

Abstract
The human gastrointestinal (GI) tract is a unique environment in which intestinal epithelial cells and non-pathogenic (commensal) bacteria coexist. It has been proposed that the microenvironment that the pathogen encounters in the commensal layer is important in determining the extent of colonization. Current culture methods for investigating pathogen colonization are not well suited for investigating this hypothesis as they do not enable co-culture of bacteria and epithelial cells in a manner that mimics the GI tract microenvironment. Here we describe a microfluidic co-culture model that enables independent culture of eukaryotic cells and bacteria, and testing the effect of the commensal microenvironment on pathogen colonization. The co-culture model is demonstrated by developing a commensal Escherichia coli biofilm among HeLa cells, followed by introduction of enterohemorrhagic E. coli (EHEC) into the commensal island, in a sequence that mimics the sequence of events in GI tract infection.
Protocol
1. Fabrication of silicon masters using standard SU-8 photolithography1 (not shown in this video).
Use standard SU-8 photolithography methods to create a SU-8 "masters" (SU-8 2050, MicroChem, Newton, MA) for fabricating the different components (PDMS membranes of different thickness, co-culture chamber) in a cleanroom. Such master molds can be fabricated at any microfabrication facility (e.g., Stanford Microfluidics Foundry; http://thebigone.stanford.edu/foundry/).
Prior to PDMS replication, expose the SU-8 masters to fluorosilane vapor in a desiccator attached to a vacuum source to facilitate easy release of PDMS from the master. Add a drop of fluorosilane to a paper towel and apply vacuum for one min. Remove the vacuum and allow 30 min for deposition of fluorosilane. Keep the SU-8 master in a closed container for future use.
2. Replica molding of PDMS from SU-8 master2
The fluidic layer and the control layer are made by replica molding of PDMS from SU-8 master.
Mix the PDMS pre-polymer and cross-linker at a 10:1 wt. ratio and degas in a desiccator for 1hour (or until air bubbles are removed from the PDMS mixture).
Place the SU-8 master in a petri dish and carefully pour the PDMS mixture on top of the SU-8 master. Degas the PDMS again.
Spin the mold at 1200 rpm for one minute to get a uniform PDMS thickness of around 100μm on the mold.
Heat the petri dish containing the PDMS and the SU-8 master mold at 80°C for one hour to cure the PDMS.
Remove the cured PDMS-covered SU-8 master from the hotplate.
Peel the PDMS mold from the SU-8 master.
Drill four holes in the PDMS mold using a 20 gauge blunt-end needle for two bacteria inlets, one outlet and one pneumatic port.
3. Bonding of Multilayer PDMS devices: This device is assembled with three layers -a top layer which contains the main channel, a middle pneumatic layer, and a bottom bacterial layer that contains the channels for bacteria. The three layers are assembled as described below.
Wet the PDMS membranes with methanol and carefully peel them from their SU-8 master mold using a clean forceps. Place membrane on a teflon sheet.
Place the pneumatic membrane and the fluidic layer on an oxygen plasma chamber. Turn on plasma for 40 sec (oxygen pressure 10 sccm, RF power 100W).
After exposure to plasma, align (within 10 minutes) the membrane on the bottom PDMS channel using forceps. Adding methanol on the membrane and the PDMS layers helps free movement of the layers, and makes alignment easier. In this case, the device is placed on a piece of gauze to allow the methanol vapors to escape.
Cure the aligned PDMS layers on a hotplate at 80°C for 8 h.
After bonding the pneumatic membrane onto the bottom layer, repeat steps 3.2 - 3.4 to bond the top channel onto the assembled composite PDMS device.
4. Assembling the PDMS device3,4
Drill holes in the inlet and outlet ports used for seeding eukaryotic cells and bacteria.
Connect Tygon tubing (0.01 inch O.D.) to the ports and operate the pneumatic layer by pulling with a 20mL syringe.
Place the PDMS model and a pre-cleaned glass slide into an oxygen plasma chamber. Turn on plasma for 20 sec (oxygen pressure 20 sccm, RF power 300W) without applying the vacuum.
In order to prevent contact between the island wall (PDMS) bonding to the glass slide, immediately connect a syringe to the device and apply vacuum by pulling the syringe and holding it in place.
Gently press the PDMS model onto the glass (within 2 minutes).
Cure the PDMS device at 80°C for 10 minutes.
5. Treating the glass surface with fibronectin
Sterilize device under UV for 15 minutes after connecting Tygon tubing to every port.
Fill and flow the sterilized PBS into the channel to remove any debris in the channel. Apply flow for 10 minutes to remove air bubbles. The air bubbles can easily escape the PDMS because of its gas permeability.
Fill a color dye solution into the pneumatic channel to prevent formation of air bubbles in the channel.
To facilitate eukaryotic cell seeding on the glass, introduce a 1 μg/mL solution of fibronectin into the device and incubate for 40 min at 37°C.
Wash the excess fibronectin with 1 mL of growth media.
6. Formation and measurement of commensal E. coli biofilm in islands
Dilute the commensal bacteria (e.g., E. coli BW25113/pCM18) in M9 minimal media to OD600~1 for forming a commensal biofilm in the closed chambers/islands.
Close the island wall by applying positive-liquid pressure. A homemade air-to-liquid pressure converter is used to prevent air bubble formation in the channel.
Introduce the commensal E. coli into the islands with the valve closed, and allow bacteria to attach for 1 hour at 32°C.
Wash the unattached bacteria and perfuse a dilute bacterial suspension (OD600~0.05) at 0.25mL/hr for 12 hours.
Rinse the biofilm with fresh DMEM growth media to remove loosely-attached bacteria and prepare for eukaryotic cell attachment.
Image the biofilm using a Leica TCS SP5 Confocal Laser Scanning Microscope (Bannockburn, IL), and visualize the biofilm architecture using the IMARIS software5.
7. Seeding eukaryotic cells around bacterial islands6-8
Trypsinize HeLa cells from tissue culture flask and resuspend in DMEM media for a seeding density of 5 x 105 cells/mL.
Introduce HeLa cells into the device at a flow rate of 2 mL/min using a Picoplus syringe pump (Harvard Apparattus, MA). Ensure that the island wall is lowered so that the bacterial biofilm is separate from the HeLa cell region. This step should be performed on the stage of a light microscope so that the uniformity of seeding can be determined.
Clamp the cell outlet to reduce the movement of the cell suspension in the channel and incubate for 6 h at 37°C in a 5% CO2 incubator.
Perfuse the device with growth medium at 0.5 mL/hr to remove unattached cells.
Refresh the DMEM media for HeLa and commensal E. coli every 6 hours.
8. Introduction of pathogenic bacteria into the island and exposure to eukaryotic cells
Prepare E. coli O157:H7 (EHEC) to infect HeLa cells at a multiplicity of infection (m.o.i) of 100:1 200:1 (number of EHEC per HeLa cell).
Rinse and wash out loosely-attached commensal bacteria.
Introduce a dilute EHEC suspension (OD600 ~ 0.1) into the islands.
Incubate device at 37°C in a 5% CO2 incubator for 6 hours without flow to allow EHEC to be exposed to signals present in the commensal biofilm.
Lift the PDMS wall to expose EHEC in the commensal islands to HeLa cells for 6 hours.
Rinse the channel with PBS and stain for live and dead cells using the Live/Dead kit (L3224, Invitrogen, CA).
Image multiple locations on a Zeiss Axiovert 200M fluorescence microscope and estimate the number of live and dead cells.
9. Representative Results 9
The sequence of events in GI tract infections was mimicked by developing a monolayer of HeLa cells and a commensal E. coli biofilm and exposing EHEC to the commensal biofilm prior to infection of HeLa cells. The steps involved in fabrication of the microfluidic co-culture model are shown in Figure 1. Figure 2A shows a 3-dimensional rendering of the co-culture device. The two regions in the co-culture device the eukaryotic cell culture chamber and the bacterial island are shown in Figure 2B with different color dyes (purple for the eukaryotic regions and green for the bacterial island). The feasibility of isolating the bacterial culture regions from the surrounding eukaryotic region is shown in Figure 2C using color dyes. Figure 3 shows representative results from co-culture of epithelial cells and bacteria. Figure 3A shows colonization of the commensal biofilm (green) island by EHEC (red). Figure 3B shows the juxtaposition of epithelial cells and commensal E. coli with EHEC in the bacterial island. The RFP expressing EHEC and GFP expressing commensal bacteria are localized in the island when the PDMS wall is lowered, illustrating the feasibility of isolating the bacterial island from epithelial cells.
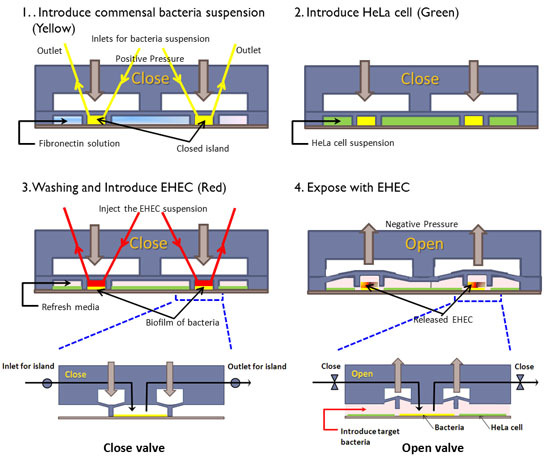 Figure 1: Cell seeding scheme in the co-culture model. (1) The PDMS wall is lowered to form an island, commensal bacteria are introduced into the island, and fibronectin is flowed around the island. (2) HeLa cells are seeded in the regions surrounding the island. (3) After HeLa cells reach confluence and the commensal biofilm has developed, EHEC is introduced into the island. (4) The PDMS wall is lifted up to expose HeLa cells surrounding the island to EHEC. Inset shows details of valve operation.
Figure 1: Cell seeding scheme in the co-culture model. (1) The PDMS wall is lowered to form an island, commensal bacteria are introduced into the island, and fibronectin is flowed around the island. (2) HeLa cells are seeded in the regions surrounding the island. (3) After HeLa cells reach confluence and the commensal biofilm has developed, EHEC is introduced into the island. (4) The PDMS wall is lifted up to expose HeLa cells surrounding the island to EHEC. Inset shows details of valve operation.
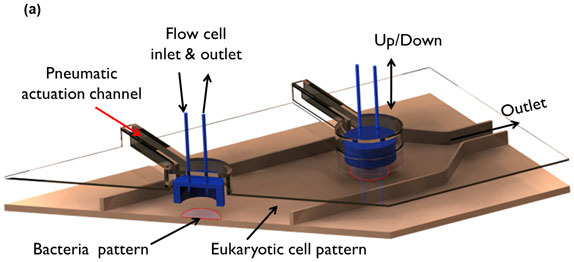
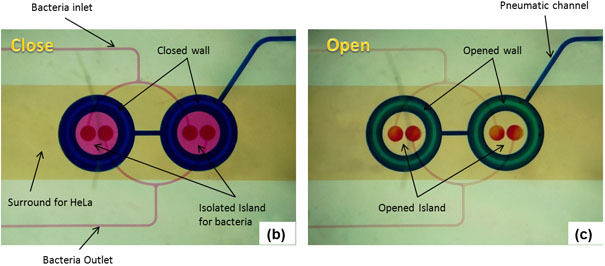 Figure 2: Microfluidic model for co-culture of epithelial cells and bacteria. (A) Three-dimensional rendition of the co-culture device showing pneumatically-actuated trapping regions for forming bacterial islands among epithelial cells. Each bacterial island (1200 μm diameter and 1000 μm apart) has a separate inlet and outlet for providing growth media and removing waste from the island. (B) Micrograph of the co-culture device with color dyes showing the different regions (epithelial cell zone, bacterial islands). (C) The fidelity of the pneumatic trapping system is shown by lowering the PDMS wall (left panel) using a pneumatically-activated channel (blue), introducing purple dye into the closed island islands, and flowing yellow dye around it for 48 h. When the PDMS wall is raised (right panel), the island region is exposed to the surrounding yellow dye. Scale bar represents 500 μm.
Figure 2: Microfluidic model for co-culture of epithelial cells and bacteria. (A) Three-dimensional rendition of the co-culture device showing pneumatically-actuated trapping regions for forming bacterial islands among epithelial cells. Each bacterial island (1200 μm diameter and 1000 μm apart) has a separate inlet and outlet for providing growth media and removing waste from the island. (B) Micrograph of the co-culture device with color dyes showing the different regions (epithelial cell zone, bacterial islands). (C) The fidelity of the pneumatic trapping system is shown by lowering the PDMS wall (left panel) using a pneumatically-activated channel (blue), introducing purple dye into the closed island islands, and flowing yellow dye around it for 48 h. When the PDMS wall is raised (right panel), the island region is exposed to the surrounding yellow dye. Scale bar represents 500 μm.
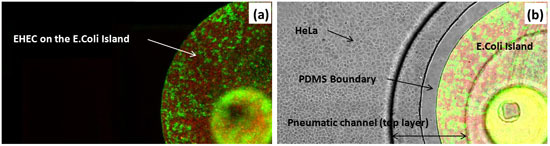 Figure 3: Co-culture of HeLa cells and bacteria. (A) Fluorescence image of RFP-expressing EHEC and GFP-expressing E. coli BW25113 in island. (B) Overlay of transmitted, green, and red fluorescence images in the device. Scale bar represents 200 μm.
Figure 3: Co-culture of HeLa cells and bacteria. (A) Fluorescence image of RFP-expressing EHEC and GFP-expressing E. coli BW25113 in island. (B) Overlay of transmitted, green, and red fluorescence images in the device. Scale bar represents 200 μm.
Discussion
Conventional assays for pathogen attachment and colonization utilize a monolayer of eukaryotic cells in tissue culture plates into which pathogens are added. These models are not physiologically relevant as they do not incorporate a commensal bacterial biofilm developed on eukaryotic cells. Simple addition of a pre-grown bacterial culture to eukaryotic cells is unlikely to lead to this conformation as biofilms are highly organized structures that develop over time, and it is extremely difficult, if not virtually impossible, to culture eukaryotic cells in the presence of bacteria for extended periods of time without significant loss in viability. Since pathogens do not navigate through a commensal biofilm in these models to attach to epithelial cells, these models do not accurately mimic the organization of epithelial cells and commensal bacteria in the GI tract. Here, we outline the development of a microfluidic device that uses pneumatically-controlled trapping for co-culture of eukaryotic cells and bacteria. Using HeLa cells as the model eukaryotic cell line and EHEC as the model pathogen, we show that the co-culture device can keep the island region isolated as well as support the cultivation and development of a HeLa cell monolayer and a commensal E. coli biofilm. The microfluidic co-culture model not only enables localization of commensal bacteria and epithelial cells, but also pre-exposes pathogens to commensal bacteria prior to encountering epithelial cells; thereby, presenting a more physiologically relevant environment during colonization. In addition, the co-culture model can be a useful tool for fundamental studies focused on investigating the role of specific signals on EHEC infectivity as well as for applications such as screening potential probiotic strains.
Acknowledgments
This work was supported in part by the National Science Foundation (CBET 0846453) and the National Institutes of Health (1R01GM089999).
References
- McDonald JC. Prototyping of microfluidic devices in poly(dimethylsiloxane) using solid-object printing. Anal Chem. 2002;74:1537–1545. doi: 10.1021/ac010938q. [DOI] [PubMed] [Google Scholar]
- Jeon NL. Design and fabrication of integrated passive valves and pumps for flexible polymer 3-dimensional microfluidic systems. Biomed Microdevices. 2002;4:117–121. [Google Scholar]
- Baek JY, Park JY, Ju JI, Lee TS, Lee SH. A pneumatically controllable flexible and polymeric microfluidic valve fabricated via in situ development. J Micromech Microeng. 2005;15:1015–1020. [Google Scholar]
- Grover WH, Ivester RH, Jensen EC, Mathies RA, A R. Development and multiplexed control of latching pneumatic valves using microfluidic logical structures. Lab Chip. 2006;6:623–631. doi: 10.1039/b518362f. [DOI] [PubMed] [Google Scholar]
- Lee J, Jayaraman A, Wood TK, K T. Indole is an inter-species biofilm signal mediated by SdiA. BMC Microbiol. 2007;7:42–42. doi: 10.1186/1471-2180-7-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu CH, Folch A. Microfluidic devices with tunable topographies. Appl Phys Lett. 2005;86:023508–023508. [Google Scholar]
- Hui EE, Bhatia SN. Micromechanical control of cell-cell interactions. Proc Natl Acad Sci USA. 2007;104:5722–5726. doi: 10.1073/pnas.0608660104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY. Integrating sensing hydrogel microstructures into micropatterned hepatocellular cocultures. Langmuir. 2009;25:3880–3886. doi: 10.1021/la803635r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Hegde M, Jayaraman A. Co-culture of epithelial cells and bacteria for investigating host-pathogen interactions. Lab-on-Chip. 2009 doi: 10.1039/b911367c. [DOI] [PubMed] [Google Scholar]