient macrophages.
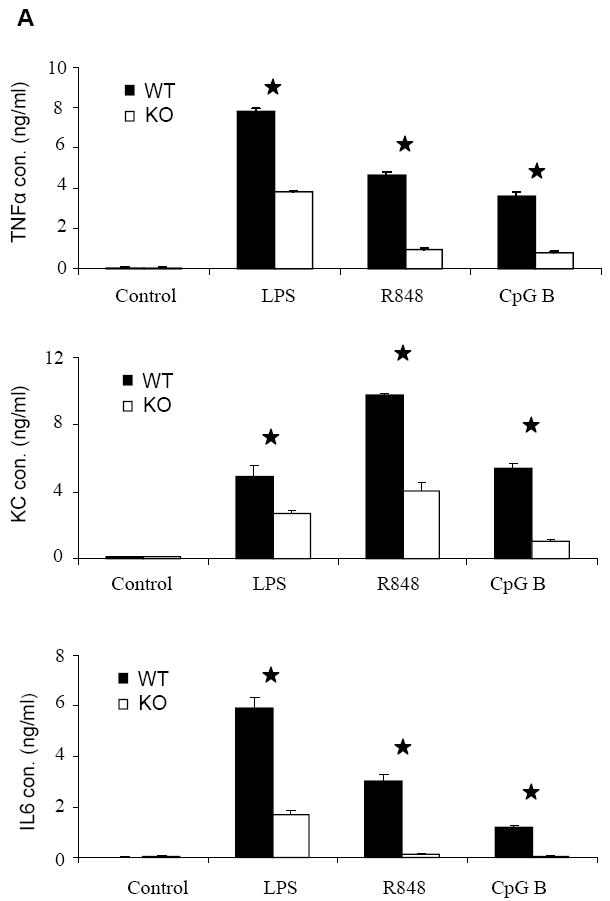
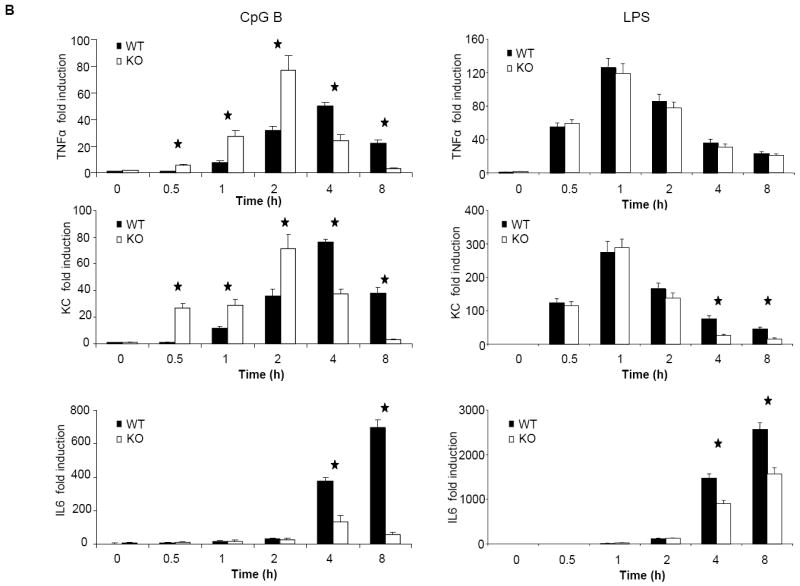
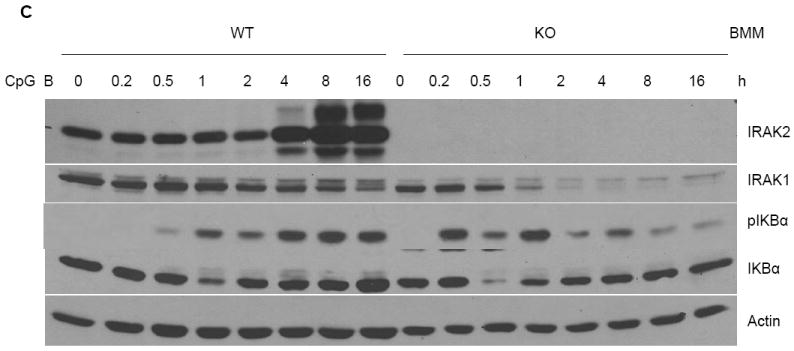
(A) Wild-type and IRAK2-deficient BM-derived macrophages were treated with LPS (1 μg/ml), R848 (1 μg/ml), or CpG B (1 μg/ml) for 24 h. TNFα, KC and IL-6 concentrations in culture media were measured by ELISA. Results shown are the means and s. d. of triplicate determinations (* p< 0.05). (B) Wild-type and IRAK2-deficient BM-derived macrophages were treated with CpG B (1 μg/ml) or LPS (1 μg/ml) for the indicated times, and total RNAs (2 μg) were analyzed by real-time PCR to examine TNFα, KC and IL-6 expression (* p< 0.05). (C) Wild-type and IRAK2-deficient BM-derived macrophages were treated with CpG B (1 μg/ml) for the indicated times. Cell lysates were analyzed by the Western method with antibodies against IRAK2, IRAK1, phospho-IKBα, IKBα, and actin.
IRAK2 deficiency reduces the efficiency of TNFα secretion
Although previous studies have shown an important role of IRAK2 at late times of NFκB activation in TLR2- and TLR7-dependent signaling, it was unclear how IRAK2 is involved in sustaining NFκB activation for up to 16 h. Many TLR ligand-induced cytokines, such as IL-1 and TNFα, can potentially activate NFκB through autocrine or paracrine pathways, thereby promoting pro-inflammatory cytokine and chemokine production through positive feedback loops. Especially, cytokines of the IL-1 family, including IL-1, IL-18 and IL-33, activate NFκB through MyD88-dependent pathways that utilize IRAKs. We thus investigated the possibility that the reduced cytokine and chemokine production in IRAK2-deficient macrophages is due to decreased responses to the IL-1 family members. However, deficiencies in the IL-1, IL-18, or IL-33 receptors had little impact on cytokine and chemokine production in response to LPS, R848 or CpG B, suggesting that these cytokines are not essential for regulating TLR ligand-induced cytokine and chemokine production (Supplementary Fig. 1).
On the other hand, previous studies demonstrated that TNFα is involved in TLR-mediated NFκB activation at late times through a positive feedback mechanism (19, 20). To investigate the involvement of TNFα in CpG B-mediated responses, we made bone marrow-derived macrophages from TNFα–deficient and wild-type mice. Compared to wild-type macrophages, TNFα-deficient macrophages indeed produced less IL-6 and KC upon CpG B stimulation (Fig. 5A), indicating that TNFα is involved in CpG B-induced cytokine and chemokine production. Specifically, TNFα deficiency resulted in decreased KC and IL-6 mRNA expression and IkBα phosphorylation at late times (Fig. 5B-C), indicating that TNFα participates in a second wave of CpG-induced pro-inflammatory cytokine and chemokine production. We then carefully examined TNFα expression and production in wild-type and IRAK2-deficient macrophages. Importantly, consistent with the negative regulatory role of IRAK2 in TLR9 signaling, IRAK2-deficient macrophages actually showed a higher percentage of TNFα-positive cells than did wild-type macrophages in response to CpG B (Fig. 6B). However, the amounts of TNFα secreted were lower in the supernatant media from IRAK2-deficient macrophages than in medium from wild-type macrophages despite the fact that wild-type and IRAK2-deficient TNFα-positive cells showed similar fluorescence intensities for TNFα staining (Fig. 6A and 6C). These results suggest that IRAK2 deficiency probably has an impact on the efficiency of TNFα secretion.
Figure 5. Impaired activation of of NFκB at late times after CpG B stimulation in TNFα-deficient macrophages.
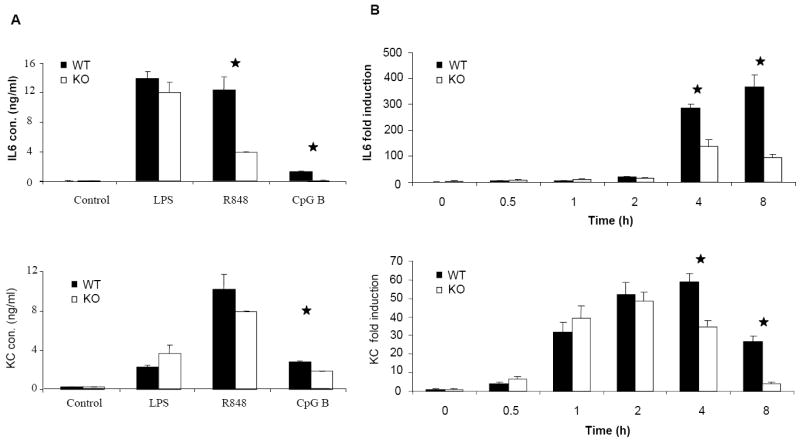
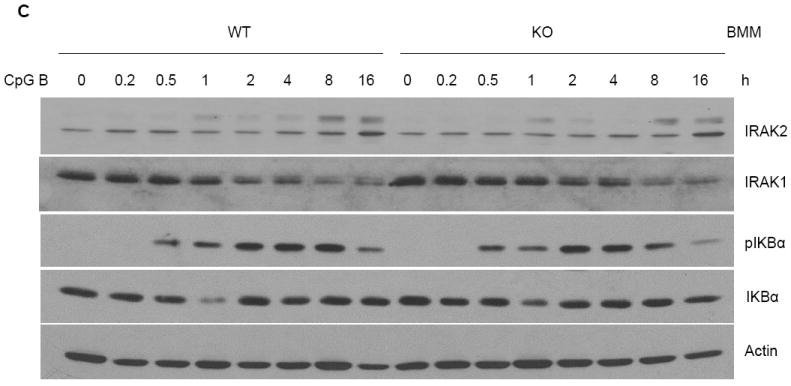
(A) Wild-type and TNFα-deficient BM-derived macrophages were treated with LPS (1 μg/ml), R848 (1 μg/ml), or CpG B (1 μg/ml) for 24 h. TNFα, KC and IL-6 concentrations were measured by ELISA. Results shown are the means and s. d. of triplicate determinations (* p< 0.05). (B) Wild-type or TNFα-deficient BM-derived macrophages were treated with CpG B (1 μg/ml) for the indicated times and total RNAs (2 μg) were analyzed by real-time PCR (* p< 0.05). (C) Wild-type or TNFα-deficient BM-derived macrophages were treated with CpG B (1 μg/ml) for the indicated times. Cell lysates were analyzed by the Western method with antibodies against IRAK2, IRAK1, phospho-IKBα, IKBα, and actin.
Figure 6. Impaired CpG B-mediated TNFα secretion in IRAK2-deficient macrophages.
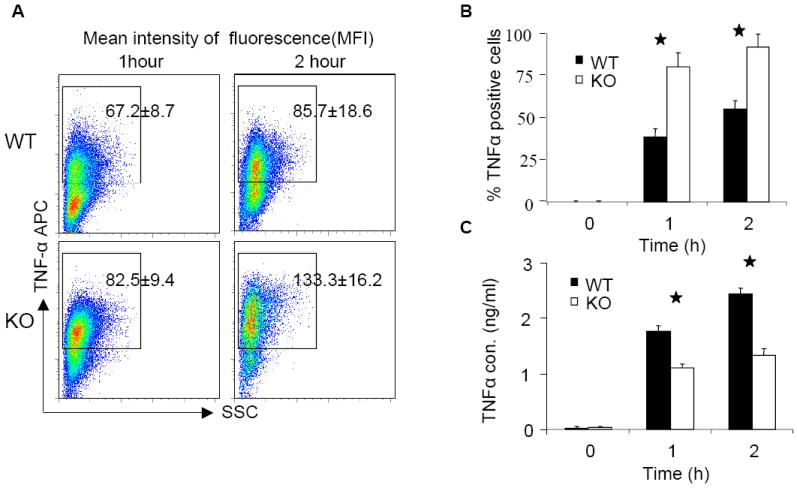
(A) Wild-type or IRAK2-deficient BM-derived macrophages were treated with CpG B (1 μg/ml) for indicated times. TNFα was assayed by intracellular staining following by flow cytometry analysis. The mean fluorescence intensities of TNFα are shown in the figures. (Similar results were obtained in three independent experiments.) (B) The percentages of TNFα positive cells are shown. Results are the means and s. d. of triplicate determinations. (* p< 0.05). (C) Wild-type and IRAK2-deficient BM-derived macrophages were treated with CpG B (1 μg/ml) or were untreated. TNFα concentrations in the supernant media were measured by ELISA. Results shown are the means and s. d. of triplicate determinations. (* p< 0.05).
Discussion
We report two novel functions of IRAK2 in TLR9-mediated signaling. While IRAK2 is a negative regulator of TLR9-mediated transcription, it is also involved in the regulation of cytokine production at the post-transcriptional level (Fig. 7). Interferon production seems mainly to be affected by the negative effect of IRAK2 at the transcriptional level, and therefore IRAK2-deficient pDCs produce more interferon upon viral infection or CpG A stimulation (Fig. 1A, 2A). For pro-inflammatory cytokine and chemokine production in macrophages, we observed both increased CpG B-induced transcription (Fig. 3B) and reduced TNFα secretion in IRAK2-deficient macrophages at early times (Fig. 6). Decreased pro-inflammatory cytokine and chemokine production in IRAK2-deficient macrophages 24 h after CpG B stimulation (Fig. 4A) demonstrates that the effect of reduced TNFα secretion is dominant at late times. Interestingly, IRAK2-deficient pDCs also produced less pro-inflammatory cytokines and chemokines than did wild-type pDCs after virus infection or CpG A stimulation (Fig. 1B, 2B), suggesting that IRAK2 might play a similar role in pDCs in regulating cytokine and chemokine production.
Figure 7. Model of the dual functions of IRAK2 in TLR9-mediated signaling.
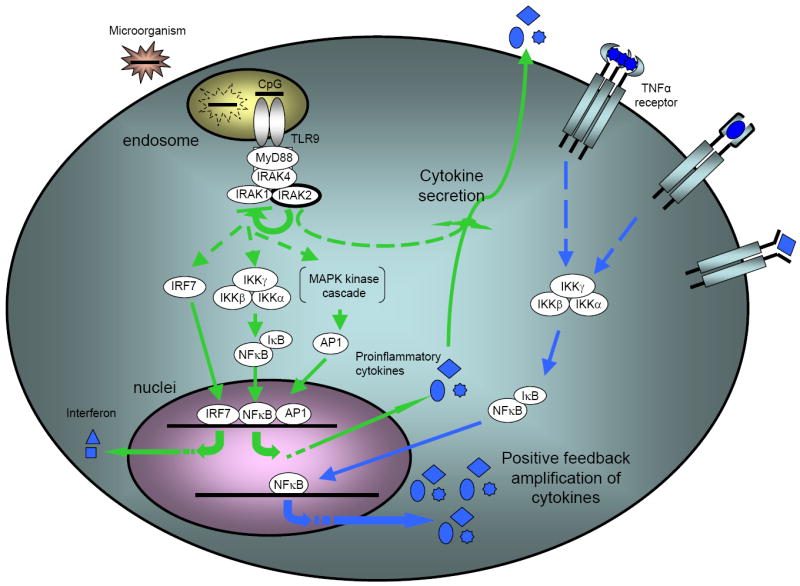
Upon stimulation of TLR9, unmodified IRAK2 inhibits the activation of downstream signaling, probably by interfering with the activation of IRAK1. Meanwhile, IRAK2 is also required for the secretion of some cytokines, such as TNFα, which is essential for the activation of NFκB at late times.
IRAK2-deficient pDCs and macrophages show increased activation of both NFκB and MAP kinase signaling cascades at early times after activation of TLR9-mediated signaling (Fig. 2B, 3A), demonstrating that the inhibitory effect of IRAK2 is at the receptor-proximal level. It is intriguing that IRAK1 is degraded faster and more completely in IRAK2-deficient macrophages than in wild-type cells (Fig. 4C). Since the modification and degradation of IRAK1 is essential for activation of TLR9-mediated signaling (34), it is conceivable that IRAK2 might compete with IRAK1 for an upstream kinase, such as IRAK4, and inhibit the activation of IRAK1. It is important to point out that the inhibitory effect of IRAK2 is specific for TLR9-mediated signaling. Interesting, while IRAK2 was modified in response to several TLR ligands, including LPS and R848, IRAK2 modification was not detected at early times after stimulation with TLR9 ligands (Figs. 2D, 3A). In addition, we only observed ligand-induced interaction between IRAK2 and MyD88 and TRAF6 in response to R848 stimulation, but not much in response to CpG B stimulation (Supplementary Fig. 2). Further study is required to map the modification sites of IRAK2 and to show how such modification affects its functions.
Many TLR-induced pro-inflammatory cytokines function not only as effectors in immune responses, but also as amplifiers of NFκB signaling, inducing pro-inflammatory cytokine and chemokine production through positive feedback mechanisms (19, 20). Our study demonstrates the importance of the production of a second wave of the cytokine TNFα for sustaining CpG-initiated NFκB activation. Consistent with the critical role of IRAK2 in TLR4-induced cytokine production at post-transcriptional levels (22), our studies show that IRAK2 deficiency affected TLR9-induced TNFα production through reduced TNFα secretion. TNFα is produced as a membrane bound form, and its secretion requires the activation of TNFα converting enzyme, which cleaves TNFα to release it (35). The activation of this enzyme is suggested to be mediated by activation of the MAP kinases ERK and p38 (36). However, the activation of ERK and p38 are not impaired in CpG-stimulated IRAK2-deficient pDCs or macrophages (Fig. 2D, 3A), suggesting that IRAK2 may mediate TNFα secretion through a different mechanism.
Although the innate immune response is crucial to suppress infection, aberrant immune responses to host antigens are frequently observed in many autoimmune diseases, such as systemic lupus erythematosus. Therefore, it is vital to keep immune responses at appropriate levels. Our study has revealed a new mechanism that regulates TLR9-mediated immune responses, in which IRAK2 plays multiple roles. IRAK2 suppresses TLR9-mediated signaling at early times after stimulation, thus increasing the threshold of immune responses induced by TLR9 ligands, and therefore potentially preventing overreaction to low-level interactions between TLR9 and host DNA. However, when TLR9-mediated pro-inflammatory cytokines are induced, IRAK2 promotes their secretion and increases cytokine production through positive feedback mechanisms, leading to an efficient immune response.
Supplementary Material
References
- 1.Medzhitov R, Preston-Hurlburt P, Janeway CA., Jr A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394–397. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 2.Rock FL, Hardiman G, Timans JC, Kastelein RA, Bazan JF. A family of human receptors structurally related to Drosophila Toll. Proc Natl Acad Sci U S A. 1998;95:588–593. doi: 10.1073/pnas.95.2.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Takeuchi O, Kawai T, Sanjo H, Copeland NG, Gilbert DJ, Jenkins NA, Takeda K, Akira S. TLR6: A novel member of an expanding toll-like receptor family. Gene. 1999;231:59–65. doi: 10.1016/s0378-1119(99)00098-0. [DOI] [PubMed] [Google Scholar]
- 4.Chuang TH, Ulevitch RJ. Cloning and characterization of a sub-family of human toll-like receptors: hTLR7, hTLR8 and hTLR9. Eur Cytokine Netw. 2000;11:372–378. [PubMed] [Google Scholar]
- 5.Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- 6.Zhang D, Zhang G, Hayden MS, Greenblatt MB, Bussey C, Flavell RA, Ghosh S. A toll-like receptor that prevents infection by uropathogenic bacteria. Science. 2004;303:1522–1526. doi: 10.1126/science.1094351. [DOI] [PubMed] [Google Scholar]
- 7.Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, Birdwell D, Alejos E, Silva M, Galanos C, Freudenberg M, Ricciardi-Castagnoli P, Layton B, Beutler B. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282:2085–2088. doi: 10.1126/science.282.5396.2085. [DOI] [PubMed] [Google Scholar]
- 8.Takeuchi O, Hoshino K, Kawai T, Sanjo H, Takada H, Ogawa T, Takeda K, Akira S. Differential roles of TLR2 and TLR4 in recognition of gram-negative and gram-positive bacterial cell wall components. Immunity. 1999;11:443–451. doi: 10.1016/s1074-7613(00)80119-3. [DOI] [PubMed] [Google Scholar]
- 9.Takeuchi O, Hoshino K, Akira S. Cutting edge: TLR2-deficient and MyD88-deficient mice are highly susceptible to Staphylococcus aureus infection. J Immunol. 2000;165:5392–5396. doi: 10.4049/jimmunol.165.10.5392. [DOI] [PubMed] [Google Scholar]
- 10.Underhill DM, Ozinsky A, Hajjar AM, Stevens A, Wilson CB, Bassetti M, Aderem A. The Toll-like receptor 2 is recruited to macrophage phagosomes and discriminates between pathogens. Nature. 1999;401:811–815. doi: 10.1038/44605. [DOI] [PubMed] [Google Scholar]
- 11.Underhill DM, Ozinsky A, Smith KD, Aderem A. Toll-like receptor-2 mediates mycobacteria-induced proinflammatory signaling in macrophages. Proc Natl Acad Sci U S A. 1999;96:14459–14463. doi: 10.1073/pnas.96.25.14459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hayashi F, Smith KD, Ozinsky A, Hawn TR, Yi EC, Goodlett DR, Eng JK, Akira S, Underhill DM, Aderem A. The innate immune response to bacterial flagellin is mediated by Toll-like receptor 5. Nature. 2001;410:1099–1103. doi: 10.1038/35074106. [DOI] [PubMed] [Google Scholar]
- 13.Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, Lipford G, Wagner H, Bauer S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science. 2004;303:1526–1529. doi: 10.1126/science.1093620. [DOI] [PubMed] [Google Scholar]
- 14.Diebold SS, Kaisho T, Hemmi H, Akira S, Reis e Sousa C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science. 2004;303:1529–1531. doi: 10.1126/science.1093616. [DOI] [PubMed] [Google Scholar]
- 15.Akira S, Takeda K, Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat Immunol. 2001;2:675–680. doi: 10.1038/90609. [DOI] [PubMed] [Google Scholar]
- 16.Cao Z, Xiong J, Takeuchi M, Kurama T, Goeddel DV. TRAF6 is a signal transducer for interleukin-1. Nature. 1996;383:443–446. doi: 10.1038/383443a0. [DOI] [PubMed] [Google Scholar]
- 17.Deng L, Wang C, Spencer E, Yang L, Braun A, You J, Slaughter C, Pickart C, Chen ZJ. Activation of the IkappaB kinase complex by TRAF6 requires a dimeric ubiquitin-conjugating enzyme complex and a unique polyubiquitin chain. Cell. 2000;103:351–361. doi: 10.1016/s0092-8674(00)00126-4. [DOI] [PubMed] [Google Scholar]
- 18.Ninomiya-Tsuji J, Kishimoto K, Hiyama A, Inoue J, Cao Z, Matsumoto K. The kinase TAK1 can activate the NIK-I kappaB as well as the MAP kinase cascade in the IL-1 signalling pathway. Nature. 1999;398:252–256. doi: 10.1038/18465. [DOI] [PubMed] [Google Scholar]
- 19.Covert MW, Leung TH, Gaston JE, Baltimore D. Achieving stability of lipopolysaccharide-induced NF-kappaB activation. Science. 2005;309:1854–1857. doi: 10.1126/science.1112304. [DOI] [PubMed] [Google Scholar]
- 20.Werner SL, Barken D, Hoffmann A. Stimulus specificity of gene expression programs determined by temporal control of IKK activity. Science. 2005;309:1857–1861. doi: 10.1126/science.1113319. [DOI] [PubMed] [Google Scholar]
- 21.Kawagoe T, Sato S, Matsushita K, Kato H, Matsui K, Kumagai Y, Saitoh T, Kawai T, Takeuchi O, Akira S. Sequential control of Toll-like receptor-dependent responses by IRAK1 and IRAK2. Nat Immunol. 2008;9:684–691. doi: 10.1038/ni.1606. [DOI] [PubMed] [Google Scholar]
- 22.Wan Y, Xiao H, Affolter J, Kim TW, Bulek K, Chaudhuri S, Carlson D, Hamilton T, Mazumder B, Stark GR, Thomas J, Li X. Interleukin-1 receptor-associated kinase 2 is critical for lipopolysaccharide-mediated post-transcriptional control. J Biol Chem. 2009;284:10367–10375. doi: 10.1074/jbc.M807822200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Honda K, Takaoka A, Taniguchi T. Type I interferon [corrected] gene induction by the interferon regulatory factor family of transcription factors. Immunity. 2006;25:349–360. doi: 10.1016/j.immuni.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 24.Gilliet M, Cao W, Liu YJ. Plasmacytoid dendritic cells: sensing nucleic acids in viral infection and autoimmune diseases. Nat Rev Immunol. 2008;8:594–606. doi: 10.1038/nri2358. [DOI] [PubMed] [Google Scholar]
- 25.Kawai T, Sato S, Ishii KJ, Coban C, Hemmi H, Yamamoto M, Terai K, Matsuda M, Inoue J, Uematsu S, Takeuchi O, Akira S. Interferon-alpha induction through Toll-like receptors involves a direct interaction of IRF7 with MyD88 and TRAF6. Nat Immunol. 2004;5:1061–1068. doi: 10.1038/ni1118. [DOI] [PubMed] [Google Scholar]
- 26.Uematsu S, Sato S, Yamamoto M, Hirotani T, Kato H, Takeshita F, Matsuda M, Coban C, Ishii KJ, Kawai T, Takeuchi O, Akira S. Interleukin-1 receptor-associated kinase-1 plays an essential role for Toll-like receptor (TLR)7- and TLR9-mediated interferon-{alpha} induction. J Exp Med. 2005;201:915–923. doi: 10.1084/jem.20042372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Honda K, Yanai H, Negishi H, Asagiri M, Sato M, Mizutani T, Shimada N, Ohba Y, Takaoka A, Yoshida N, Taniguchi T. IRF-7 is the master regulator of type-I interferon-dependent immune responses. Nature. 2005;434:772–777. doi: 10.1038/nature03464. [DOI] [PubMed] [Google Scholar]
- 28.Kim TW, Staschke K, Bulek K, Yao J, Peters K, Oh KH, Vandenburg Y, Xiao H, Qian W, Hamilton T, Min B, Sen G, Gilmour R, Li X. A critical role for IRAK4 kinase activity in Toll-like receptor-mediated innate immunity. J Exp Med. 2007;204:1025–1036. doi: 10.1084/jem.20061825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Verthelyi D, Ishii KJ, Gursel M, Takeshita F, Klinman DM. Human peripheral blood cells differentially recognize and respond to two distinct CPG motifs. J Immunol. 2001;166:2372–2377. doi: 10.4049/jimmunol.166.4.2372. [DOI] [PubMed] [Google Scholar]
- 30.Krieg AM. Therapeutic potential of Toll-like receptor 9 activation. Nat Rev Drug Discov. 2006;5:471–484. doi: 10.1038/nrd2059. [DOI] [PubMed] [Google Scholar]
- 31.Ishii KJ, Akira S. Innate immune recognition of, and regulation by, DNA. Trends Immunol. 2006;27:525–532. doi: 10.1016/j.it.2006.09.002. [DOI] [PubMed] [Google Scholar]
- 32.Hemmi H, Kaisho T, Takeda K, Akira S. The roles of Toll-like receptor 9, MyD 88, and DNA-dependent protein kinase catalytic subunit in the effects of two distinct CpG DNAs on dendritic cell subsets. J Immunol. 2003;170:3059–3064. doi: 10.4049/jimmunol.170.6.3059. [DOI] [PubMed] [Google Scholar]
- 33.Guiducci C, Ott G, Chan JH, Damon E, Calacsan C, Matray T, Lee KD, Coffman RL, Barrat FJ. Properties regulating the nature of the plasmacytoid dendritic cell response to Toll-like receptor 9 activation. J Exp Med. 2006;203:1999–2008. doi: 10.1084/jem.20060401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xiao H, Qian W, Staschke K, Qian Y, Cui G, Deng L, Ehsani M, Wang X, Qian YW, Chen ZJ, Gilmour R, Jiang Z, Li X. Pellino 3b negatively regulates interleukin-1-induced TAK1-dependent NF kappaB activation. J Biol Chem. 2008;283:14654–14664. doi: 10.1074/jbc.M706931200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Black RA, Rauch CT, Kozlosky CJ, Peschon JJ, Slack JL, Wolfson MF, Castner BJ, Stocking KL, Reddy P, Srinivasan S, Nelson N, Boiani N, Schooley KA, Gerhart M, Davis R, Fitzner JN, Johnson RS, Paxton RJ, March CJ, Cerretti DP. A metalloproteinase disintegrin that releases tumour-necrosis factor-alpha from cells. Nature. 1997;385:729–733. doi: 10.1038/385729a0. [DOI] [PubMed] [Google Scholar]
- 36.Fan H, Derynck R. Ectodomain shedding of TGF-alpha and other transmembrane proteins is induced by receptor tyrosine kinase activation and MAP kinase signaling cascades. EMBO J. 1999;18:6962–6972. doi: 10.1093/emboj/18.24.6962. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.