

The intracellular levels and activity of RhoA and RhoC, two closely related homologues of the RhoGTPase family, are interdependent, and their cross-talk requires RhoGDIά. This dynamic balance regulates the activation of the ROCK downstream effector and the expression of several genes, including NAG-1, that alter tumorigenesis in vitro and in vivo.
Abstract
RhoGTPases are key signaling molecules regulating main cellular functions such as migration, proliferation, survival, and gene expression through interactions with various effectors. Within the RhoA-related subclass, RhoA and RhoC contribute to several steps of tumor growth, and the regulation of their expression affects cancer progression. Our aim is to investigate their respective contributions to the acquisition of an invasive phenotype by using models of reduced or forced expression. The silencing of RhoC, but not of RhoA, increased the expression of genes encoding tumor suppressors, such as nonsteroidal anti-inflammatory drug–activated gene 1 (NAG-1), and decreased migration and the anchorage-independent growth in vitro. In vivo, RhoC small interfering RNA (siRhoC) impaired tumor growth. Of interest, the simultaneous knockdown of RhoC and NAG-1 repressed most of the siRhoC-related effects, demonstrating the central role of NAG-1. In addition of being induced by RhoC silencing, NAG-1 was also largely up-regulated in cells overexpressing RhoA. The silencing of RhoGDP dissociation inhibitor α (RhoGDIα) and the overexpression of a RhoA mutant unable to bind RhoGDIα suggested that the effect of RhoC silencing is indirect and results from the up-regulation of the RhoA level through competition for RhoGDIα. This study demonstrates the dynamic balance inside the RhoGTPase network and illustrates its biological relevance in cancer progression.
INTRODUCTION
The small GTPases of the Rho family are key intermediates in transducing signals arising from soluble mediators as well as those originating from clustered cell adhesion receptors. They drive several vital aspects of cell physiology, such as cytoskeleton organization and cell survival, proliferation, migration, and differentiation (Jaffe and Hall, 2005). These signaling molecules function as binary molecular switches: on the activity of guanine nucleotide exchange factors (GEFs) at the plasma membrane, they switch from an inactive, GDP-bound state to a GTP-bound form that can bind and activate effector molecules. The RhoGTPases return to an inactivated state by hydrolysis of the GTP into GDP by catalysis of the intrinsic GTPase activity by a GTPase-activating protein (GAP). They are sequestrated under an inactivate state in the cytoplasm by forming 1:1 complexes with RhoGDP dissociation inhibitor (RhoGDI). Although there are many GEFs and GAPs, only three RhoGDI proteins have been identified in humans. RhoGDIα is the most ubiquitously expressed member of the family. Quantitation of RhoGDI levels in several cell types has shown that the molar amount of RhoGDI is roughly equal to the total level of RhoGTPases (Michaelson et al., 2001). RhoGDIs are being appreciated as pivotal regulators of the RhoGTPase function.
Among the RhoGTPase family, the Rho subclass includes RhoA, the most-studied member, and two closely related homologues, RhoC and RhoB. These three proteins share several properties, including the regulation of actin stress fibers formation. However, their role in cancer progression is clearly not redundant. RhoB is a short-lived protein displaying antitumorigenic properties (Liu et al., 2001; Mazieres et al., 2004, 2005), whereas RhoA and RhoC are reported as protumorigenic. Despite their high degree of homology, RhoA and RhoC display different affinity for their downstream effectors (Sahai and Marshall, 2002) and exert distinct functions in several key steps of cancer progression, such as epithelial-to-mesenchymal transition (Bellovin et al., 2006) and invasion of breast carcinoma cells (Simpson et al., 2004). Whereas RhoA is ubiquitously expressed, increasing evidences suggest that RhoC expression is associated with highly aggressive cancers, where it promotes metastasis (Clark et al., 2000; Kleer et al., 2002; Shikada et al., 2003; Wang et al., 2004, 2008; Yao et al., 2006). In particular, this was observed in prostate cancer cell lines and tissues where RhoC protein expression was associated with a metastatic phenotype (Iiizumi et al., 2008).
Of interest, it has been established that small GTPases of the Rho family are also integral components of signaling pathways regulating transcription (Coso et al., 1995; Hill et al., 1995). For example, potential RhoA- and RhoC-responsive genes have been identified in NIH3T3 and MCF10A cells, respectively (Teramoto et al., 2003; Wu et al., 2004). More recently, a comparison of the gene expression profiles in cells transfected by constitutively active RhoA, RhoB, or RhoC has been reported (Berenjeno et al., 2007). Such studies, although providing valuable information, have some limitations. Overexpression of Rho proteins, especially for constitutively active mutants, creates conditions that are not fully relevant to physiological situations. Similarly, cell lines used as models need to be chosen carefully for their ability to regulate and to respond efficiently to the pathways involving the RhoGTPase network.
Here, a model of PC-3 cells—a prostate cancer cell line expressing significant levels of endogenous RhoA and RhoC—was used to evaluate the respective contributions of these two RhoGTPases to the acquisition of a transformed phenotype (Yao et al., 2006). Our data suggest that levels and activity of RhoA and RhoC are mutually dependent and that their cross-talk involves RhoGDIα. This dynamic balance regulates the activation of the downstream effector Rho kinase (ROCK) and the expression of several genes, including nonsteroidal anti-inflammatory drug–activated gene 1 (NAG-1), that alter tumorigenesis in vitro and in vivo. This study demonstrates that cross-talks between individual members of the RhoGTPase regulatory network, including RhoGDI, are central for cell regulation and tumor formation, seem to be preferentially established in a hierarchical order between related homologues, and are highly sensitive to slight modifications of the relative abundance of each individual partner.
RESULTS
Silencing RhoC, but not RhoA, significantly decreases the anchorage-independent growth of PC-3 and LnCaP cells
The specific contributions of RhoA and RhoC in the PC-3 cell tumoral phenotype were evaluated by using a small interfering RNA (siRNA) approach. As recommended in an editorial in Nature Cell Biology (Editors, 2003) our results were validated by using specific controls (ctrA and ctrC) designed by introducing two nucleotide changes in the siRNA specific for RhoA and RhoC, respectively (Ho et al., 2008). These two mutations completely abrogated the silencing activity of the siRNAs, even at concentrations as high as 60 nM (Figure 1, A and B). The effect of RhoA or RhoC silencing on the in vitro tumorigenic properties of PC-3 cells was tested in an anchorage-independent growth model. Immediately after transfection, PC-3 cells were seeded in soft agar, and the total number of colonies formed after 2 wk was counted. It is worth noting that even after 2 wk of culture in soft agar, the siRNAs were still efficient, as a significant inhibition of the targeted RhoGTPase was observed (Figure 1, C and D), and that the ability of PC-3 cells to grow in this model was altered neither by the transfection agent alone (mock transfected) nor by transfection with control siRNA (Figure 1, E). The silencing of RhoA failed to alter anchorage-independent growth of PC-3 in soft agar, whereas RhoC siRNA (siRhoC) efficiently repressed it (Figure 1F). Similar data were obtained using LnCaP prostate carcinoma cells (Figure 1G), suggesting that this regulation is not limited to a single cell type. To address concerns about potential “off-target” effects, experiments were repeated with additional distinct siRNAs targeting RhoA (siRhoA#2) and RhoC (siRhoC#2). As illustrated in Supplemental Figure S1, these two additional siRNAs repressed their specific target as efficiently as siRhoA and siRhoC, respectively. Similar to the first siRhoC, transfection with siRhoC#2 also strongly reduced anchorage-independent growth of PC-3 (Figure 1H), whereas the silencing of RhoA with siRhoA#2 slightly increased the number of colonies. We previously reported that silencing RhoA or RhoC induced the expression of RhoB (Ho et al., 2008). Cosilencing experiments revealed that RhoB is not involved in the repression of anchorage-independent growth following RhoC silencing (Supplemental Figure S2). As an additional characterization of our model, it was determined that RhoA or RhoC silencing did not significantly modify the expression or the activity of Cdc42 or Rac1 (Supplemental Figure S3). These data demonstrate a specific effect of RhoC silencing on the anchorage-independent growth properties of prostate cancer cells.
FIGURE 1:

Silencing RhoC but not RhoA repressed anchorage-independent growth. Increasing concentrations (2–60 nM) of Scr, ctrA, and siRhoA (A) or of Scr, ctrC, and siRhoC (B) were used to transfect PC-3 cells. At 48 h posttransfection, cells were lysed and analyzed by immunoblotting with specific antibodies to RhoA, RhoC, and Erk1/2. Strong and specific inhibitions of expression were observed with siRhoA and siRhoC at concentration as low as 2 nM. In sharp contrast, no effect was detected for the controls even at the highest concentrations. PC-3 cells were transfected with 20 nm of ctrC and siRhoC (C) or ctrA and siRhoA (D) and plated in soft agar as described in Materials and Methods. After 2 wk, cellular proteins were extracted and analyzed by immunoblotting with specific antibodies to RhoA, RhoC, and Erk1/2. siRhoA and siRhoC are still efficient after 2 wk of culture in soft-agar. PC-3 (E, F, and H) and LnCaP (G) transfected with 20 nM of the indicated siRNA were plated in soft agar as described in Materials and Methods. SiRhoA#2 and siRhoC#2 were designed and used to assess the specificity of the effects observed with siRhoA and siRhoC, respectively (H). After 2 wk of culture, colonies were counted in the whole-culture dishes. Three independent experiments were performed. Results are expressed in percentage of value obtained with untransfected cells (E), ctrC (F, G), or Scr (H). Only siRhoC is able to significantly reduce the number of colonies. *p < 0.05 and ***p < 0.001, analysis of variance (ANOVA), followed by Tukey–Kramer analysis.
RhoC silencing regulates genes involved in cell proliferation and survival
To identify mechanisms involved in the regulation of PC-3 anchorage-independent growth, comparative analyses of gene expression profiles following silencing of RhoA or RhoC were carried out with Affimetryx microarrays. An additional analysis using siRhoC#2 was performed to validate the regulations observed in the first experiment. The most significant differences are listed in Table 1. Several genes were up-regulated upon RhoC silencing while remaining unchanged or being down-regulated in RhoA-silenced PC-3. Among them, NAG-1, p21Cip1, p57Kip2, growth arrest and DNA damage gene 153 (GADD153), activating factor 3 (ATF-3), growth arrest and DNA damage gene 34, p8 protein, tribbles homologue 3, cystathionase, and homocysteine-inducible, endoplasmic reticulum stress–inducible, ubiquitin-like domain member 1 are involved in signaling networks controlling cell growth arrest and apoptosis and in the endoplasmic reticulum stress response. Microarray results were confirmed by real-time PCR (Figure 2) and, at the protein level, by Western blot analysis (Figure 3A, lanes 1–4, and Supplemental Figure S4). Of interest, the expression level of SPARC (also called osteonectin), an extracellular matrix component potentially involved in tumorigenesis (Hooi et al., 2006), was repressed upon RhoC silencing and up-regulated upon RhoA silencing (Table 1). This antagonistic regulation was confirmed by real-time PCR (Figure 2) and Western blot analysis (Figure 3A). Our observations revealed the regulation of genes potentially involved in the alteration of the cell phenotype following RhoC silencing.
TABLE 1:
Selected genes regulated in PC-3 cells following RhoA or RhoC silencing.
| Fold change | ||||||
|---|---|---|---|---|---|---|
| siRhoC vs. Scr | siRhoC#2 vs. Scr | siRhoA vs. Scr | ||||
| Array 1 | Array 2 | Array 1 | Array 2 | |||
| ras homologue gene family, member C | RHOC | −7 | −4.6 | −3 | 1.2 | 1.1 |
| ras homologue gene family, member A | RHOA | 1 | −1.1 | −1.1 | −5.2 | −4.9 |
| Up-regulated | ||||||
| NSAID-activated gene 1/growth differentiation factor 15 | NAG-1/GDF15 | 2.5 | 2.5 | 2.6 | −1.1 | −1.3 |
| Cyclin-dependent kinase inhibitor 1A (p21, Cip1) | p21Cip1/CDKN1A | 3.2 | 3 | 1.1 | 1.1 | 1 |
| Growth arrest and DNA damage gene 153 | GADD153/DDIT3 | 4 | 4.6 | 2.8 | −1.4 | −1.3 |
| Cyclin-dependent kinase inhibitor 1C (p57, Kip2) | p57Kip2/CDKN1C | 2.1 | 1.7 | 1.2 | 1.3 | 1.6 |
| Activating transcription factor 3 | ATF3 | 2.1 | 2.3 | 1.9 | −1.2 | −1.4 |
| Cystathionase (cystathionine γ-lyase) | CTH | 2.1 | 2.8 | 1.6 | −1.3 | −1.1 |
| Homocysteine-inducible, endoplasmic reticulum stress–inducible, ubiquitin-like domain member 1 | HERPUD1 | 2.3 | 1.9 | 1.9 | −1.1 | 1 |
| Growth arrest and DNA damage gene 34 | GADD34 | 3.5 | 3.2 | 1.5 | 1.1 | 1.1 |
| Tribbles homologue 3 | TRIB3 | 2 | 1.7 | 1.7 | −1.9 | −1.7 |
| Asparagine synthetase | ASNS | 2 | 1.5 | 1.9 | −1.1 | −1.2 |
| CCAAT/enhancer-binding protein B | CEBPB | 2.8 | 1.9 | 1.7 | −1.3 | −1.2 |
| p8 protein (candidate of metastasis 1) | p8 | 2.8 | 2.3 | 2.6 | −1.4 | −1.6 |
| Sequestosome 1 | SQSTM1 | 2.6 | 3 | 3 | 1.1 | 1.3 |
| ATPase H+-transporting lysosomal 42-kDa V1 subunit C1 | ATP6V1C1 | 2 | 2 | 1.7 | −1.2 | 1.4 |
| UDP-N-acetyl-α-d-galactosamine:polypeptide | GALNT3 | 2.5 | 3 | 1.9 | −1.1 | 1 |
| N-Acetylgalactosaminyltransferase 3 (GalNAc-T3) | ||||||
| Arginase type II | ARG2 | 4.3 | 3.7 | 1.5 | 1.2 | 1 |
| S100 calcium-binding protein P | S100P | 2.6 | 2.8 | 1.5 | −2.8 | −2.6 |
| GTP-binding protein overexpressed in skeletal muscle | GEM | 2.1 | 2.8 | 1.9 | −1.1 | −1.1 |
| Tripeptidyl peptidase I | TPP1 | 2.5 | 2.5 | 1.6 | 1.1 | 1.1 |
| Brain-expressed X-linked-like 1 | BEXL1 | 2.5 | 2 | 1.9 | −1.2 | −1.4 |
| HSPB (heat shock 27-kDa)–associated protein 1 | HSPBAP1 | 3 | 2.1 | 1.5 | 1.2 | −1.1 |
| Cell cycle progression 1 | CCPG1 | 2 | 2.1 | 1.5 | 1.3 | 1.1 |
| Syntaxin 3 | STX3 | 2.1 | 1.6 | 2.1 | 1.1 | −1.1 |
| Neutrophil cytosolic factor 2 (65 kDa) | NCF2 | 4 | 7 | 4 | −2 | −1.4 |
| Metallothionein 1F (functional) | MT1F | 1.9 | 2.5 | 2 | −1.4 | −1.4 |
| Metallothionein 1X | MT1X | 1.9 | 3.5 | 1.9 | −1.3 | 1.3 |
| DnaJ (Hsp40) homologue subfamily C, member 12 | DNAJC12 | 1.9 | 2.1 | 1.7 | 1.1 | 1 |
| Down-regulated | ||||||
| Secreted protein, acidic, cysteine-rich (osteonectin) | SPARC | −3 | −5.7 | −2.1 | 2.3 | 2.3 |
| Parathyroid hormone–like hormone | PTHLH | −1.9 | −1.6 | −2 | 1.3 | 1.3 |
| Interleukin 1 β | IL1B | −4.3 | −2.3 | −2.3 | −1.1 | −1.1 |
| Angiopoietin-like 4 | ANGPTL4 | −2.5 | −4.3 | −2.8 | 1.1 | 1.1 |
| Chromosome 1 open reading frame 165 | C1orf165 | −3 | −2.1 | −2.3 | 1.1 | −1.1 |
| Plasminogen activator tissue | PLAT | −2.5 | −2 | −1.9 | 1 | −1.1 |
| COBL-like 1 | COBLL1 | −2.3 | −2 | −1.9 | −1.9 | −1.2 |
| Very low density lipoprotein receptor | VLDLR | −2.3 | −2.1 | −2 | 1.1 | −1.1 |
| Opsin 3 (encephalopsin, panopsin) | OPN3 | −2.3 | −2 | −1.6 | 1 | −1.2 |
| Retinol-binding protein 4 plasma | RBP4 | −2.3 | −2.8 | −1.9 | 1.1 | −1.1 |
| Urotensin 2 | UTS2 | −2 | −3.2 | −1.7 | 1.9 | 1.2 |
| Solute carrier family 12 (sodium/potassium/chloride transporters), member 2 | SLC12A2 | −2.1 | −2.8 | −2.1 | 1 | 1 |
| Phosphofructokinase, platelet | PFKP | −2.1 | −1.9 | −2.1 | −1.4 | −1.2 |
| T-cell receptor β constant 1 | TRBC1 | −2.5 | −2 | −2.1 | −1.5 | −1.1 |
| Solute carrier family 2, member 1 | SLC2A1 | −1.5 | −2.1 | −2.5 | 1 | 1 |
| Procollagen-proline 2-oxoglutarate 4-dioxygenase (proline 4-hydroxylase) α polypeptide II | P4HA2 | −1.6 | −2 | −2 | −1.3 | −1.4 |
NSAID, nonsteroidal anti-inflammatory drug.
FIGURE 2:

Regulation of the expression of genes involved in growth arrest and of SPARC following RhoC or RhoA silencing. Real-time quantitative RT-PCR analysis was performed with total RNA purified from PC-3 cells 48 h after transfection with 20 nM of the various siRNA, either controls (ctrA and ctrC) or specific (siRhoA and siRhoC). Results are expressed as the mean ± SD of three independent experiments and confirms the microarray data reported in Table 1. *p < 0.05, **p < 0.01, ***p < 0.001, ANOVA, followed by Tukey–Kramer analysis.
FIGURE 3:

(A, B) Western blot analyses of regulation induced by silencing RhoA or RhoC. (A) The regulation of NAG-1, p21Cip1, and SPARC expression was analyzed at the protein level. PC-3 cells were transfected with 20 nM of the indicated control and specific siRNA. At 48 h posttransfection, cells were lysed and analyzed by immunoblotting with specific antibodies to NAG-1, p21Cip1, SPARC, RhoA, RhoC, and Erk1/2. (B) The induction of NAG-1 was also observed in LnCaP and MCF-7 cells upon RhoC silencing by either siRhoC or siRhoC#2, again demonstrating that these regulations are not cell specific. At 48 h posttransfection, cells were lysed and analyzed by immunoblotting with specific antibodies to NAG-1, RhoC, and Erk1/2. (C, D) NAG-1 is involved in the regulation of anchorage-independent growth following RhoC silencing. (C) PC-3 cells or (D) LnCaP cells were transfected with 20 nM of the indicated control and specific siRNA. At 24 h after transfection cells were plated in soft agar as described in Materials and Methods. Results are reported as percentages of values obtained with the ctrC and are mean ± SD of three independent experiments. *p < 0.05, **p < 0.01, ***p< 0.001, ANOVA, followed by Tukey–Kramer analysis.
Repression of NAG-1 reverses most of the gene regulation and the inhibition of anchorage-independent growth induced by RhoC silencing
NAG-1 was reported to regulate the expression of genes controlling proliferation and apoptosis (Kim et al., 2005). To address its role in the modulation of gene expression induced by RhoC silencing, a simultaneous repression of RhoC and NAG-1 was performed. As illustrated in Figure 3A, both proteins were efficiently silenced. In these conditions, the induction of p21Cip1, as evaluated at the mRNA (Figure 4) and protein levels (Figure 3A), was no longer observed. The expression of all the other genes up-regulated by RhoC silencing was sharply reduced by the simultaneous NAG-1 down-regulation (Figure 4), except for ATF-3 and p57Kip2, which were not significantly repressed (Supplemental Figure S5). The down-regulation of SPARC induced by RhoC silencing was also dependent on NAG-1 since cosilencing restored SPARC expression (Figure 3A). As NAG-1 appears to play a central role in the regulation of the PC-3 cell phenotype upon RhoC silencing, its expression was also tested in LnCaP and MCF-7 cells. As illustrated in Figure 3B, the silencing of RhoC in both cell types with siRhoC or siRhoC#2 significantly increased NAG-1 expression, demonstrating that this regulation applies to other cell types.
FIGURE 4:

The up-regulation of the expression of several genes involved in growth arrest following RhoC silencing depends on NAG-1. Real-time quantitative PCR analysis of the expression of the indicated mRNA was performed with total RNA extracted from PC-3 cells 48 h after transfection with 20 nM (or 40 nM) of specific controls for siRhoC (ctrC or ctrC[2X]), with 20 nM of siRhoC, or with 20 nM siRhoC + 20 nM of siNAG-1 (siRhoC + siNAG1). Results are the mean ± SD of three independent experiments. *p < 0.05, **, p<0.01, ***p < 0.001, ANOVA, followed by Tukey–Kramer analysis.
The role of NAG-1 in the repression of anchorage-independent growth induced by RhoC silencing was addressed by transfecting PC-3 cells with siRhoC alone or together with siNAG-1 and tested for colony formation in soft agar. As illustrated in Figure 3C, the cosilencing of NAG-1 suppressed the inhibitory effect of RhoC repression. Similar results were obtained using LnCaP, although the reversal induced by silencing NAG-1 was not complete (Figure 3D).
Altogether, these results demonstrate that NAG-1 is an essential mediator of the cell regulation induced by RhoC silencing.
RhoA overexpression leads to a similar phenotype to RhoC silencing
As clearly illustrated in Figure 1, an up-regulation of RhoA upon silencing of RhoC and the converse were consistently observed. This compensatory mechanism seems to be limited to the closely related members of the RhoA subclass (Supplemental Figure S3). It extends to other RhoGTPases, such as Rac1, for example, only if RhoA and RhoC are silenced simultaneously (Supplemental Figure S6). To assess whether the inhibition of anchorage-independent growth of PC-3 cells by siRhoC resulted from the direct repression of RhoC or from an up-regulation of RhoA, we generated clones expressing RhoA (PC-3/TR/RhoA) or RhoC (PC-3/TR/RhoC) in a doxycycline-dependent way. Western blot analysis revealed that RhoA overexpression decreased RhoC and conversely, whereas Rac1 and Cdc42 levels and activities remained unchanged (Figure 5A and Supplemental Figure S7). Similar to RhoC silencing, overexpression of RhoA induced up-regulation of NAG-1 and p21Cip1 protein levels and repression of SPARC (Figure 5B). These effects were correlated to the level of RhoA overexpression (compare the levels of expression for clones 6 and 5). Moreover, the transient overexpression of RhoA in MCF-7 cells also induced the expression of NAG-1, suggesting that the correlation between RhoC silencing and RhoA overexpression is not cell type specific (Figure 5C). At a cellular level, RhoC overexpression did not affect the ability of three different clones to form colonies in soft agar (Figure 6A). By contrast, overexpression of RhoA significantly decreased the anchorage-independent growth of PC-3 (Figure 6B) in a dose-dependent manner (Figure 5B, clones 6 and 5). These data highlight the physiological consequences of the interdependence of RhoA and RhoC expression.
FIGURE 5:

(A) Cross-regulation of the levels of RhoA and RhoC was evaluated by using PC-3 clones overexpressing RhoA (PC-3/TR/RhoA; clone 5) or RhoC (PC-3/TR/RhoC; clone 1) in a doxycycline-dependent way in the presence (+dox) or absence of doxycycline. After 48 h of culture in these conditions, cells were lysed and analyzed by immunoblotting with specific antibodies to RhoA, RhoC, Rac1, Cdc42, and Erk1/2. Representative blots of at least three independent experiments are shown. (B, C) Regulation of gene expression by RhoA overexpression. (B) Two clones of PC-3 cells overexpressing RhoA in a doxycycline-dependent way (PC-3/TR/RhoA) and a control clone (PC-3/TR/TO) were used. Cells were supplemented or not with doxycycline (100 ng/ml). After 48 h of culture, cells were lysed and analyzed by immunoblotting with specific antibodies to SPARC, p21Cip1, NAG-1, RhoA, RhoC, and Erk1/2. (C) MCF-7 cells were transiently transfected with control vector (pcDNA4/TO) or with RhoA expression vector (pcDNA4/TO/RhoA). At 48 h after transfection, cells were lysed and analyzed with specific antibodies to NAG-1, RhoA, and Erk1/2. Representative blots of at least three independent experiments are shown.
FIGURE 6:

Overexpression of RhoA, but not of RhoC, inhibits anchorage-independent growth. PC-3 clones overexpressing RhoC (A) or RhoA (B) and a control clone (PC-3/TR/T0) (B) were plated in soft agar as described in Materials and Methods. Cells were supplemented (+dox) or not with weekly renewed doxycycline (100 ng/ml). After 2 wk of culture, colonies were counted in the whole dishes. Results are reported on the unsupplemented condition and are expressed as mean ± SD. Representative results of three independent experiments are shown. ***p < 0.001, ANOVA, followed by Tukey–Kramer analysis.
The regulation of PC-3 phenotype induced by down-regulation of RhoC, but not by overexpression of RhoA, depends on RhoGDIα
The silencing of RhoC increases the level of NAG-1, possibly as a result of RhoA accumulation. This effect is strongly dependent on the presence of RhoGDIα, as illustrated in cells cotransfected with siRhoC and siRhoGDIα (Figure 7A). Furthermore, interactions between RhoA and RhoGDIα are actually increased in PC-3 cells silenced for RhoC, as demonstrated by coimmunoprecipitation experiments (Figure 7D). These data suggest that reduction of RhoC levels makes the pool of cytoplasmic RhoGDIα more available for interacting and stabilizing RhoA. The cross-talks between RhoA and RhoC were further investigated in cells overexpressing RhoGDIα. Overexpression of RhoGDIα increases the basal levels of RhoA and RhoC (Supplemental Figure S8), whereas further induction of RhoA accumulation following RhoC silencing was clearly attenuated when RhoGDIα was overexpressed (Supplemental Figure S9), again suggesting that the limited availability of RhoGDIα takes part in the regulation of the cross-talk between RhoA and RhoC. The role of RhoGDIα was further tested on the regulation of the anchorage-independent growth of PC-3 cells. Silencing RhoGDIα alone did not affect the colony formation in soft agar by control PC-3 cells, but it significantly reversed the inhibition induced by RhoC silencing (Figure 7C) illustrating its function as a regulator of cell phenotype. By contrast, inhibition of RhoGDIα did not influence the expression of NAG-1 when recombinant RhoA was overexpressed upon induction by doxycycline (Figure 7B). To decouple the regulation of RhoA from that of RhoC, PC-3 cells overexpressing, in an inducible way, a RhoA mutant (RhoAR68E) unable to bind RhoGDIα but still able to activate the downstream effectors of wtRhoA were created. The overexpression of this mutant did not affect the level of RhoC but significantly increased the expression of NAG-1 and repressed the anchorage-independent growth of PC-3 cells (Figure 8). Altogether, these data suggest that the effect of RhoC silencing in our model is indirect and results from the accumulation of RhoA through an increased availability of RhoGDIα for its stabilization. The regulations of cell phenotype are mediated by an increase of RhoA signaling rather than a decrease of RhoC signaling.
FIGURE 7:
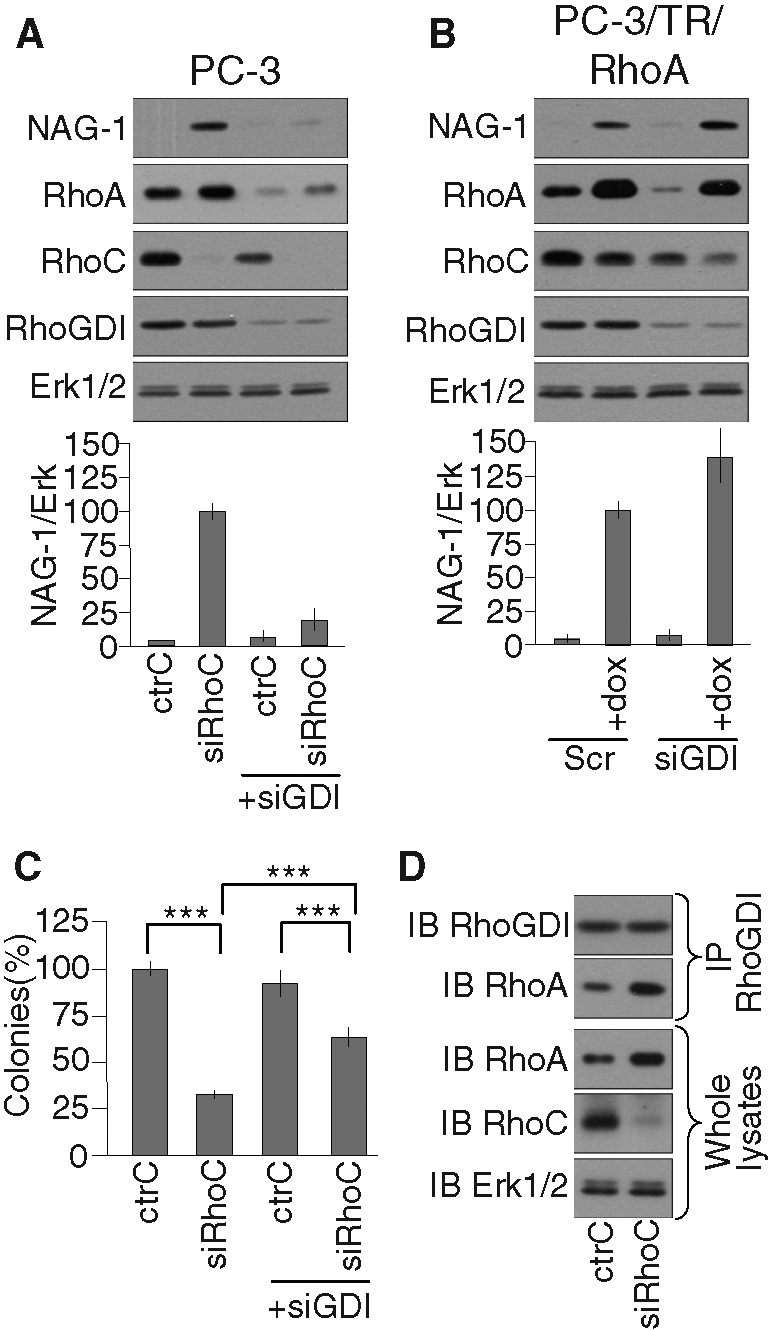
(A, B) The induction of NAG-1 expression following RhoC silencing, but not following RhoA overexpression, depends on RhoGDIα. (A) PC-3 cells or (B) PC-3/TR/RhoA were transfected with 20 nM of the indicated control and specific siRNA. Cells were supplemented (+dox) or not with 100 ng/ml doxycycline for 48 h and processed for Western blot analysis with specific antibodies to NAG-1, RhoA, RhoC, RhoGDIα, and Erk1/2. Representative analyses are illustrated. Bottom, the mean ± SD of densitometric analysis of three independent experiments. (C) RhoGDIα contributes to the inhibition of anchorage-independent growth following RhoC silencing. PC-3 cells were transfected with 20 nM of the indicated control and specific siRNA. 24 h posttransfection, cells were plated in soft agar as described in Materials and Methods. After 2 wk of culture, colonies were counted in the whole dishes. Results are reported on the ctrC condition and are mean ± SD of three independent experiments. ***p < 0.001, ANOVA, followed by Tukey–Kramer analysis. (D) Increased RhoA-RhoGDIα association following RhoC silencing. At 48 h after transfection with 20 nM of the indicated siRNA, PC-3 cells were lysed. Clarified lysates were immunoprecipitated with specific antibodies to RhoGDIα. The whole lysates and the immunoprecipitates were subjected to Western blot analysis with specific antibodies to RhoA, RhoC, RhoGDIα, and Erk1/2. Representative analyses out of three independent experiments are shown.
FIGURE 8:

The regulation of NAG-1 and the inhibition of anchorage-independent growth are mediated by the up-regulation of RhoA independent of the repression of RhoC expression. (A–C) PC-3/TR/RhoAR68E was supplemented (+dox) or not with 100 ng/ml doxycycline for 48 h and processed for Western blot analysis with specific antibodies to NAG-1, RhoA, RhoC, and Erk1/2. Representative analyses are illustrated. (B, C) The mean ± SD of densitometric analysis of three independent experiments. (D) PC-3/TR/RhoAR68E cells were plated in soft agar as described in Materials and Methods. Cells were supplemented (+dox) or not with weekly renewed doxycycline (100 ng/ml). After 2 wk of culture, colonies were counted in the whole dishes. Results are reported on the unsupplemented condition and are expressed as mean ± SD Representative results of three independent experiments are shown. **p < 0.01, ANOVA, followed by Tukey–Kramer analysis.
The regulation of NAG-1 following either RhoC silencing or RhoA overexpression depends on Rho kinase and p38 mitogen–activated kinase
As illustrated in Figure 9A, RhoC silencing resulted in an accumulation of RhoA but also in an increase of its activity as measured by pull-down assay. One main effector downstream of RhoA is Rho kinase. Its specific inhibition antagonized the induction of NAG-1 upon RhoC silencing (Figure 9B) and RhoA overexpression (Figure 9C). The inhibition of the p38 mitogen–activated kinase (p38MAPK) pathway also repressed the induction of NAG-1 in both conditions (Figure 9, D and E). Thus, Rho kinase and p38MAPK are necessary components of the signaling pathway mediating the regulation of NAG-1.
FIGURE 9:

Regulation of NAG-1 depends on the RhoA–Rho kinase pathway and on p38MAPK but not on Erk1/2. (A) RhoC silencing increased RhoA activity. PC-3 cells were transfected with 20 nM of ctrC or with 20 nM of siRhoC. At 48h posttransfection, cells were harvested and processed for Western blot and pull-down experiments. An aliquot of each lysate was denatured in SDS–PAGE loading buffer to analyze the concentration of RhoC and Erk1/2 with specific antibodies. RhoA activity was determined as the amount of GST-RBD–bound RhoA (RhoA-GTP) normalized to Erk1/2 in whole-cell lysates. (B) PC-3 cells were transfected with 20 nM of ctrC or siRhoC and then treated or not with Y-27632 (10 µM) during 48 h. (C) PC-3/TR/RhoA cells were treated or not with doxycycline (100 ng/ml) and with Y-27632 (10 μM) or the vehicle alone during 48 h. (D) PC-3 cells were transfected with 20 nM of ctrC or siRhoC and then treated or not with the indicated concentrations of U0126 or SB203580 or vehicle alone during 48 h. (E)PC-3/TR/RhoA cells were treated or not with doxycycline (100 ng/ml) and with indicated concentration of SB203580 or SB202190 or vehicle alone during 48 h. Cells were lysed and analyzed by immunoblotting with specific antibodies to NAG-1, RhoA, RhoC, and Erk1/2. Representative blots of at least three independent experiments are shown. *p < 0.05, **p < 0.01, ANOVA, followed by Tukey–Kramer analysis.
Silencing RhoC or overexpressing RhoA represses PC-3 cell migration
To further evaluate the relevance of the balance between RhoA and RhoC on cellular functions involved in tumorigenesis, we evaluated the effect of the modulation of RhoA and RhoC expression on the migratory properties of PC-3 cells in an in vitro scratch wound-healing assay. It clearly showed that silencing RhoC, but not RhoA, decreased the migratory properties of PC-3 cells. NAG-1 is not required for this effect since its cosilencing together with RhoC did not restore the migratory capacities to PC-3 cells. Moreover, its sole overexpression did not modulate the migratory properties of PC-3 cells (Figure 10). A similar assay was performed using cells inducible for RhoA or RhoC overexpression. In these conditions, high levels of RhoA clearly inhibited cell migration, whereas overexpression of RhoC had no significant effect. Moreover, the overexpression of RhoAR68E significantly reduced migration (Figure 11). Altogether, these data demonstrate that migration is also regulated by the RhoA/RhoC balance but independent of NAG-1 expression.
FIGURE 10:

Silencing RhoC but not RhoA inhibited PC-3 cell migration independent of NAG-1. (A) PC-3 cells were transfected with 20 nM of ctrA, siRhoA, ctrC, or siRhoC or 20 nM siRhoC + 20 nM of siNAG-1. Immediately after transfection, cells were seeded in six-well plates. Twenty-four hours later, the monolayers were scratched. Phase-contrast microscopy photographs were taken immediately after wounding (0 h) and 48 h after wounding (48h). (B, D) Quantification of wound closure. The mean ± SD of at least four independent experiments. (C) PC-3/TR/NAG-1 cells were seeded in six-well plates. Twenty-four hours later, the monolayers were scratched and treated (+dox) or not (–dox) with 100 ng/ml doxycycline. Phase-contrast microscopy photographs were taken immediately after wounding (0 h) and 48 h after wounding (48 h). Bar, 250 μm. n.s., not significant; *p < 0.05, **p < 0.01, ANOVA, followed by Tukey–Kramer analysis.
FIGURE 11:

PC-3 cell migration is inhibited by the up-regulation of RhoA independent of the repression of RhoC expression. Confluent monolayers of PC-3/TR/RhoA (A), PC-3/TR/RhoC (B), or PC-3/TR/RhoAR68E (C) cells were scratched and treated (+dox) or not (−dox) with 100 ng/ml doxycycline. Phase-contrast microscopy photographs were taken immediately after wounding (0 h) and 48 h after wounding (48 h). Right, quantification of wound closure. The results are expressed as mean ± SD of at least four independent experiments. Bar, 250 μm. ***p < 0.001, ANOVA, followed by Tukey–Kramer analysis.
Intratumoral injection of siRNA targeting RhoC delays PC-3 tumor growth, a process reversed by coinjecting siRNA targeting NAG-1
The role of NAG-1 in in vivo tumorigenesis was tested on PC-3 tumor development in nude mice. Three weeks after subcutaneous injection of PC-3 cells, developing tumors (50–100 mm3) were locally treated with siRNA (ctrC, siRhoC, or siRhoC + siNAG-1) as described in Materials and Methods. The treatment was repeated 12 and 24 d later. Injection of siRhoC induced a strong inhibition of tumor growth as compared to the tumors receiving the ctrC (Figure 12). This effect was completely abolished by coinjecting siNAG-1, supporting a role for NAG-1 in the regulation of tumorigenic properties in vivo as observed in vitro.
FIGURE 12:

The in vivo antitumorigenic effect of the siRNA targeting RhoC is inhibited by coadministration of siRNA targeting NAG-1. After induction of tumor formation by subcutaneous injection of 2 × 106 PC-3 cells, 50 μl of a solution containing 10 μM of a specific control for siRhoC (ctrC), 10 μM of the first siRNA targeting RhoC (siRhoC), or 10 μM of the first siRNA targeting RhoC + 10 μM of the siRNA targeting NAG-1 (siRhoC + siNAG1) mixed with atelocollagen as described in Materials and Methods was injected into the tumor region on days 21, 33, and 45. (A) Representative tumors photographed after sacrifice of the mice at day 48. (B) Tumor volume was calculated from tumor biaxial diameter measurement at regular interval up to day 48. Results represent the means ± SEM (n = 12 tumors). *p < 0.05, **p < 0.01, ***p < 0.001, ANOVA, followed by Tukey–Kramer analysis.
DISCUSSION
RhoA, RhoB, and RhoC are the three members of the same Rho subclass. Their differential contribution to cancer progression is mediated by their specific effects on various cellular functions. RhoGTPases are well known for their action on the actin cytoskeleton, but they are also involved in the regulation of other cellular activities, including gene expression. For instance, the serum responsive factor can be activated through Rho-mediated cytoskeletal changes (Miralles et al., 2003), and RhoA can regulate c-jun through the ROCK-JNK pathways independent of its effect on the cytoskeleton (Marinissen et al., 2004). Three microarray analyses using cells overexpressing RhoA, RhoB, and/or RhoC are available (Teramoto et al., 2003; Wu et al., 2004; Berenjeno et al., 2007). It was shown, however, that a similar cellular transformation program was elicited by constitutively active RhoA, RhoB, or RhoC overexpression (Berenjeno et al., 2007). Although informative, these experimental strategies are not fully relevant to the biological program specifically activated by the different endogenous Rho proteins. In our work, the specific contributions of RhoA and RhoC in the tumorigenic phenotype were investigated by an inverse approach, by silencing RhoA and RhoC in cellular models expressing significant amounts of both RhoGTPases. Regarding the methodological aspects, our results were validated by silencing each target with two different siRNAs and by using both irrelevant scrambled sequence and specific controls made of the siRNA sequence bearing two mutations that impede their silencing effect. Previous studies suggested that inhibition of RhoC activity by expressing a dominant-negative RhoC mutant or of RhoA by short hairpin RNA affected the activation level of Rac1 (Simpson et al., 2004; Yao et al., 2006). This was not observed in our model, suggesting that such indirect effect does not contribute to the regulation observed here. This discrepancy could be related to the experimental model. These previous studies used cells obtained after treatment with a selection agent. It cannot be ruled out that the time required for the selection of transfected cells was sufficient to induce compensatory mechanisms or to favor the counterselection of cells with the higher proliferative potential. By contrast, our cells were efficiently transfected with siRNA immediately before use, avoiding such potential side effect.
Our experimental strategy was based on the analysis of gene expression profiles in RhoA- and RhoC-silenced tumoral prostate cancer cells. These investigations were completed by assessing the relevance of the gene regulation to models of in vitro tumorigenic phenotype, that is, growth in soft agar and cell migration, and to in vivo tumor growth. Several genes specifically up-regulated by RhoC silencing were most attracting since they are involved in growth arrest and apoptosis and reported to be cross-regulated (Jiang et al., 2004; Kim et al., 2005; Carracedo et al., 2006; Zu et al., 2006; Clark et al., 2008). Among them, NAG-1 seems to play a pivotal role in this network, as we demonstrated that it acts upstream by controlling the expression of most of the other siRhoC-regulated genes. A similar regulation of its expression was observed in two other cancer cell lines, one of prostatic and one of mammary lineage, suggesting that our observations are not restricted to a single cell type. NAG-1 is generally considered as part of the antitumoral repertoire of the cells (Bauskin et al., 2006). Here we showed that it contributes to the repression of the tumorigenic properties of PC-3 and LnCaP cells upon RhoC silencing, as its inhibition restored, at least partly, the anchorage-independent growth of both cell types. It also reversed the up-regulation of genes involved in cell growth arrest and the repression of SPARC (or osteonectin), a component of the extracellular matrix displaying protumorigenic properties in prostatic cancer (Hooi et al., 2006; Chen et al., 2007). Finally, the biological relevance of NAG-1 in Rho-mediated pathways was further demonstrated in vivo in a model of tumorigenesis.
We and others recently reported direct cross-regulation within the Rho subclass (Simpson et al., 2004; Ho et al., 2008; Tillement et al., 2008; Steffan et al., 2009). As a result, the phenotypic modifications induced by RhoC silencing in PC-3 cells might be due to either the repression of RhoC or the concurrent induction of RhoA or RhoB. Of interest, the use of inducible clones overexpressing RhoA recapitulates most of the effects elicited by silencing RhoC, including down-regulation of RhoC. This demonstrates that RhoA and RhoC are in equilibrium and cannot be studied separately. Shifting this balance toward RhoA, either by overexpressing RhoA or repressing RhoC, reduces migration and anchorage-independent growth. In parallel, NAG-1 and p21CIP1 are induced, whereas SPARC is repressed, through a Rho kinase–dependent pathway. It was noted that RhoB protein level is also regulated via cross-talks within the Rho subclass (Ho et al., 2008). However, the lack of correlation between RhoB expression level and the modulations of PC-3 cells phenotype (unpublished data), as well as the lack of effect of RhoB silencing on the inhibition of anchorage-independent growth following RhoC inhibition, does not support its involvement in the regulations reported here. RhoGDIα, a key regulator of RhoGTPases cycling, prevents RhoGTPase degradation. Its knockout was reported to indeed decrease RhoA, Rac1, and Cdc42 protein levels (Bielek et al., 2009). Previous work in our laboratory suggested a role for RhoGDIα in the cross-regulation observed within the Rho subfamily (Ho et al., 2008) that was recently confirmed and extended by Boulter et al. (2010). These cross-regulations between RhoGTPases are driven by the competition for binding and stabilization by RhoGDIα, which is present in limited amount in the cell (Michaelson et al., 2001). This mechanism was confirmed here. Our data show that overexpression or silencing of RhoGDIα is accompanied, respectively, by increased or decreased levels of RhoA and RhoC and demonstrate that the dynamic balance between RhoA and RhoC is also regulated by the RhoGDIα availability. This hypothesis was further confirmed by using a RhoA mutant unable to bind RhoGDIα. Overexpression of this mutant did not modify the level of RhoC. In sharp contrast, NAG-1 was induced, and both migration and anchorage-independent growth were repressed. Altogether, our data indicate that the effects of RhoC silencing are indirect and result from the regulation of RhoA level through competition for RhoGDIα (Figure 13). In this study, as in previously published reports (Deroanne et al., 2003, 2005; Ho et al., 2008), we did not observe cross-regulation with other RhoGTPases such as Rac1 or Cdc42. In cell types expressing significant levels of RhoA and RhoC, a regulation of the expression of one of them automatically modifies the level of the other. This dynamic balance inside the Rho subgroup likely prevents the additional interactions between RhoGDIα and Rac1 (or Cdc42) as described by Boulter et al. (2010) in their models, where the level of RhoC is likely lower. According to this hypothesis, we observed that simultaneous knockdowns of RhoA and RhoC are required to make RhoGDIα available for Rac1, thus increasing its stability and decreasing its activation level, likely through inhibition of the interaction between Rac1 and its GEFs. Our data also suggest that “preferential” RhoGDIα-mediated cross-talks take place between closely related homologues. The specific mechanisms regulating this type of cross-talk remained to be firmly identified. They may depend on different intrinsic affinities for RhoGDIα, on the relative abundance of the different RhoGTPases, or on the regulating events able to modulate the affinity of RhoGDIα toward RhoGTPases through phosphorylation, thus adding one level of complexity in these networks (DerMardirossian et al., 2006; Dovas et al., 2010; Fei et al., 2010). Other, more indirect regulation also could be involved. It is worth noting, for example, that silencing RhoC increases the expression of Ccpg1 (Table 1). Ccpg1 is known to interact with a RhoGEF and to decrease its activity toward RhoA (Kostenko et al., 2006). In this way, the overexpression of Ccpg1 could indirectly and specifically favor the interaction between RhoA and RhoGDIα. Although RhoA and RhoC can likely regulate cellular functions independent of each other, these cross-talks must be taken into account in drawing conclusions from experiments using tools interfering with RhoGTPase function. They are also crucial for a better understanding of the physiological regulations induced by miRNA targeting RhoA and RhoC (Ma et al., 2007; Kong et al., 2008; Chiba et al., 2009; Jiang et al., 2010), for characterizing the effect of bacterial toxins that can inhibit the interaction between RhoGTPases and RhoGDIα (Huelsenbeck et al., 2007), and for determining the consequences of RhoGTPases up- or down-regulation in various diseases, especially cancers.
FIGURE 13:

Schematic overview of the RhoGDIα-mediated balance between RhoA and RhoC, resulting in modifications of the signaling pathway and leading to cancer cell growth and migration. (1) In basal condition, the activation of RhoA and RhoC is not sufficient to induce NAG-1 expression via ROCK. Cancer cells proliferate and migrate efficiently. (2) On RhoC silencing, the amount of RhoA-GDP stabilized by RhoGDIα and available for activation by specific GEF is increased. This leads to an enhanced level of RhoA-GTP, which drives NAG-1 gene expression via ROCK. RhoA overexpression induces a similar phenotype. The shift of the balance toward RhoA also inhibits cell migration but through an NAG-1–independent pathway.
MATERIALS AND METHODS
Reagents and cells
Y-27632 and U0126 were from Calbiochem (Hull, United Kingdom) and SB203580 was from Alexis (Zandhoven, Belgium). Mouse anti-RhoA (sc-418), rabbit anti-RhoB (sc-180), goat anti-RhoC (sc-26480), rabbit anti-RhoGDIa (sc-360), rabbit anti-p21 (sc-397), rabbit anti-SPARC (sc-25574), goat anti–NAG-1 (sc-10603), and donkey anti–goat immunoglobulin G (IgG) (sc-2020) were from Santa Cruz Biotechnology (Boechout, Belgium). Rabbit anti–extracellular signal-regulated kinase (Erk) 1/2 (M-5670) was from Sigma-Aldrich (St. Louis, MO). The secondary horseradish peroxidase–conjugated rabbit anti-mouse IgG (P0260) and swine anti–rabbit IgG (P0217) were from DAKO (Heverlee, Belgium). PC-3 human prostate adenocarcinoma cells were cultured in F-12 Kaighn's medium (Invitrogen, Merelbeke, Belgium) supplemented with 7% fetal bovine serum (FBS) (Lonza, Verviers, Belgium), human prostate carcinoma cells (LnCaP) were cultured in RPMI 1640 (Lonza) supplemented with 7% FBS, and human breast adenocarcinoma cells (MCF-7) were cultured in DMEM (Lonza) supplemented with 10% FBS.
siRNA transfection
The chemically synthesized, desalted, deprotected, and PAGE-purified 21-nucleotide-long siRNAs were from Eurogentec (Liège, Belgium). The sequences of the siRNA targeting RhoA (siRhoA and siRhoA#2) and RhoC (siRhoC and siRhoC#2), of the irrelevant siRNA used as control (siScr), and of the specific controls for siRhoA and siRhoC (ctrA and ctrC) were described previously (Ho et al., 2008). The siRNAs used to silence NAG-1 were as previously described (Shim and Eling, 2005). Each pair of oligoribonucleotides was annealed at a concentration of 20 μM in 50 mM NaCl, 1 mM EDTA, 10 mM Tris-HCl, pH 7.5. siRNA transfection was carried out as previously described (Deroanne et al., 2003). Briefly, calcium phosphate–mediated transfection was performed overnight (14–16 h) on subconfluent cells at a final concentration of 20 nM siRNA. Cells were washed twice with phosphate-buffered saline (PBS) and once with complete medium. This last step was defined as time 0 posttransfection. Cells were lysed for Western blot or RT-PCR analysis 48 h posttransfection.
Microarray analysis
The effect of RhoA and RhoC silencing on the gene expression profile of PC-3 cells was assessed by microarray analysis using the Affymetrix (Santa Clara, CA) HG-U133 Plus 2.0 chip containing 22,000 probe sets. Total RNA was isolated from siRNA-transfected cells using the High Pure RNA Isolation Kit (Roche Molecular Biochemical, Vilvoorde, Belgium). The integrity of the RNA was checked with the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). Probe synthesis hybridization, washing protocols, and signals scanning were performed at the GenoTranscriptomics facility of the Groupe Interdisciplinaire de Génoprotéomique Appliquée, University of Liège.
Real-time quantitative PCR
Total RNA was isolated from siRNA-transfected cells using the High Pure RNA Isolation Kit. A total of 1 μg of RNA was reversed transcribed using SuperScript II Reverse Transcriptase (Invitrogen). Real-time PCR was performed in a final volume of 20 μl containing 2 μl of cDNA (corresponding to 10 ng of total RNA for GDF-15, p21Cip1, GADD153, ATF-3, SPARC, and p57Kip2 amplification and corresponding to 0.1 ng of total RNA for glyceraldehyde-3-phosphate dehydrogenase [GAPDH] amplification), 300 nM of each primer, and 10 μl of the qPCR MasterMix Plus for SYBR green (Eurogentec) in the Abi Prism 7000 Sequence Detection system (Applied Biosystems, Halle, Belgium). The results were normalized to the GAPDH transcript.
Creation of PC-3 clones expressing RhoA, RhoC, RhoAR68E, RhoGDIα, or NAG-1 under the dependence of doxycycline
The clones expressing RhoA in a doxycycline-dependent way and the expression vector for RhoAR68E were described previously (Ho et al., 2008). The entire coding sequences of human RhoC, human RhoGDIα, and human NAG-1 were amplified by RT-PCR, cloned into pcDNA4/TO (Invitrogen), and sequenced. Clones of PC-3 cells expressing a high level of tetracycline repressor (PC-3/TR) isolated as previously described (Ho et al., 2008) were transfected with pcDNA4/TO/RhoC, pcDNA4/TO/RhoAR68E, pcDNA4/TO/RhoGDIα, or pcDNA4/TO/NAG-1 and selected in medium supplemented with 1 μg/ml blasticidin + 200 μg/ml Zeocin. Several clones expressing RhoAR68E, RhoC, RhoGDIα, or NAG-1 in a doxycycline-dependent way (PC-3/TR/RhoAR68E, PC-3/TR/RhoC, PC-3/TR/RhoGDIα, PC-3/TR/NAG-1) were isolated and used in this study.
Western blotting
Cells were lysed in SDS–PAGE lysis buffer, and proteins were separated by PAGE. Proteins were transferred to a polyvinylidene fluoride transfer membrane (NEN Life Science Products, PerkinElmer, Waltham, MA). Membranes were blocked for 1 h with 3% dry milk in PBS–0.05% Tween 20 and incubated for 4 h with the diluted primary antibody. Membranes were then washed three times, incubated in the diluted secondary horseradish peroxidase–conjugated antibody for 1 h, and revealed by chemiluminescence using the ECL Kit (Amersham Biosciences, GE Healthcare, Piscataway, NJ) and x-ray film exposure. The membranes were reprobed with anti–Erk1/2 antibodies to control protein loading.
GTPase activity assay
The assay was carried out as previously described (Sander et al., 1999; Deroanne et al., 2003). Briefly, cells were chilled on ice and lysed in ice-cold buffer containing 1% Triton X-100, 25 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, pH 7.3, 150 mM NaCl, 4% glycerol, 0.1 mM 4-(-2-aminoethyl)benzenesulfonyl fluoride hydrochloride (AEBSF), and 4 μg/ml aprotinin. Lysates were centrifuged for 6 min at 16,000 × g. Supernatants were immediately frozen in liquid nitrogen and stored at –80°C until used. An aliquot of each supernatant collected before freezing was denatured in SDS–PAGE lysis buffer to measure the total RhoGTPase content by Western blotting. For pull-down assays, supernatants were incubated for 30 min with 30 μg of GST-PBD protein containing the Cdc42- and Rac-binding region of PAK-1B or 30 μg of GST-RBD protein containing the RhoA-binding region of Rhotekin, both affinity linked to glutathione–Sepharose beads. The beads were washed four times in lysis buffer and boiled in 60 μl of SDS–PAGE lysis buffer.
Immunoprecipitation
PC-3 cells were chilled on ice and lysed in buffer containing 0.5% Nonidet NP-40, 50 mM Tris/HCl, pH 7.4, 50 mM NaCl, 1 mM MgCl2, 50 mM NaF, 10% glycerol, 0.1 mM AEBSF, and 4 μg/ml aprotinin. Lysates were centrifuged for 5 min at 16,000 × g. An aliquot of each supernatant was denatured in SDS–PAGE lysis buffer to measure the total RhoGTPase content by Western blotting. Supernatants were incubated with 2 μg of rabbit anti-RhoGDIα. After 2 h of incubation at 4°C, 30 μl of protein A–Sepharose beads was added for 1 h to capture immune complexes. Beads were washed four times in ice-cold lysis buffer and boiled in 60 μl of SDS–PAGE lysis buffer.
Soft agar assay
Anchorage-independent growth was determined by soft agar assay (Laboisse et al., 1981). A total of 2 × 103 siRNA-transfected PC-3 cells was plated in 60-mm dishes in growth medium containing 0.3% agar, on top of a 0.6% agar gel. After 15 d, colonies were counted in the whole dishes using an inverted phase-contrast microscope. Each assay was done in triplicate. Results are the means ± SD of three independent experiments.
Migration assay
Confluent monolayers of PC-3 cells were scraped with a 1-ml pipette tip to create a wound. Immediately after wounding and 48 h later, different fields of each wound were photographed with a phase-contrast microscope. Each experiment was repeated at least four times. Cell migration was quantified by image analysis with the Quantity One software from Bio-Rad (Hercules, CA). The measurements of the cell-free surface and of the total surface covered by the cells were used to calculate the percentage of wound closure in each condition. Representative photographs of each condition are shown.
Tumorigenicity studies in nude mice
A volume of 200 μl of serum-free medium containing 2 × 106 PC-3 cells was inoculated into both flanks of 4-wk-old male nude (athymic) mice by using a 27-gauge needle (n = 6). After 3 wk, the tumors had reached an average volume of 50–100 mm3. They were then injected with 50 μl of siRNA (10 μM) mixed with atelocollagen (2.5 mg/ml) in PBS at days 21, 33, and 45. Tumor growth was assessed by measuring the length and width of tumors, and the volume was determined by using the following formula: (length) × (width)2 × 0.4. Data are presented as means ± SEM. This experimental protocol was approved by the Animal Ethics Committee of the University of Liège.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge the Groupe Interdisciplinaire de Génoprotéomique Appliquée GenoTranscriptomics facility. This work was partly supported by grants from the Belgian Fonds Nationale de la Recherche Scientifique (3.4514.07) to C.F.D., from the Belgian Federation against Cancer to A.C.C., and from the Belgian Federal Science Policy Office/Programme for the Development of Scientific Experiments and European Space Agency to B.V.N. T.T.G.H. and A.S. were fellows of the Belgian Fonds Nationale de la Recherche Scientifique (Fonds pour la Recherche dans l'Industrie et l'Agriculture and Télévie). A.C.C. is a Senior Research Associate and C.F.D is a Research Associate at the Belgian Fonds de la Recherche Scientifique–Fonds Nationale de la Recherche Scientifique.
Abbreviations used:
- AEBSF
4-(-2-aminoethyl)benzenesulfonyl fluoride hydrochloride
- ATF-3
activating factor 3
- Erk
extracellular signal-regulated kinase
- GADD153
growth arrest and DNA damage gene 153
- GAP
GTPase-activating protein
- GEF
guanine nucleotide exchange factor
- NAG-1
nonsteroidal anti-inflammatory drug–activated gene 1
- p38MAPK
p38 mitogen-activated kinase
- RhoGDI
RhoGDP dissociation inhibitor
- ROCK
Rho kinase
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E11-01-0020) on July 14, 2011.
REFERENCES
- Bauskin AR, Brown DA, Kuffner T, Johnen H, Luo XW, Hunter M, Breit SN. Role of macrophage inhibitory cytokine-1 in tumorigenesis and diagnosis of cancer. Cancer Res. 2006;66:4983–4986. doi: 10.1158/0008-5472.CAN-05-4067. [DOI] [PubMed] [Google Scholar]
- Bellovin DI, Simpson KJ, Danilov T, Maynard E, Rimm DL, Oettgen P, Mercurio AM. Reciprocal regulation of RhoA and RhoC characterizes the EMT and identifies RhoC as a prognostic marker of colon carcinoma. Oncogene. 2006;25:6959–6967. doi: 10.1038/sj.onc.1209682. [DOI] [PubMed] [Google Scholar]
- Berenjeno IM, Nunez F, Bustelo XR. Transcriptomal profiling of the cellular transformation induced by Rho subfamily GTPases. Oncogene. 2007;26:4295–4305. doi: 10.1038/sj.onc.1210194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielek H, Anselmo A, Dermardirossian C. Morphological and proliferative abnormalities in renal mesangial cells lacking RhoGDI. Cell Signal. 2009;21:1974–1983. doi: 10.1016/j.cellsig.2009.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulter E, Garcia-Mata R, Guilluy C, Dubash A, Rossi G, Brennwald PJ, Burridge K. Regulation of Rho GTPase crosstalk, degradation and activity by RhoGDI1. Nat Cell Biol. 2010;12:477–483. doi: 10.1038/ncb2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carracedo A, et al. The stress-regulated protein p8 mediates cannabinoid-induced apoptosis of tumor cells. Cancer Cell. 2006;9:301–312. doi: 10.1016/j.ccr.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Chen N, Ye XC, Chu K, Navone NM, Sage EH, Yu-Lee LY, Logothetis CJ, Lin SH. A secreted isoform of ErbB3 promotes osteonectin expression in bone and enhances the invasiveness of prostate cancer cells. Cancer Res. 2007;67:6544–6548. doi: 10.1158/0008-5472.CAN-07-1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba Y, Tanabe M, Goto K, Sakai H, Misawa M. Down-regulation of miR-133a contributes to up-regulation of Rhoa in bronchial smooth muscle cells. Am J Respir Crit Care Med. 2009;180:713–719. doi: 10.1164/rccm.200903-0325OC. [DOI] [PubMed] [Google Scholar]
- Clark DW, Mitra A, Fillmore RA, Jiang WG, Samant RS, Fodstad O, Shevde LA. NUPR1 interacts with p53, transcriptionally regulates p21 and rescues breast epithelial cells from doxorubicin-induced genotoxic stress. Curr Cancer Drug Targets. 2008;8:421–430. doi: 10.2174/156800908785133196. [DOI] [PubMed] [Google Scholar]
- Clark EA, Golub TR, Lander ES, Hynes RO. Genomic analysis of metastasis reveals an essential role for RhoC. Nature. 2000;406:532–535. doi: 10.1038/35020106. [DOI] [PubMed] [Google Scholar]
- Coso OA, Chiariello M, Yu JC, Teramoto H, Crespo P, Xu N, Miki T, Gutkind JS. The small GTP-binding proteins Rac1 and Cdc42 regulate the activity of the JNK/SAPK signaling pathway. Cell. 1995;81:1137–1146. doi: 10.1016/s0092-8674(05)80018-2. [DOI] [PubMed] [Google Scholar]
- DerMardirossian C, Rocklin G, Seo JY, Bokoch GM. Phosphorylation of RhoGDI by Src regulates Rho GTPase binding and cytosol-membrane cycling. Mol Biol Cell. 2006;17:4760–4768. doi: 10.1091/mbc.E06-06-0533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deroanne C, Vouret-Craviari V, Wang B, Pouyssegur J. EphrinA1 inactivates integrin-mediated vascular smooth muscle cell spreading via the Rac/PAK pathway. J Cell Sci. 2003;116:1367–1376. doi: 10.1242/jcs.00308. [DOI] [PubMed] [Google Scholar]
- Deroanne CF, Hamelryckx D, Ho TT, Lambert CA, Catroux P, Lapiere CM, Nusgens BV. Cdc42 downregulates MMP-1 expression by inhibiting the ERK1/2 pathway. J Cell Sci. 2005;118:1173–1183. doi: 10.1242/jcs.01707. [DOI] [PubMed] [Google Scholar]
- Dovas A, Choi Y, Yoneda A, Multhaupt HA, Kwon SH, Kang D, Oh ES, Couchman JR. Serine34 phosphorylation of RHO guanine dissociation inhibitor (RHOGDIalpha) links signaling from conventional protein kinase C to RHO GTPase in cell adhesion. J Biol Chem. 2010;285:23296–23308. doi: 10.1074/jbc.M109.098129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Editors Whither RNAi? Nat Cell Biol. 2003;5:489–490. doi: 10.1038/ncb0603-490. [DOI] [PubMed] [Google Scholar]
- Fei F, Kweon SM, Haataja L, De Sepulveda P, Groffen J, Heisterkamp N. The Fer tyrosine kinase regulates interactions of Rho GDP-dissociation inhibitor alpha with the small GTPase Rac. BMC Biochem. 2010;11:48. doi: 10.1186/1471-2091-11-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill CS, Wynne J, Treisman R. The Rho family GTPases RhoA, Rac1, and CDC42Hs regulate transcriptional activation by SRF. Cell. 1995;81:1159–1170. doi: 10.1016/s0092-8674(05)80020-0. [DOI] [PubMed] [Google Scholar]
- Ho TT, Merajver SD, Lapiere CM, Nusgens BV, Deroanne CF. RhoA-GDP regulates RhoB protein stability. Potential involvement of RhoGDIalpha. J Biol Chem. 2008;283:21588–21598. doi: 10.1074/jbc.M710033200. [DOI] [PubMed] [Google Scholar]
- Hooi CF, et al. ST7-mediated suppression of tumorigenicity of prostate cancer cells is characterized by remodeling of the extracellular matrix. Oncogene. 2006;25:3924–3933. doi: 10.1038/sj.onc.1209418. [DOI] [PubMed] [Google Scholar]
- Huelsenbeck J, Dreger SC, Gerhard R, Fritz G, Just I, Genth H. Upregulation of the immediate early gene product RhoB by exoenzyme C3 from Clostridium limosum and toxin B from Clostridium difficile. Biochemistry. 2007;46:4923–4931. doi: 10.1021/bi602465z. [DOI] [PubMed] [Google Scholar]
- Iiizumi M, et al. RhoC promotes metastasis via activation of the Pyk2 pathway in prostate cancer. Cancer Res. 2008;68:7613–7620. doi: 10.1158/0008-5472.CAN-07-6700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe AB, Hall A. Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol. 2005;21:247–269. doi: 10.1146/annurev.cellbio.21.020604.150721. [DOI] [PubMed] [Google Scholar]
- Jiang HY, Wek SA, McGrath BC, Lu D, Hai T, Harding HP, Wang X, Ron D, Cavener DR, Wek RC. Activating transcription factor 3 is integral to the eukaryotic initiation factor 2 kinase stress response. Mol Cell Biol. 2004;24:1365–1377. doi: 10.1128/MCB.24.3.1365-1377.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L, Liu X, Kolokythas A, Yu J, Wang A, Heidbreder CE, Shi F, Zhou X. Downregulation of the Rho GTPase signaling pathway is involved in the microRNA-138-mediated inhibition of cell migration and invasion in tongue squamous cell carcinoma. Int J Cancer. 2010;127:505–512. doi: 10.1002/ijc.25320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JS, Baek SJ, Sali T, Eling TE. The conventional nonsteroidal anti-inflammatory drug sulindac sulfide arrests ovarian cancer cell growth via the expression of NAG-1/MIC-1/GDF-15. Mol Cancer Ther. 2005;4:487–493. doi: 10.1158/1535-7163.MCT-04-0201. [DOI] [PubMed] [Google Scholar]
- Kleer CG, van Golen KL, Zhang Y, Wu ZF, Rubin MA, Merajver SD. Characterization of RhoC expression in benign and malignant breast disease: a potential new marker for small breast carcinomas with metastatic ability. Am J Pathol. 2002;160:579–584. doi: 10.1016/S0002-9440(10)64877-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong W, Yang H, He L, Zhao JJ, Coppola D, Dalton WS, Cheng JQ. MicroRNA-155 is regulated by the transforming growth factor beta/Smad pathway and contributes to epithelial cell plasticity by targeting RhoA. Mol Cell Biol. 2008;28:6773–6784. doi: 10.1128/MCB.00941-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostenko EV, Olabisi OO, Sahay S, Rodriguez PL, Whitehead IP. Ccpg1, a novel scaffold protein that regulates the activity of the Rho guanine nucleotide exchange factor Dbs. Mol Cell Biol. 2006;26:8964–8975. doi: 10.1128/MCB.00670-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laboisse CL, Augeron C, Potet F. Growth and differentiation of human gastrointestinal adenocarcinoma stem cells in soft agarose. Cancer Res. 1981;41:310–315. [PubMed] [Google Scholar]
- Liu A, Cerniglia GJ, Bernhard EJ, Prendergast GC. RhoB is required to mediate apoptosis in neoplastically transformed cells after DNA damage. Proc Natl Acad Sci USA. 2001;98:6192–6197. doi: 10.1073/pnas.111137198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma L, Teruya-Feldstein J, Weinberg RA. Tumour invasion and metastasis initiated by microRNA-10b in breast cancer. Nature. 2007;449:682–688. doi: 10.1038/nature06174. [DOI] [PubMed] [Google Scholar]
- Marinissen MJ, Chiariello M, Tanos T, Bernard O, Narumiya S, Gutkind JS. The small GTP-binding protein RhoA regulates c-jun by a ROCK-JNK signaling axis. Mol Cell. 2004;14:29–41. doi: 10.1016/s1097-2765(04)00153-4. [DOI] [PubMed] [Google Scholar]
- Mazieres J, Antonia T, Daste G, Muro-Cacho C, Berchery D, Tillement V, Pradines A, Sebti S, Favre G. Loss of RhoB expression in human lung cancer progression. Clin Cancer Res. 2004;10:2742–2750. doi: 10.1158/1078-0432.ccr-03-0149. [DOI] [PubMed] [Google Scholar]
- Mazieres J, Tillement V, Allal C, Clanet C, Bobin L, Chen Z, Sebti SM, Favre G, Pradines A. Geranylgeranylated, but not farnesylated, RhoB suppresses Ras transformation of NIH-3T3 cells. Exp Cell Res. 2005;304:354–364. doi: 10.1016/j.yexcr.2004.10.019. [DOI] [PubMed] [Google Scholar]
- Michaelson D, Silletti J, Murphy G, D'Eustachio P, Rush M, Philips MR. Differential localization of Rho GTPases in live cells: regulation by hypervariable regions and RhoGDI binding. J Cell Biol. 2001;152:111–126. doi: 10.1083/jcb.152.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miralles F, Posern G, Zaromytidou AI, Treisman R. Actin dynamics control SRF activity by regulation of its coactivator MAL. Cell. 2003;113:329–342. doi: 10.1016/s0092-8674(03)00278-2. [DOI] [PubMed] [Google Scholar]
- Sahai E, Marshall CJ. ROCK and Dia have opposing effects on adherens junctions downstream of Rho. Nat Cell Biol. 2002;4:408–415. doi: 10.1038/ncb796. [DOI] [PubMed] [Google Scholar]
- Sander EE, ten Klooster JP, van Delft S, van der Kammen RA, Collard JG. Rac downregulates Rho activity: reciprocal balance between both GTPases determines cellular morphology and migratory behavior. J Cell Biol. 1999;147:1009–1022. doi: 10.1083/jcb.147.5.1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shikada Y, Yoshino I, Okamoto T, Fukuyama S, Kameyama T, Maehara Y. Higher expression of RhoC is related to invasiveness in non-small cell lung carcinoma. Clin Cancer Res. 2003;9:5282–5286. [PubMed] [Google Scholar]
- Shim M, Eling TE. Protein kinase C-dependent regulation of NAG-1/placental bone morphogenic protein/MIC-1 expression in LNCaP prostate carcinoma cells. J Biol Chem. 2005;280:18636–18642. doi: 10.1074/jbc.M414613200. [DOI] [PubMed] [Google Scholar]
- Simpson KJ, Dugan AS, Mercurio AM. Functional analysis of the contribution of RhoA and RhoC GTPases to invasive breast carcinoma. Cancer Res. 2004;64:8694–8701. doi: 10.1158/0008-5472.CAN-04-2247. [DOI] [PubMed] [Google Scholar]
- Steffan JJ, Snider JL, Skalli O, Welbourne T, Cardelli JA. Na/H Exchangers and RhoA regulate acidic extracellular pH-induced lysosome trafficking in prostate cancer cells. Traffic. 2009;10:737–753. doi: 10.1111/j.1600-0854.2009.00904.x. [DOI] [PubMed] [Google Scholar]
- Teramoto H, Malek RL, Behbahani B, Castellone MD, Lee NH, Gutkind JS. Identification of H-Ras, RhoA, Rac1 and Cdc42 responsive genes. Oncogene. 2003;22:2689–2697. doi: 10.1038/sj.onc.1206364. [DOI] [PubMed] [Google Scholar]
- Tillement V, Lajoie-Mazenc I, Casanova A, Froment C, Penary M, Tovar D, Marquez R, Monsarrat B, Favre G, Pradines A. Phosphorylation of RhoB by CK1 impedes actin stress fiber organization and epidermal growth factor receptor stabilization. Exp Cell Res. 2008;314:2811–2821. doi: 10.1016/j.yexcr.2008.06.011. [DOI] [PubMed] [Google Scholar]
- Wang W, Wu F, Fang F, Tao Y, Yang L. Inhibition of invasion and metastasis of hepatocellular carcinoma cells via targeting RhoC in vitro and in vivo. Clin Cancer Res. 2008;14:6804–6812. doi: 10.1158/1078-0432.CCR-07-4820. [DOI] [PubMed] [Google Scholar]
- Wang W, Yang LY, Huang GW, Lu WQ, Yang ZL, Yang JQ, Liu HL. Genomic analysis reveals RhoC as a potential marker in hepatocellular carcinoma with poor prognosis. Br J Cancer. 2004;90:2349–2355. doi: 10.1038/sj.bjc.6601749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Wu ZF, Kumar-Sinha C, Chinnaiyan A, Merajver SD. RhoC induces differential expression of genes involved in invasion and metastasis in MCF10A breast cells. Breast Cancer Res Treat. 2004;84:3–12. doi: 10.1023/B:BREA.0000018426.76893.21. [DOI] [PubMed] [Google Scholar]
- Yao H, Dashner EJ, van Golen CM, van Golen KL. RhoC GTPase is required for PC-3 prostate cancer cell invasion but not motility. Oncogene. 2006;25:2285–2296. doi: 10.1038/sj.onc.1209260. [DOI] [PubMed] [Google Scholar]
- Zu K, Bihani T, Lin A, Park YM, Mori K, Ip C. Enhanced selenium effect on growth arrest by BiP/GRP78 knockdown in p53-null human prostate cancer cells. Oncogene. 2006;25:546–554. doi: 10.1038/sj.onc.1209071. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.