

Abstract
DNA or RNA aptamers have gained attention as the next generation antibody-like molecules for medical or diagnostic use. Conventional secondary structure prediction tools for nucleic acids play an important role to truncate or minimize sequence, or introduce limited chemical modifications without compromising or changing its binding affinity to targets in the design of improved aptamers selected by Systematic Evolution of Ligands by EXponential enrichment (SELEX). We describe a novel software package, ValFold, capable of predicting secondary structures with improved accuracy based on unique aptamer characteristics. ValFold predicts not only the canonical Watson-Crick pairs but also G-G pairs derived from G-quadruplex (known structure for many aptamers) using the stem candidate selection algorithm.
Availability
The database is available for free at http://code.google.com/p/valfold/
Keywords: aptamer, SELEX, aptamer truncation, secondary structure prediction
Background
Aptamers are well-known nucleic acid molecules that tightly bind to their targets by recognizing their three-dimensional structures. Aptamers are usually obtained by the SELEX (Systematic Evolution of Ligands by EXponential enrichment) process [1, 2]. The aptamer sequence data show the following four unique characteristics compared to the usual genomic, RNA or other natural sequences analysis. The first is that the length of each aptamer is usually shorter than 150 bases. The second is that the sequence data sets from one selection process usually consist of multiple sequence groups. The third unique characteristic was that at least two structurally different forms are predicted depending on the existence of their targets which is well-known in riboswitch studies. Riboswitches are one of the natural aptamers found in micro-organisms and plants [3]. The fourth is that multiple aptamer sequences usually contain some redundant motifs. Experimental truncation approaches of aptamers retaining the redundant sequence part could sometimes successfully minimize an aptamer length without significant loss of its binding activity [4]. It is worthwhile in considering the third and the forth characteristics of aptamers to verify the strategy of a secondary structure prediction algorithm. The widely-used secondary structure prediction software for RNA are well established for the universal prediction of RNA molecules, but they have no capability to predict the structural changes between the free-form and the target molecule-bound form, which, we think, are important aspects when the aptamer secondary structure is predicted.
Methodology
The ValFold program is developed using Java (JDK 1.6.0). The mainstream of ValFold is a secondary structure prediction algorithm to minimize the free energy. We expand this algorithm to predict the G-quartet structure, which is generally known as the unique structure of the telomere part of all chromosomes [5], and also reported in many aptamers binding to target molecules [6]. ValFold algorithm is based on the optimization concept for all of the stem candidate combinations. It also predicts the G-quartet structure because the G-quartet structure candidate list is combined with the stem candidate list at the same time. Each G-quartet structure candidate has a report with Gibbs free energy parameter [7]. The ValFold algorithm process uses this energy parameter as an indicator to find out the G-quartet structure candidates or the usual stem candidates. The “CastOff” function is a characteristic feature of ValFold. Aptamers are generally known to bind to target molecules on single stranded regions known as “bulge” or “loop” [8]. CastOff uses this hypothetical feature in aptamers to predict the aptamer binding motif from single sets of SELEX sequences.
The practical process of ValFold algorithm is as follows (Figure 1A) : First, the suboptimal secondary structures among all of the sequence sets of SELEX process are predicted by main function of ValFold (Step 1). A flowchart of this process is represented in Figure 2. All of the single stranded regions such as “bulge” or “loop” candidates are extracted from the suboptimal secondary structures, except for the primer compliment sequences (Step 2). The maximum length of the motif candidates and the number of allowed mismatches in the motif candidates are input for the calculation process (Step 3). Then CastOff algorithm finds the conserved scores for each of the motif candidates according to the number of the motif conserved sequences. If one of the single stranded region candidates possesses a motif candidate, CastOff determines that the sequence is a conserved motif candidate (Step 4). A flowchart of these steps 2 to 4 is represented in Figure 3.
Figure 1.

Truncation procedure of ValFold. Secondary structures were drawn by using PseudoViewer [11], Turner model [12] and SantaLucia model [13] are used for the calculation of free energies of RNA and DNA aptamers.
Figure 2.

Flowchart of ValFold 1 (Figure 1A Step 1).
Figure 3.
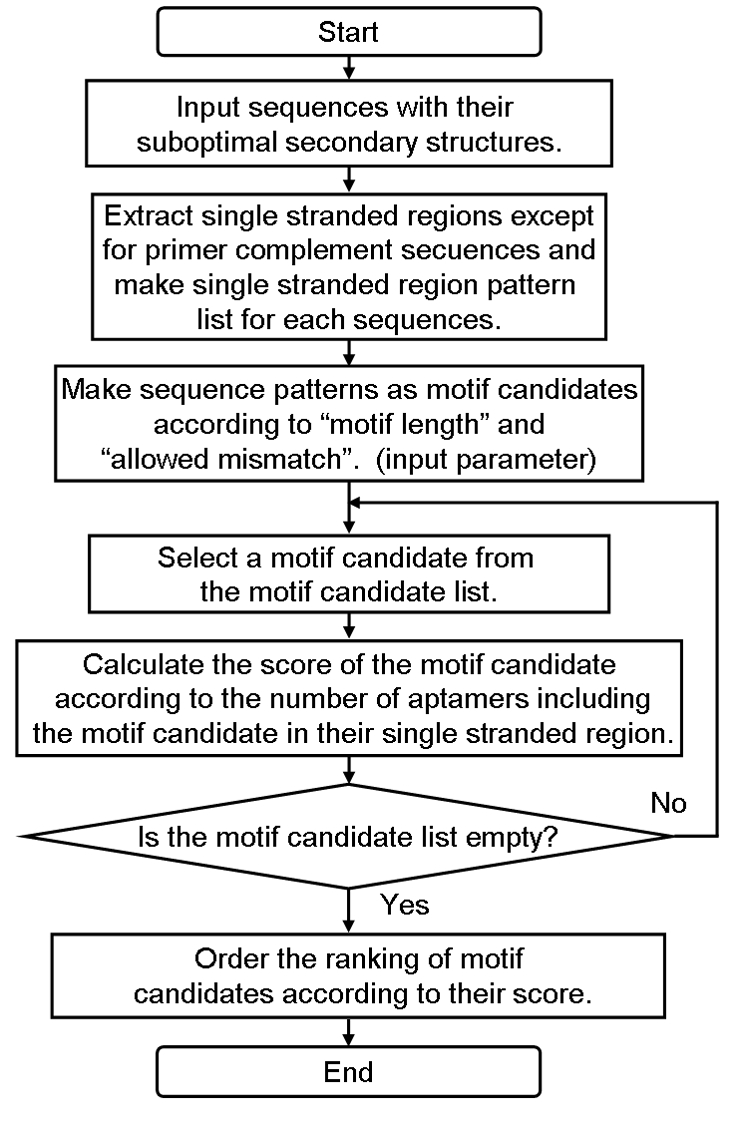
Flowchart of ValFold 2 (Figure 1A Step 2, 3, 4).
CastOff determines the highest scoring motif as the most possible motif. The filtering of secondary structure candidates with CastOff generates results including cases where the motif candidates are found only in a bulge and loop structures. Therefore, it is possible to select the aptamer secondary structures that can bind with the target molecules as a binding conformation. If the candidate motif is found on “bulge” or “loop” structure on the binding conformation, it is worthwhile to attempt to retain those regions in a truncate aptamer candidates and subject for further binding assays by researchers. The example is shown in Figure 1C.
Software Input and output
ValFold supports FASTA format as an input. Users can add the primer region information for each aptamer sequence to use the CastOff function. Calculation results of ValFold are displayed with GUI (Figure 1B). Figure 1B shows the result of a RNA aptamer bound to sphingosylphosphorylcholine (SPC) shown as an examples [9]. “UUA” was predicted as target binding motif in the loop region in this example. The result was subsequently validated as the truncated aptamer, m12, is shown to bind SPC with good affinity (Figure 1C).
Caveat and future development
We have developed the aptamer truncation program by using a binding conformation prediction. The truncation strategy worked very well in some cases such as SPC [9] and rabbit immunoglobulin G [10] aptamers which are based on 96 well based sequencers. At this moment, ValFold does not support large data sets from next-generation sequencing equipments because the current version is computationally slow. So, we need to develop the next version of ValFold to adapt the large sequence data sets which will enable us to find better aptamers.
Acknowledgments
The authors thank Drs. Penmetcha K. R. Kumar, Hiroshi Mizuno, Fumiko Nishikawa and Satoshi Nishikawa for scientific discussions. The development of ValFold was partially supported by the Bio-oriented Technology Research Advancement Institution (BRAIN).
Footnotes
Citation:Akitomi et al, Bioinformation 7(1): 38-40 (2011)
References
- 1.C Tuerk, L Gold. Science. 1990;249:505. [Google Scholar]
- 2.AD Ellington, JW Szostak. Nature. 1990;346:818. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- 3.E Nudler, AS Mironov. Trends Biochem Sci. 2004;29:11. doi: 10.1016/j.tibs.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 4.SD Jayasena. Clin Chem. 1999;45:1628. [PubMed] [Google Scholar]
- 5.JR Williamson, et al. Cell. 1989;59:871. [Google Scholar]
- 6.LC Bock, et al. Nature. 1992;355:564. doi: 10.1038/355564a0. [DOI] [PubMed] [Google Scholar]
- 7.N Sugimoto, et al. Nucleosides & Nucleotides. 1996;15:559. [Google Scholar]
- 8.T Hermann, DJ Patel. Science. 2000;287:820. [Google Scholar]
- 9.K Horii, et al. Molecules. 2010;15(8):5742. doi: 10.3390/molecules15085742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Y Yoshida, et al. Anal Biochem. 2008;375(2):217. doi: 10.1016/j.ab.2008.01.005. [DOI] [PubMed] [Google Scholar]
- 11.K Han, et al. Nucleic Acids Res. 2003;31(13):3432. doi: 10.1093/nar/gkg539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.DH Mathews, et al. J Mol Biol. 1999;288(5):911. doi: 10.1006/jmbi.1999.2700. [DOI] [PubMed] [Google Scholar]
- 13.JJr SantaLucia. Proc Natl Acad Sci U S A. 1998;95:1460. [Google Scholar]