

Abstract
Enantioenriched allenylsilanes are used in three component propargylation reactions with aldehydes and silyl ethers to form syn-homopropargylic ethers that contain an imbedded azide. These materials then undergo thermally induced intramolecular 1,3-dipolar cycloaddition reactions, resulting in unique fused ring systems containing 1,2,3-triazoles. The ability to modify all three components of the reaction allows for expedient access to compounds containing significant structural and stereochemical variation.
Recent advancements in the Huisgen 1,3-dipolar cycloaddition reaction have led to a renewed interest in its application in the synthesis of 1,2,3-triazoles.1 Small molecules containing a triazole functionality have been shown to exibit a range of biological function, including antitumor, antibacterial, antiparasitic, and antiviral activity (Figure 1).2
Figure 1.

Representative examples of biologically active molecules containing 1,2,3-triazoles.
Historically terminal and symmetrically substituted alkynes have been used predominantly in these reactions, since unsymmetrical alkynes often result in diminished reaction rates and poor regioselectivity.3 One solution to this problem is a tandem dipolar cycloaddition/cross coupling reaction, which can access fully substituted triazoles.4 Another approach is an intramolecular cycloaddition, where the regioselectivity is determined by the initial positions of the azide and alkyne in the starting material.5 While both of these strategies have proven to be useful, methods for the formation of fully functionalized triazoles remain underdeveloped.
We have recently reported a three component reaction of enantioenriched allenylsilanes with aldehydes and silyl ethers, resulting in highly functionalized syn-homopropargylic ethers.6 In our continued interest in developing these allenes as chiral carbon nucleophiles,7 we sought to further expand the use of the resulting homopropargylic ethers by incorporating a pendant azide moiety which will further react to provide heterocycles containing fused 1,2,3-triazoles. In this paper we describe the development of a three component propargylation/cycloaddition reaction to give access to densely functionalized fused triazole ring systems.
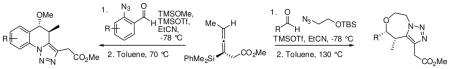
The propargylation of enantioenriched allenylsilane (Ra)-4a with 2-azidobenzaldehyde8 and methoxytrimethylsilane (TMSOMe), promoted by TMSOTf, resulted in formation of alkyne 5 in 71% yield as a single observed diastereomer (Scheme 1). Heating alkyne 5 in toluene at 110 ºC promoted the desired dipolar cyclization reaction, resulting in the formation of triazole 6a in 90% yield. As anticipated, a single regioisomer of the desired triazole was observed.
Scheme 1.

Two step triazole formation
a Isolated yields after purification over silica gel. b Diastereomeric ratios determined by 1H NMR analysis on crude material.
A similar experiment was conducted with 2-azidoacetaldehyde dimethylacetal (Scheme 1). Due to concerns with volatility and stability, the preformed acetal was used instead of the aldehyde. The propargylation reaction provided homopropargylic ether 7, and heating this product in toluene gave desired triazole 8 in 90 % isolated yield. This reaction sequence demonstrates that the methodology can be expanded to aliphatic systems using acetals with imbedded azides.
When this strategy was applied to more substituted azidobenzaldehydes we observed that some of the triazole product was formed in the crude propargylation reaction mixture.8 Attempts to purify these propargylation products proved difficult, as the triazole product would be observed in post-chromatographic NMR spectra. This led to the development of a two step sequence where the propargylation reaction was quenched, and the crude product was heated to 70 ºC in toluene, to directly produce the desired triazole product.
This modified procedure gave the desired tricyclic system with an imbedded triazole 6 in good yield and high diastereoselectivity (Scheme 2). The reactions were tolerant to a range of functionality on the aromatic ring, including electron donating and withdrawing groups. When achiral allenylsilane 4b was subjected to the same reaction conditions, products 6f and 6g were formed in moderate yield.
Scheme 2.

One-pot propargylation/cycloaddition
a Isolated yields after purification over silica gel. b Diastereomeric ratios determined by 1H NMR analysis on crude material. c Reaction run using achiral allenylsilane 4b.
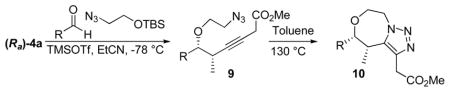
Further structural variation was achieved by using silyl ether reaction partners containing an azide functionality (Table 1). The silyl protected 2-azidoethanol could be prepared from 2-bromoethanol in two steps using a known procedure.9 The TBS ether was explored after the TMS version proved to be difficult to work with due to it’s inherent volatility.
Table 1.
Propargylations with TBS 2-azidoethanol
 | |||
|---|---|---|---|
| entry | aldehyde (R) | product 9 (yielda; drb) | product 10 (yielda) |
| 1 |
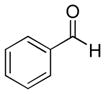
|
9a (72%; 18:1) | 10a (71%) |
| 2 |
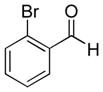
|
9b (89%; >20:1) | 10b (75%) |
| 3 |
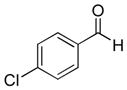
|
9c (76%; 15:1) | 10c (76%) |
| 4 |
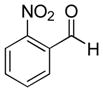
|
9d (87%; >20:1) | 10d (74%) |
| 5 |
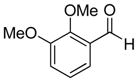
|
9e (74%; >20:1) | 10e (66%) |
| 6 |
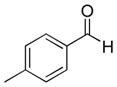
|
9f (76%; 15:1) | 10f (74%) |
| 7 |
|
9g (60%; >20:1) | 10g (75%) |
| 8 |
|
9h (37%; 5:1) | 10h (57%) |
| 9 |
|
9i (16%, >20:1) | 10i (80%) |
Isolated yields after purification over silica gel.
Diastereomeric ratios determined by 1H NMR analysis on crude material.
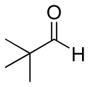
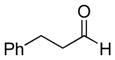
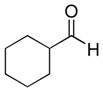
The propargylation reaction of allenylsilane (Ra)-4 with aldehydes and silyl protected 2-azidoethanol resulted in the formation of syn-homopropargylic ethers 9a-i. High yields and selectivities were obtained for a variety of aromatic aldehydes (entries 1–6). The use of aliphatic aldehydes in this reaction sequence resulted in lower yields (entries 7–9), and lower selectivity for the unbranched system (entry 8).
For all of the examples studied, heating the reaction products to 130 ºC in toluene gave the desired triazoles 10a-i in good yield.10 None of the triazole products were observed after the propargylation reaction, as the alkyne products were stable at room temperature, and as expected, the dipolar cyclizations proceeded with complete regioselectivity.
To expand the scope of this methodology, we synthesized four additional azidosilyl ethers (Scheme 3) which would allow access to fused triazole systems with greater complexity.11 Silyl ether 11a was prepared in four steps from (S)-ethyl lactate, and contains a chiral center on the carbon with the silyl ether. Silyl ether 11b, derived from the epoxide opening of (R)-styrene oxide, has a chiral center on the carbon containing the azide. TBS protected 3-azidopropanol 11c was prepared from 3-bromopropanol using the same procedure as TBS protected 2-azidoethanol. Finally, the 3-carbon silyl ether containing a chiral center at C2 (11d) was accessed using commercially available (S)-methyl-3-hydroxy-2-methylpropanpoate in four steps. These procedures are all general and relatively straight forward, allowing for the rapid incorporation of stereochemical variation into the synthesis of the desired fused triazole systems.
Scheme 3.

Silyl ether synthesis
Propargylation reactions with 11a proceeded in good yield, but with a significant loss of diastereoselctivity when the (Ra) enantiomer of allenylsilane 4a was used (Scheme 4, 12a+b). When the (Sa) enantiomer of the allenylsilane was used the diasteroselectivity was substantially higher (Scheme 4, 12c+d), exhibiting a “matched pair” of reaction partners in a double stereodifferentiating reaction. The cycloadditions proceeded with moderate to high yield, although the products of matched cases (13c+d) showed higher yields than the mismatched cases (13a+b).12
Scheme 4.

Propargylations and dipolar cycloadditions with substituted silyl ethers
a All yields refer to isolated products after purification over silica gel. b All diastereomeric ratios were determined by 1H NMR analysis on crude material.
c The (Sa) enantiomer of the allenylsilane 1 was used for the propargylation reaction. dCycloaddition reaction run in chlorobenzene at 150 ºC.
When the silyl ether’s chiral center was located on the carbon baring the azide the diastereoselectivity of the reaction products was not effected by switching the enantiomer of the allene. Propargylations with silyl ether 11b gave high yields and diastereoselectivities with both enantiomers (Scheme 4, 12e–h). The cycloadditions also proceeded in similar yields for the different diastereomers (Scheme 4, 13e–h), with moderate yields obtained in all cases.
Propargylations with the three-carbon silyl ether 11c proceeded with good yield and excellent diastereoselectivity (Scheme 4, 12i+j). While the reactions were sluggish in toluene, switching the solvent to chlorobenzene at 150 ºC resulted in significantly higher yields for 13i+j. Use of silyl ether 11d, which contained a stereocenter at C2, had no effect on the selectivity, as the products were formed in good yields as a single observed diasteromer regardless of which enantiomer of allenylsilane 4a was used (Scheme 4, 12k+l).
In conclusion we have reported a tandem propargylation/dipolar cycloaddition sequence to rapidly construct structurally and stereochemically diverse fused ring systems containing 1,2,3-triazoles. Continued exploration of these ring systems, along with preliminary biological evaluation, is currently in progress.
Supplementary Material
Acknowledgments
Financial support was obtained from the NIH CA 53604 and through an AstraZeneca graduate fellowship to RAB.
Footnotes
Supporting Information Available Experimental data and selected spectral data for all new compounds. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.(a) Huisgen R. In: 1,3-Dipolar Cycloaddition Chemistry. Padwa A, editor. Wiley; New York: 1984. [Google Scholar]; (b) Kolb HC, Finn MG, Sharpless KB. Angew Chem Int Ed. 2001;40:2004–2021. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]; (c) Rostovtsev VV, Green LG, Fokin VV, Sharpless KB. Angew Chem Int Ed. 2002;41:2596–2599. doi: 10.1002/1521-3773(20020715)41:14<2596::AID-ANIE2596>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]; (d) Kelly AR, Wei J, Kesavan S, Marié JC, Windmon N, Young DW, Marcaurelle LA. Org Lett. 2009;11:2257–2260. doi: 10.1021/ol900562u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Spiteric C, Moses JE. Angew Chem Int Ed. 2009. p. 48. [DOI] [Google Scholar]
- 2.(a) Lewis WG, Green LG, Grynszpan F, Radić Z, Carlier PR, Taylor P, Finn MG, Sharpless KB. Angew Chem Int Ed. 2002;41:1053–1057. doi: 10.1002/1521-3773(20020315)41:6<1053::aid-anie1053>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]; (b) Tron GC, Pirali T, Billington RA, Canonico PL, Sorba G, Genazzani AA. Med Res Rev. 2008;28:278–308. doi: 10.1002/med.20107. [DOI] [PubMed] [Google Scholar]; (c) Lee T, Cho M, Ko SY, Youn HJ, Baek DJ, Cho WJ, Kang CY, Kim S. J Med Chem. 2007;50:585–589. doi: 10.1021/jm061243q. [DOI] [PubMed] [Google Scholar]; (d) Maurya SK, Gollapalli DR, Kirubakaran S, Zhang M, Johnson CR, Benjamin NN, Hedstrom L, Cuny GD. J Med Chem. 2009;52:4623–4630. doi: 10.1021/jm900410u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Kolb HC, Sharpless KB. Drug Discovery Today. 2003;8:1128–1137. doi: 10.1016/s1359-6446(03)02933-7. [DOI] [PubMed] [Google Scholar]; (f) Puig-Basagoiti F, Qing M, Dong H, Zhang B, Zou G, Yuan Z, Shi PY. Antiviral Research. 2009;83:71–79. doi: 10.1016/j.antiviral.2009.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Zhang L, Chen X, Xue P, Sun HHY, Williams ID, Sharpless KB, Fokin VV, Jia G. J Am Chem Soc. 2005;127:15998–15999. doi: 10.1021/ja054114s. [DOI] [PubMed] [Google Scholar]; (b) Majireck MM, Weinreb SM. J Org Chem. 2006;71:8680–8683. doi: 10.1021/jo061688m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Ackermann L, Potukuchi HK, Landsberg D, Vicente R. Org Lett. 2008;10:3081–3084. doi: 10.1021/ol801078r. [DOI] [PubMed] [Google Scholar]; (b) Chowdhury C, Mukherjee S, Das B, Achari B. J Org Chem. 2009;75:3612–3615. doi: 10.1021/jo900428j. [DOI] [PubMed] [Google Scholar]
- 5.(a) Pearson WH, Bergmeier SC, Chytra JA. Synthesis. 1990:156–159. [Google Scholar]; (b) Oliva AI, Christmann U, Font D, Cuevas F, Ballester P, Buschmann H, Torrens A, Yenes S, Pericás MA. Org Lett. 2008;10:1617–1619. doi: 10.1021/ol800291t. [DOI] [PubMed] [Google Scholar]; (c) Balducci E, Bellucci L, Petricci E, Taddei M, Tafi A. J Org Chem. 2008;74:1314–1321. doi: 10.1021/jo802463r. [DOI] [PubMed] [Google Scholar]
- 6.Brawn RA, Panek JS. Org Lett. 2007;9:2689–2692. doi: 10.1021/ol070936d. [DOI] [PubMed] [Google Scholar]
- 7.For a recent review on chiral organosilanes in diversity oriented synthesis see: Shaw J. Nat Prod Rep. 2009;26:11–26. doi: 10.1039/b814468k.
- 8.For the synthesis of functionalized 2-azidobenzaldehydes see: Pelkey ET, Gribble GW. Tetrahedron Lett. 1997;38:5603–5606.Main CA, Petersson HA, Rahman SS, Hartley RC. Tetrahedron. 2008;64:901–914.Cuevas JC, De Mendoza J, Prados P. J Org Chem. 1988;53:2055–2066.
- 9.Smith RH, Mehl AF, Shantz DL, Chmurny GN, Michejda CJ. J Org Chem. 1988;53:1467–1471. [Google Scholar]
- 10.All of the dipolar cycloaddition reactions were run in a sealed tube. No reactivity was observed using the copper (I) catalyst systems which have been previously reported.
- 11.For the synthesis of these silyl ethers: see Supporting Information.
- 12.Masamune S, Choy W, Petersen JS, Sita LR. Angew Chem, Int Ed. 1985;24:1. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.