

Abstract
The biosynthesis of the C ring of the anti-tumor antibiotic agent, tomaymycin, is proposed to proceed through five enzyme-catalyzed steps from L-tyrosine. The genes encoding these enzymes have recently been cloned and their functions tentatively assigned, but there is limited biochemical evidence supporting the assignments of the last three steps. One enzyme, TomN, shows 58% pairwise sequence similarity with 4-oxalocrotonate tautomerase (4-OT), an enzyme found in a catabolic pathway for aromatic hydrocarbons. The TomN sequence includes three amino acids (Pro-1, Arg-11, and Arg-39) that have been identified as critical catalytic residues in 4-OT. However, the proposed substrate for TomN is very different from the one processed by 4-OT. In order to establish the function and mechanism of TomN and its relationship to 4-OT, kinetic, mutagenic, and structural studies have been carried out. The kinetic parameters for TomN, and four alanine mutants, P1A, R11A, R39A, and R61A, were determined using 2-hydroxymuconate, the substrate for 4-OT. The TomN-catalyzed reaction using this substrate compares favorably to that of 4-OT. In addition, the kinetic parameters for the P1A, R11A, and R39A mutant of TomN parallel the trends observed for the corresponding 4-OT mutants, implicating an analogous mechanism. A high resolution crystal structure (1.4 Å) of TomN shows that the overall structure and the active site region are highly similar to those of 4-OT with an RMS deviation of 0.81 Å. Moreover, key active site residues are positionally conserved. The combined results suggest that the tentative assignment for TomN and the proposed sequence of events in the biosynthetic pathway leading to the formation of the C ring of tomaymycin might not be correct. An alternative pathway that awaits biochemical confirmation is proposed.
The pyrrolo[1,4]benzodiazepine (PBD) natural products such as anthramycin, sibiromycin, and tomaymycin (1, Scheme 1) are noted for their anti-tumor and antibiotic activities, resulting from sequence-specific DNA alkylation (1,2). Interest in their anticancer properties has spawned the design, synthesis, and characterization of numerous PBD derivatives and the recent cloning of the biosynthetic gene clusters for sibiromycin and tomaymycin (3). Biochemical characterization of the individual enzymes making up these biosynthetic pathways and manipulation of the corresponding genes can expand the repertoire of PBD analogues to include synthetically inaccessible ones (1-3).
Scheme 1.
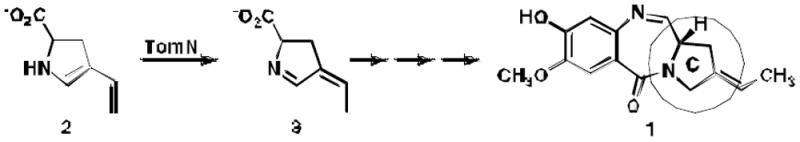
The proposed TomN-catalyzed reaction, a putative step in the biosynthesis tomaymycin C ring.
One step in the biosynthesis of the tomaymycin C ring is catalyzed by the 4-oxalocrotonate tautomerase (4-OT) homologue designated TomN (1). It is proposed that TomN converts 4-vinyl-2,3-dihydropyrrole-2-carboxylic acid (2) to 4-ethylidene-3,4-dihydropyrrole-2-carboxylic acid (3). The putative TomN substrate is very different from the one processed in the canonical 4-OT-catalyzed reaction, which involves the conversion of 2-hydroxy-2,4-hexadienedioate or 2-hydroxymuconate (4, Scheme 2) to 2- oxo-3-hexenedioate (5) (4,5). Moreover, TomN is found in a biosynthetic pathway whereas 4-OT is part of a catabolic pathway in Pseudomonas putida mt-2, Pseudomonas sp. CF600, and other soil bacteria (6). These two observations led to our interest in the enzymes of the tomaymycin and related biosynthetic pathways.
Scheme 2.
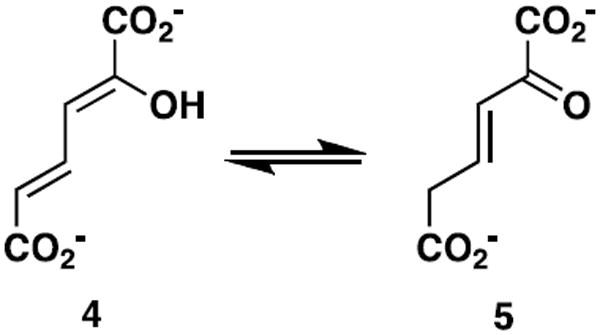
The canonical 4-OT-catalyzed reaction.
Kinetic, mechanistic, and structural studies identified Pro-1, Arg-11, Arg-39, and Phe-50 as key players in the 4-OT-catalyzed conversion of 4 to 5 (7-15). In the proposed mechanism, Pro-1, which has a pKa of ~6.4, transfers a proton from the 2-hydroxy group of 4 to C-5 of 5 (7-10). The interaction between Arg-11 and the C-6 carboxylate group is proposed to bind the substrate and draw electron density to C-5 to facilitate protonation at this position (11,12,14,15). Arg-39 may interact with the 2-hydroxy group and a carboxylate oxygen of C-1. Mutagenesis results suggest that its role is primarily catalytic, where the positively charged guanidinium moiety might stabilize the developing carbanionic character after deprotonation of the 2-hydroxy group. Phe-50 is a major contributor to a hydrophobic pocket near the prolyl nitrogen of Pro-1 (13,15). The proximity of the pocket is largely responsible for the unusually low pKa of Pro-1.
The TomN sequence has 66 amino acids (with Pro-1, Arg-11, and Arg-39 conserved, but Phe-50 replaced by tryptophan) and shows ~58% similarity (~33% identity) to that of 4-OT (from P. putida mt-2) (1). In view of the high degree of similarity and the presence of the conserved key residues, notably Arg-11 and Arg-39, it is somewhat odd that the proposed substrate for TomN has one carboxylate group, whereas the known substrate for 4-OT has two carboxylate groups. This observation raises two possibilities: the proposed substrate for TomN is not correct or our understanding of the functions played by the two arginine residues in the canonical 4-OT-catalyzed reaction is not complete.
In order to address these issues, TomN was characterized using 42. It was found that 4 is a substrate for TomN with kinetic parameters that are comparable to those of 4-OT (using 4). Mutagenesis studies implicate Pro-1, Arg-11, and Arg-39 as critical residues in this TomN-catalyzed reaction, suggesting an analogous mechanism to that of 4-OT. Finally, a crystal structure (1.40 Å resolution) indicates that the fold and active site of TomN are very similar to those of 4-OT, with an RMS deviation of 0.81 Å. Our results suggest that the proposed TomN substrate might not be correct, and that the actual substrate might resemble a molecule with the 2-hydroxymuconate framework (i.e., 4). These observations impact the tentatively assigned functions of the enzymes involved in the biosynthesis of the C ring of tomaymycin.
EXPERIMENTAL PROCEDURES
Materials
Chemicals, biochemicals, buffers, and solvents were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO), Fisher Scientific Inc. (Pittsburgh, PA), Fluka Chemical Corp. (Milwaukee, WI), or EM Science (Cincinnati, OH). The syntheses of 2-hydroxymuconate (4) (4), 5-(methyl)-2-hydroxymuconate (6) (16), 5-(carboxymethyl)-2-hydroxymuconate (8) (17), 2-hydroxy-2,4-pentadienoate (9) (18), 2- hydroxy-2,4-heptadiene-1,7-dioate (12) (19), and 2-oxo-3-pentynoate (17) (20) are reported in the indicated references. Recombinant 4-OT was purified by previously reported procedures (15,21). Sequencing grade endoproteinase Glu-C (protease V-8) from Staphylococcus aureus was obtained from Sigma-Aldrich Chemical Co. The Phenyl Sepharose 6 Fast Flow and DEAE-Sepharose resins and the prepacked PD-10 Sephadex G-25 columns were obtained from GE Healthcare (Piscataway, NJ). The Econo-Column® chromatography columns and Freeze ‘N Squeeze units were obtained from Bio-Rad Laboratories, Inc. (Hercules, CA). Enzymes and reagents used for molecular biology procedures were obtained from New England Biolabs, Inc. (Ipswich, MA).
Bacterial Strains, Plasmids, and Growth Conditions
E. coli strain DH5α was obtained from Invitrogen (Carlsbad, CA). E. coli strain BL21-Gold(DE3) was obtained from Stratagene (La Jolla, CA). The pET vectors were obtained from Novagen (Madison, WI). Plasmids were isolated from cell cultures using the Sigma GenElute Plasmid Miniprep Kit or the Qiagen QIAprep Spin Miniprep Kit. Cells for cloning and overproduction were grown on Luria-Bertani (LB) agar plates or in LB media supplemented with kanamycin (Kn) (30 μg/mL). The sources for the components of Luria-Bertani media are reported elsewhere (22).
General Methods
Techniques for restriction enzyme digestion, ligation, transformation, and other standard molecular biology manipulations were based on methods described elsewhere (22). Oligonucleotide primers were synthesized by Sigma-Aldrich. DNA sequencing was performed at the DNA core facility in the Institute for Cellular and Molecular Biology (ICMB) at the University of Texas at Austin. Mass spectral data were obtained on an LCQ electrospray ion-trap mass spectrometer (Thermo, San Jose, CA) in the ICMB Protein and Metabolite Analysis Facility at the University of Texas at Austin. Samples were prepared as described elsewhere (23). A Voyager-DE Pro matrix-assisted laser desorption/ionization (MALDI) mass spectrometer (PerSeptive Biosystems, Framingham, MA) was used to determine peptide masses. Kinetic data were obtained at 24 °C on an Agilent 8453 diode-array spectrophotometer. Nonlinear regression data analysis was performed using the program Grafit (Erithacus Software Ltd., Staines, U.K.) obtained from Sigma-Aldrich. Protein concentrations were determined by the method described by Waddell (24). Protein was analyzed by tricine sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) on 15%T/2%C gels on a Bio-Rad Mini-Protean II gel electrophoresis apparatus (25). Nuclear magnetic resonance (NMR) spectra were recorded in 100% H2O on a Varian Unity INOVA-500 spectrometer as reported previously (15,23).
Cloning of the TomN Gene into the pET24 Vector
The tomN gene was previously cloned into the pRSF Duet 1 vector between the NdeI and EcoRV restriction sites with a unique XhoI site downstream. The pRSF Duet 1 vector containing tomN and an aliquot of pET24 were digested with the NdeI and XhoI restriction enzymes. The two digestion mixtures were electrophoresed on an agarose gel and the appropriate pieces recovered using a Freeze ‘N Squeeze unit followed by ethanol precipitation and centrifugation. After ligation with T4 DNA ligase, the product was used to transform E. coli DH5α cells by electroporation using a BioRad MicroPulser Electroporation unit. The transformation reaction was plated on LB/Kn plates and grown overnight at 37°C. Following PCR screening, two positive colonies were used to start individual overnight cultures from which plasmid was isolated and sequenced.
Construction of the TomN Mutants
Four TomN mutants (P1A, R11A, R39A, and R61A) were constructed. Two mutants (R11A and R61A) were constructed by a PCR method that is based on the Stratagene QuikChange and New England Biolabs Phusion site-directed mutagenesis systems. The wild type TomN in the pRSF Duet 1 vector served as the template with the following primer pairs (forward and reverse, respectively), where the mutation site is underlined. For the R11A mutant, primer 1 was 5’-CCTTGCTGGAGGGCGCGTCGCCGCAGGAGGTG-3’ and primer 2 was 5’-CACCTCCTGCGGCGACGCGCCCTCCAGCAAGG-3’. For the R61A mutant, primer 3 was 5’-GGCCGAGCGGGCGGCCTCCCCCTCG-3’ and primer 4 was 5’-CGAGGGGGAGGCCGCCCGCTCGGCC-3’.
For the R11A mutant, the PCR mixture (50 μL) contained tomN in the pRSF plasmid (50 ng), primers 1 and 2 (125 ng each), 5× Phusion GC buffer supplied with the polymerase (10 μL), dNTPs (0.2 mM of each) and NEB Phusion DNA polymerase (1 unit). The reaction was processed in a heated top PCR thermocycler using a protocol that consisted of an initial 30-s denaturation cycle at 95° C, followed by 18 cycles of 95 °C for 30 s, 55 °C for 1 min, and 72 °C for 7 min, and ending with a hold at 4 °C. Following the PCR, DpnI restriction enzyme (1 μL of a 20 unit/ μL solution) was added to the mixture to digest the methylated parent plasmid. After the mixture incubated for 2 h at 37 °C, the DNA was precipitated, suspended in 10 mM Tris chloride buffer (pH 8.5), and added to E. coli DH5α cells (40 μL) for electroporation. The transformed cells were selected by overnight growth at 37 °C on LB/Kn plates. Two colonies were chosen and grown overnight in LB/Kn media at 37 °C. The plasmid was isolated and sequenced to confirm that only the desired mutation was present. Subsequently, the gene was transferred to pET24 in the same manner as described above for the wild type. The R61A mutant of TomN was constructed similarly except the DpnI-digested product was recovered and treated with T4 DNA ligase for 10 min (in the ligase buffer). The DNA was recovered and added to the E. coli DH5α cells for electroporation.
The P1A and R39A mutants were constructed by overlap extension PCR, as follows and described elsewhere (26). The wild type TomN was amplified from the pET24 construct using primers 5 and 6 (5’-CATGAGCCCGAAGTGGCGAGC-3’ and 5’-GCTAGTTATTGCTCAGCGG-3’, respectively), where primer 5, (the forward primer), is specific for a region slightly upstream of the T7 promoter sequence and primer 6, (the reverse primer) is complementary to a region immediately upstream of the T7 terminator region of the pET24 plasmid. The PCR mixture (50 μL) contained wild type TomN in the pET24 plasmid (62 ng), primers 5 and 6 (0.2 μM each), 10× Thermo buffer supplied with the polymerase (5 μL), dNTPs (0.2 mM each) and NEB Taq polymerase (1.5 units). The reaction was processed in the heated top PCR thermocycler using a protocol that consisted of an initial 2-min denaturation cycle at 94° C, followed by 29 cycles of 94 °C for 1 min, 55 °C for 2 min, and 72 °C for 3 min, and ended with an elongation step at 72°C for 10 min and a hold at 4 °C. The product was treated with DpnI restriction enzyme, the reaction mixture was electrophoresed, and the desired band was excised and processed through a Freeze ‘N Squeeze unit to recover the TomN coding region. The individual pieces required for overlap extension for the P1A and R39A mutants were generated in separate PCR reactions. Each PCR mixture (50 μL) contained the TomN coding region (1 μL from the Freeze ‘N Squeeze unit), the pair of primers indicated below (0.2 μM each), the 10× Thermo buffer (5 μL), dNTPs (0.2 mM of each) and NEB Taq polymerase (1.5 units). Each reaction was carried out using the PCR conditions described above. For the P1A mutant, primer 7 was 5’-GATATACATATGGCGCTCATCCGCGTCAC-3’, primer 8 was 5’-GTGACGCGGATGAGCGCCATATGTATATC-3’, and primer 9 was 5’-5’GATCCCGCGAAATTAATACGAC-3’. For the R39A mutant, primer 10 was 5’-GGCCGTCGCGGTGATCGTGGAGGAG-3’, primer 11 was 5’-CTCCTCCACGATCACCGCGACGGCC-3’, and primer 6 (above). The 5’ piece for the P1A mutant was constructed using primers 8 and 9. The 3’ piece was constructed using primers 6 and 7. The 5’ piece for the R39A mutant was constructed using primers 9 and 11. The 3’ piece was constructed using primers 6 and 10. Primer 9 is specific for part of the T7 promoter sequence and immediately upstream of that sequence in pET24. Each reaction mixture was electrophoresed, the desired band excised and processed through a Freeze ‘N Squeeze unit, and used in subsequent PCR mixtures, as follows. The PCR mixture (50 μL) for overlap extension contained the 5’ piece eluted from the Freeze ‘N Squeeze (1 μL), the 3’ piece eluted from the Freeze ‘N Squeeze (3 μL for the P1A mutant and 1 μL for the R39A mutant), primers 6 and 9 (0.2 μM each), 10× Thermo buffer (5 μL), dNTPs (0.2 mM of each) and NEB Taq polymerase (1.5 units). The reaction was processed in the heated top PCR thermocycler and the overlap product recovered as described above. In order to generate sufficient material for insertion into the pET24 vector, each overlap product was further amplified in four 50-μL PCRs using overlap product (1 μL) as template. The reaction products were pooled and the products recovered as described above, followed by ethanol precipitation and centrifugation. Each sequence (containing the mutation) was cloned into the pET24 vector between the NdeI and XhoI restriction sites following the procedures described for the cloning of the wild type gene into the pET24 vector. For each mutant, one colony was grown for plasmid, which was sequenced and subsequently used to transform E. coli BL21-Gold(DE3) cells for protein expression.
Expression and Purification of TomN and the TomN Mutants
Several colonies (i.e., a streak) of the pET vector construct (coding TomN or a TomN mutant) transformed in E. coli BL21-Gold(DE3) cells were used to inoculate LB/Kn media (~20 mL). After shaking overnight at 37 °C, an aliquot was used to inoculate LB/Kn media (~550 mL) in a 2-L flask so that the initial OD600 reading was ~0.05. Cells were allowed to grow at 37 °C with shaking until the OD600 reached ~1 (~2 h), and then induced by the addition of isopropyl-β-d-thiogalactoside (IPTG) (final concentration of 0.5 mM). After growing for an additional 3 h, the cells were collected by centrifugation (15 min at 11,000g) at 4 °C, and stored at -80 °C. Typically, ~11.8 g of cells were collected from eight 550-mL cultures.
The purification protocol was adapted from previously described ones and modified as follows (15,21). In a typical procedure, cells (~13.4 g) were thawed and suspended in ~40 mL of 20 mM HEPES buffer (pH 7.6) containing 0.1 M KCl (Buffer A). The cells were sonicated and centrifuged as described (15,21), to yield ~38 mL of supernatant. The supernatant was diluted to 120 mL with Buffer A, placed on ice and set stirring while (NH4)2SO4 (~31g) was added slowly to make a 45% saturated solution. After stirring for 30 min, the sample was centrifuged (10 min at 10,000 rpm) and the pellet was discarded. The supernatant was loaded onto a Phenyl-Sepharose column (8 mL of resin) equilibrated with 45% saturated (NH4)2SO4 in 20 mM HEPES buffer, pH 7.6. After the sample loaded, the column was washed with the equilibrating buffer (40 mL) followed by a linear gradient [160 mL total, 80 mL of 45% saturated (NH4)2SO4 in 20 mM HEPES buffer, pH 7.6, to 80 mL of buffer with no (NH4)2SO4]. Fractions (~0.8 mL) were collected and analyzed for activity using 2-hydroxymuconate (4). Fractions containing the highest activity were pooled and diluted 20-fold with 20 mM HEPES buffer (pH 7.6). The resulting solution was loaded onto a DEAE-Sepharose column (8 mL of resin) that had been equilibrated with 20 mM HEPES buffer (pH 7.6) containing 0.1 M KCl. After washing the column with 5 column volumes of the equilibrating buffer, a linear gradient (160 mL total, 0.1-0.5 M KCl in 20 mM HEPES buffer, pH 7.6) was used to elute the protein. Active fractions were identified by their activity with 4 and examined for purity using tricine SDS-PAGE. Typically, this procedure yields ~19 mg of TomN purified to >95% homogeneity (as assessed by SDS-PAGE). The enzyme or mutant enzyme was exchanged into the assay buffer using a PD-10 column, per the manufacturer’s instructions.
Enzyme Assays and Kinetic Studies Using TomN and TomN Mutants
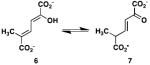
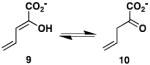
The activities of the wild type TomN were examined using 4, 6, 8, 9, 12, and 15 (Scheme 3). TomN processed all except for 8. Accordingly, the kinetic parameters were measured for the substrates processed in 10 mM potassium phosphate buffer, pH 7.3, as follows, and are reported in Table 1. The ketonization of 4 to 5 (by TomN) was measured by following the increase in absorbance at 236 nm (ε = 6580 M-1 cm-1) using substrate concentrations ranging from 20-200 μM with an enzyme concentration of 4 nM (4). The ketonization of 6 to 7 (by TomN and 4-OT) was measured by following the increase in absorbance at 236 nm (ε = 18,500 M-1 cm-1) using substrate concentrations ranging from 40-400 μM with enzyme concentrations of 317 nM (TomN) and 18 nM (4-OT) (4). For 6, the composition of an equilibrium mixture [43% 6, 24% 7, and 33% of 5-(methyl)-2-oxo-4-hexenedioate] was determined by 1H NMR spectroscopy. The ketonization of 9 to 10 was measured by following the decrease in absorbance at 265 nm (ε = 12,700 M-1 cm-1) using substrate concentrations ranging from 20-160 μM with enzyme concentrations of 3000 nM (TomN) or 18 nM (4-OT). The ketonization of 12 to 13 was measured by following the decrease in absorbance at 276 nm (ε = 15,900 M-1 cm-1) using substrate concentrations ranging from 20-100 μM with an enzyme concentration of 92 nM (19) (data not shown). The ketonization of 15 to 16 was measured by following the decrease in absorbance at 284 nm (ε = 11,500 M-1 cm-1) using substrate concentrations ranging from 10-100 μM with an enzyme concentration of 160 nM (27). Stock solutions (20 mM) of 2-hydroxymuconate (4), 5-(methyl)-2-hydroxymuconate (6), 2-hydroxy-2,4-pentadienoate (9), 2-hydroxy-2,4-heptadiene-1,7-dioate (12), and phenylenolpyruvate (15) were made up in ethanol. Reactions were initiated by the addition of substrate. The TomN mutants were assayed following the ketonization of 4 to 5 (at 236 nm) using substrate concentrations ranging from 20-200 μM with enzyme concentrations of 2.2 μM (P1A), 0.73 μM (R11A), 7.6 μM (R39A), and 40 nM (R61A) and are reported in Table 2.
Scheme 3.
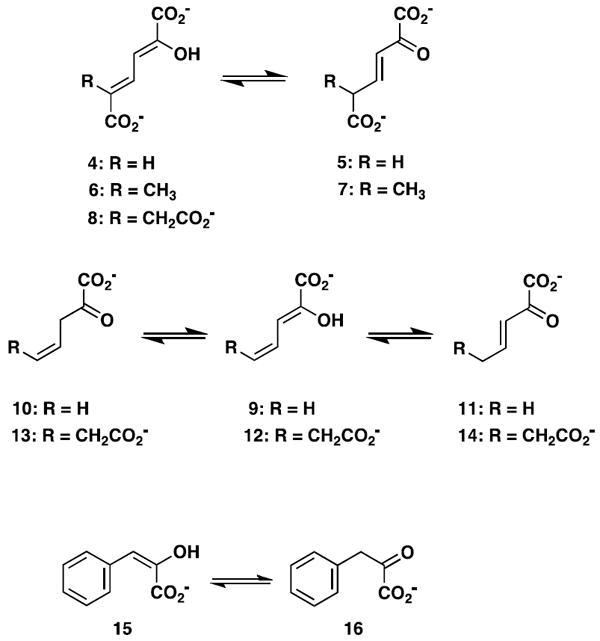
Known tautomerase superfamily substrates examined as substrates for TomN.
Table 1.
Kinetic Parameters for TomN, 4-OT, and hh4-OT Using Dienol and Enol Substratesa
| Reaction | Enzyme | kcat (s-1) | Km (μM) | kcat/Km (M-1 s-1) |
|---|---|---|---|---|
|
TomNb | 1850 ± 630 | 512 ± 225 | 3.6 × 106 |
| 4-OTc | 4000 ± 182 | 62 ± 8 | 6.5 × 107 | |
| hh4-OTc | 3000 ± 100 | 70 ± 8 | 4.3 × 107 | |
|
TomN | 8.8 ± 0.7 | 306 ± 45 | 2.9 × 104 |
| 4-OT | 111 ± 6 | 125 ± 17 | 8.9 × 105 | |
|
TomN | 0.9 ± 0.13 | 231 ± 41 | 3.9 × 103 |
| 4-OT | 176 ± 20 | 160 ± 26 | 1.1 × 106 | |
|
|
TomN | 16 ± 3.0 | 334 ± 90 | 4.8 × 104 |
| 4-OT | 73 ± 6 | 199 ± 23 | 3.7 × 105 | |
| hh4-OTc | 13 ± 1 | 121 ± 20 | 1.1 × 105 |
The steady-state kinetic parameters were determined under the conditions described in the text.
Errors are standard deviations.
The kinetic parameters are from reference 15.
Table 2.
Kinetic Parameters for TomN, 4-OT, and Mutants using (4)a.
| enzyme | kcat (s-1) | Km (μM) | kcat/Km (M-1 s-1) |
|---|---|---|---|
| TomNb | 1850 ± 630 | 512 ± 225 | 3.6 × 106 |
| 4-OT | 4000 ± 182 | 62 ± 8 | 6.5 × 107 |
| P1A-TomN | 1.5 ± 0.1 | 180 ± 10 | 8.3 × 103 |
| P1A-4-OTc | 60 ± 3 | 100 ± 12 | 6.0 × 105 |
| R11A-TomN | 14 ± 3 | 1050 ± 240 | 1.3 × 104 |
| R11A-4-OTd | 40 ± 6 | 1600 ± 300 | 2.5 × 104 |
| R39A-TomN | 0.20 ± 0.02 | 440 ± 60 | 4.5 × 102 |
| R39A-4-OTd | 28 ± 2 | 290 ± 40 | 9.7 × 104 |
| R61A-TomN | 360 ± 5.0 | 780 ± 155 | 4.6 × 105 |
| R61A-4-OTd | 3500 ± 240 | 290 ± 40 | 1.2 × 107 |
The ketonization of 9 to 11 and 12 to 14 in the presence of TomN were examined under conditions that gave complete conversion of substrate to product (i.e., 9 to 11 and 12 to 14) using 4-OT. Accordingly, the enzyme (~40 μg) was added to a cuvette containing 10 mM potassium phosphate buffer, pH 7.3. The reaction was initiated by the addition of a 7-μL aliquot of 9 or 12 from a 20 mM stock solution, and followed for 10 min (9) or 2 min (12).
Inactivation of TomN by 2-Oxo-3-pentynoate (17)
2-Oxo-3-pentynoate (17, Scheme 4, 1.9 mg) was dissolved in 0.3 mL of 100 mM Na2HPO4 buffer (pH ~ 9.5) and then diluted by the addition of water (0.7 mL). After the addition of 17, the pH of the reaction mixture decreased to ~ 7. A 50-μL aliquot of this stock solution was added to a 10 mL solution of TomN (~1 mg/mL), the solution inverted, and the resulting mixture was incubated at 4 °C for ~ 15 min. An aliquot (2 μL) was removed and assayed for activity using 4. Subsequently, an additional 25-μL aliquot of the inhibitor stock solution was added to the enzyme solution, the solution was inverted, and the resulting mixture was incubated at 4 °C for 15 min. An aliquot (2 μL) was removed and assayed for activity using 4. A final 15-μL aliquot of the inhibitor stock solution was added, and the mixture was treated as described above. The activity assay showed that TomN was inactivated by a total of 90 μL of the inhibitor solution. The inactivated enzyme was concentrated to 1 mL in an Amicon stirred cell concentrator (1,000 MW cutoff). The resulting solution was desalted by a PD-10 Sephadex G-25 gel filtration column pre-washed with ~50 mL of 10 mM NH4HCO3 buffer (pH ~8). Fractions (0.3 mL) were tested for protein content using the Bradford reagent (28). Fractions containing the highest protein concentrations were combined and an aliquot directly infused in the ESI mass spectrometer.
Scheme 4.
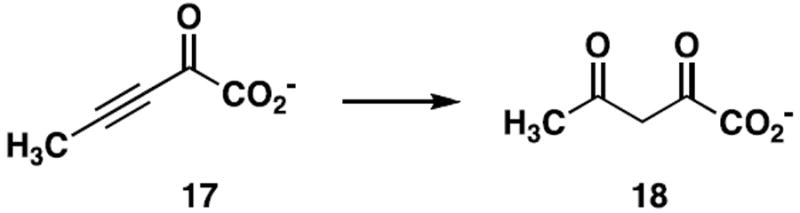
The conversion of 2-oxo-3-pentynoate to acetopyruvate, a diagnostic reaction for tautomerase superfamily members with a cationic Pro-1.
Peptide Mapping and MALDI-MS Analysis
A quantity of TomN (200 μL of a 5.5 mg/mL solution in 10 mM KH2PO4 buffer, pH 7.3) was incubated with 17 (50 μL of a 7 mg/mL solution in 100 mM Na2HPO4 buffer, pH ~7) for 30 min. An aliquot (50 μL) of a NaBH4 solution (100 mg in 100 mM Na2HPO4 buffer, pH ~10) was added. After 1 h, the solution was concentrated and exchanged into 100 mM NaH2PO4 buffer, pH 7.0, (a total of 3×), using an Amicon Ultra (3,000 MW cutoff) centrifugal filter unit. The residual liquid was diluted to 200 μL with 100 mM NaH2PO4 buffer, pH 7.0, and a second aliquot (50 μL) of the solution of 17 was added. After 30 min, 50 μL of the NaBH4 solution was added. After 1 h, the mixture was desalted as described above and analyzed by electrospray ion-trap spectrometry (ESI-MS) to verify covalent modification of the protein. Subsequently, samples of unmodified or modified TomN (50 μL each) were concentrated to dryness using a Speed Vac concentrator and the residue was dissolved in 3 μL of a saturated guanidine hydrochloride solution. After a 1 h incubation period at 37 °C, the individual mixtures were diluted to 30 μL with 10 mM NaH2PO4 buffer, pH 4.0, and treated with 20 μL of a protease V-8 solution (50 μg dissolved in 50 μL of 10 mM NaH2PO4 buffer, pH 4.0). After a 48 h incubation period at 37 °C, the resulting peptide mixtures were analyzed by MALDI-MS, as previously described (14).
Crystallization and Structural Determination of TomN
TomN was purified as described above, and then concentrated to 10 mg/mL in 20 mM HEPES buffer, pH 7.6, containing 300 mM KCl. Several crystallization conditions were identified by the sitting drop method of vapor diffusion. The two conditions resulting in the best diffraction-quality crystals used in this specific study were 100 mM magnesium acetate/100 mM sodium acetate, 5%-21% PEG 8000, pH 4.5, and 100 mM calcium acetate/100 mM sodium acetate, 1%-13% PEG 4000, pH 4.5. A crystal from the crystallization drop was transferred to mother liquor with 30% (v/v) glycerol as cryoprotectant before being frozen in liquid nitrogen for data collection. The X-ray diffraction data were collected at the wavelength of 1 Å at ~100K on beam line 8.3.1 of Advanced Light Sources (ALS) (Berkeley, CA). The data were processed and scaled using the software HKL2000 (29). The data collection statistics are summarized in Table 3.
Table 3.
Data Collection and Refinement Statistics for TomN
| Data Collection | |
|---|---|
| Space Group | I4132 |
| Cell Dimensions | |
| a, b, c (Å) | 117.637, 117.637, 117.637 |
| α, β, γ (°) | 90, 90, 90 |
| Resolution (Å) | 83.2-1.4 (1.42 -1.40) |
| Rsym (%) | 7.5 (22.5)a |
| I/σI | 76.9 (1.8)a |
| Completeness (%) | 100 (100)a |
|
| |
| Refinement | |
|
| |
| Resolution (Å) | 83.2-1.4 |
| No. Reflections | 26052 |
| Rwork/Rfree (%) | 22.4/25.6 |
| Number of atoms | |
| Protein | 964 |
| Water | 172 |
| B factors (Å2) | |
| Protein | 18.33 |
| Water | 35.07 |
| RMS Deviation | |
| Bond lengths (Å) | 0.028 |
| Bond angles (°) | 2.34 |
| Ramachandran Plot (%) | |
| Residues in most favored regions | 96.2 |
| Residues in additional allowed regions | 3.8 |
| Residues in generously allowed regions | 0 |
| Residues in disallowed regions | 0 |
The last resolution shell is shown in parentheses.
The TomN structure was determined by molecular replacement (MR) using the 4-OT model (PDB Code: 1BJP) as the search model. MR solutions were refined by REFMAC in the CCP4 Program Suite (30). The final models were evaluated by PROCHECK (31). Refinement statistics are summarized in Table 3. The Figures were prepared with PyMol (32), as noted in the Figure legends.
RESULTS
Identification of TomN as a Tautomerase Superfamily Member
The TomN sequence was uncovered by a BLAST search of the NCBI database using the sequence of the α- subunit of the heterohexamer 4-OT (hh4-OT) from Chloroflexus aurantiacus J-10-fl as the query sequence (15)3. Tom N shows 48% sequence identity with the α-subunit of hh4-OT (65% similarity) and 33% sequence identity with the P. putida mt-2 4-OT (58% similarity) (15). The TomN sequence has Pro-1, Arg-11, and Arg-39, which have been identified as critical residues for 4-OT and hh4-OT activity. Like the hh4-OT, a tryptophan residue replaces Phe-50 of 4-OT in TomN (15).
TomN is most similar to the α-subunits of the six previously identified hh4-OT homologues (38-50% sequence identity and 51-69% sequence similarity), followed by the 4-OT isozymes from P. putida mt-2 and P. sp CF600 (30% sequence identity and 59% sequence similarity with the P. sp CF600) (15). TomN has less similarity with the β-subunits of these hh4-OT homologues (27-37% sequence identity and 41-48% sequence similarity). TomN also showed similarity with a 4-OT domain in a putative indigoidine synthase (IndC) from Streptomyces clavuligerus (27% sequence identity and 38% sequence similarity) and a related hypothetical protein (designated PAU_02638) from the human pathogen, Photothabdus asymbiotica, (33% sequence identity and 67% sequence similarity) (33,34). Indigoidine is a blue pigment that may protect the host organism from oxidative stress and pathogenicity. Finally, TomN has 29% sequence identity and 47% sequence similarity with the α-subunit of CaaD and no detectable identity or similarity with the β-subunit of trans-3-chloroacrylic acid dehalogenase (CaaD), another known heterohexamer in the tautomerase superfamily (35).
Kinetic Characterization of TomN
The proposed substrate for TomN (i.e., 2 in Scheme 1) is not available (1). Hence, the TomN protein was examined for activity with known tautomerase superfamily substrates including 2-hydroxymuconate (4, Scheme 3), 5-(methyl)-2-hydroxymuconate (6), 5-(carboxymethyl)-2-hydroxymuconate (8), 2-hydroxy-2,4-pentadienoate (9), 2-hydroxy-2,4-heptadiene-1,7-dioate (12), and phenylenolpyruvate (15) (36,37). TomN showed 1,5-keto-enol tautomerization activity with 4 and 6, and 1,3-keto-enol tautomerization activities with 6 (data not shown) 9, and 15 (Table 1). The enzyme does not process 8. TomN processes 12 to 13, but the kinetic data could not be fit to the Michaelis-Menten equation because consistent results could not be obtained at higher substrate concentrations.
The catalytic efficiency of TomN with 4 is slightly less than those of the canonical 4-OT and the hh4-OT. The Km value is elevated and kcat value is reduced resulting in 18-fold (4-OT) and 12-fold lower kcat/Km values (4-OT and hh4-OT, respectively) (15). Likewise, the catalytic efficiency of TomN with 6 is slightly less than that of 4-OT. The Km value is elevated (2.5-fold) and kcat value is reduced (12.6-fold) resulting in a 31-fold lower kcat/Km value. The catalytic efficiency of 4-OT using 9, and converting it to 10, is ~280-fold higher than that of TomN. The difference is primarily due to the 195-fold higher kcat value measured for 4-OT. TomN does not process 9 to 11 or 12 to 14 at concentrations where the analogous 4-OT-catalyzed reactions have converted all substrate to product. The catalytic efficiency of TomN with 15 is comparable to that of the hh4-OT, but ~8-fold less than that of 4-OT. The difference is due primarily to the 5-fold higher kcat value measured for 4-OT.
Kinetic Characterization of the TomN Mutants
Our previous work on 4-OT and hh4-OT indicated that Pro-1, Arg-11, and Arg-39 (βPro-1, αArg-12, and αArg-40 in hh4-OT) played key roles in the enzyme-catalyzed reaction using 4 (7-12,15). Hence, these residues were replaced by alanine in TomN individually, and the kinetic parameters of the resulting mutant enzymes were determined using 4 as substrate (Table 2). There are significant effects on the kcat and kcat/Km values. Replacing Pro-1, Arg-11, and Arg-39 with alanines results respectively in 1230-fold, 130-fold, and 9250-fold decreases in kcat. Less significant changes are seen in the Km values (a 2.3-fold decrease for the P1A mutant, a 2-fold increase for the R11A mutant, and a comparable value for the R39A mutant). As a result, the kcat/Km values for the three mutant enzymes are down 430-fold, 280-fold, and 8000-fold, and are a reflection (primarily) of the decreased kcat. The most notable effects are the increase in the Km value for the R11A mutant and the marked decrease in the kcat value for the R39A mutant.
The R61A mutant of TomN was also constructed and examined for activity using 4. Our previous work on 4-OT did not implicate this residue in the mechanism (11,20), but the position of Arg-61 in the TomN crystal structure (vide infra) suggested a possible contribution. However, the changes in the kinetic parameters for the R61A mutant are not significant: there is a 5-fold decrease in kcat and little change in Km value. The overall result is an 8-fold reduction in the kcat/Km value. This result suggests that Arg-61 plays a relatively marginal role in catalysis even though it is in close proximity to the active site. This analysis does not rule out a more direct role for Arg-61 with the biological substrate.
Inactivation of TomN by 2-Oxo-3-pentynoate (17)
Previous work showed that the reaction of 17 (Scheme 4) with tautomerase superfamily members reflects the ionization state of Pro-1 (36). The compound is a potent active-site-directed irreversible inhibitor of 4-OT (20), whereas CaaD, cis-3-chloroacrylic acid dehalogenase (cis-CaaD), and malonate semialdehyde decarboxylase (MSAD) catalyze the hydration of 17 to yield acetopyruvate (18) (Scheme 4) (23,36,37). CaaD, cis-CaaD, and MSAD are found in a pathway for the catabolism of 1,3-dichloropropene (37). The different reactivities of 17 correlate with the different pKa values of Pro-1. In 4-OT, Pro-1 has a pKa of ~6.4 so that it functions largely as a base and attacks 17 at C-4 in a Michael-type reaction (20). In the three other enzymes, Pro-1 is largely cationic and cannot function as a nucleophile (23,36,37). Instead, the compound is hydrated because the active site is designed to carry out a hydration reaction. We find that TomN is inactivated by 17. Moreover, MALDI-MS analysis of the peptide mixture resulting after proteolysis (using protease V-8) indicates that Pro-1 is the site of covalent modification. The spectrum shows two signals: one signal corresponds to the unmodified peptide Pro-1-Glu-9 (1053.52 Da) and the other signal corresponds to the same peptide modified after incubation with 17 (1169.55 Da). The mass difference between the two peaks is 116.03 Da, which is consistent with the covalent modification of TomN by 17 at Pro-1 (20). These observations indicate that TomN, like 4-OT, functions as a tautomerase, and that Pro-1 likely has a pKa value comparable to that of Pro-1 in 4-OT.
Crystal Structure of TomN
The structure shows that TomN is constructed from the signature tautomerase superfamily β–α–β fold (Figure 1A) (7,37,38). Two TomN monomers align in an inverted manner to form a dimer (Figure 1A). The quaternary structure can be viewed as a trimer of dimers where the inner core of the hexamer is formed by the parallel β-strands that dimerize at the interface (Figure 1B). The long α–helices (α1 from each dimer) constitute the outer wall of the hexamer and have short C-terminal α–helices (α2 from each monomer) that extend to the neighboring dimer and provide a lid to the active site for the neighboring molecule (Figure 1C). The β–sheets from two monomers form a four-stranded anti-parallel β–sheet on one side while two anti-parallel α–helices are on the other side. The hydrogen bonds formed by the two anti-parallel β–sheets stabilize the dimer. The core structure is comparable to that of 4-OT and the hh4-OT (15,20).
Figure 1.
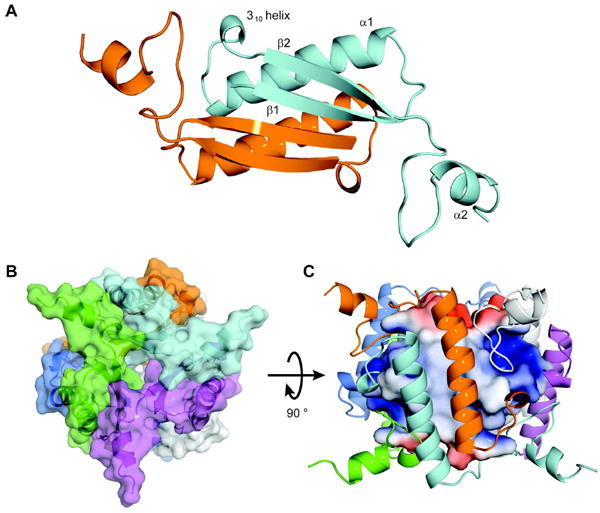
Structural Overview of TomN. (A) Model of the TomN dimer showing the antiparallel β-sheets in front of the two α-helices. The two monomers are colored orange and cyan, respectively. The β–α–β signature fold for one monomer is shown as β1, α1 and β2. The short 310 helix that precedes the β2 strand and the C-terminal α2 helix are also shown. (B) Overall architecture of the TomN hexamer. A top view of the TomN hexamer where the six monomers making up the homohexamer are colored orange, cyan, blue, green, white, and magenta (counter-clockwise). (C) A side view of the TomN hexamer (90° rotation from B) with the outer α-helices colored as in B. The inner β-strands are shown as the electrostatic surface potentials, where blue indicates positive charge, red indicates negative charge, and white indicates hydrophobic regions. The figures were generated in PyMoL (32). The surfaces were generated with the Adaptive Poisson-Boltzman Solver (APBS) (39) using the AMBER forcefield (APBS Tools 2.1 PyMoL plugin, Michael G. Lerner) and the conversion program PDB2PQR (40,41).
The active site in both TomN (Figure 2A) and 4-OT (Figure 2B) is a cavity on the surface created by the ends of the β–strands of two neighboring dimers. In TomN, the active site contains Pro-1 and Arg-39 from one monomer, Arg-11’, Arg-61’, and Trp-50’ from the neighboring monomer of the dimer, and Arg-39” from the neighboring dimer (Figure 2A)4. This active site configuration mirrors that of 4-OT (with the exception of Trp-50’ and Arg-61’). Each active site is polarized with the positively charged arginine residues at either end (i.e., Arg-11’ at one end and Arg-39 at the other end) and the catalytic Pro-1 roughly spaced in between. Importantly, the highly similar structure and orientation of the active sites are entirely consistent with the preceding sequence, kinetic, and mutagenic analysis.
Figure 2.
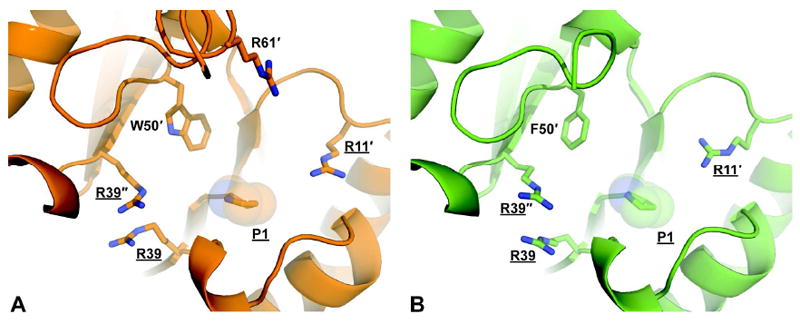
A comparison of the TomN (orange) and 4-OT (green) active sites. (A) The active site of TomN showing the catalytically required residues (underlined) from three different monomers (designated by primes) as sticks. (B) The active site of 4-OT showing the catalytically required residues (underlined) from three different monomers (designated by primes) as sticks.
A detailed examination of the active sites shows that the residues are mostly identical or very similar (Figures 3A and 3B). The larger bulk of Trp-50 (Phe-50 in 4-OT) is accommodated by the replacement of Met-45 (4-OT) with Thr-45. However, one difference (Val-52 in place of Ile-52) may render the TomN active site slightly more accommodating than that of 4-OT (Figures 3A and 3B). Overall, these changes suggest subtle differences in the active sites. The electrostatic potential surface shows that the charge distribution (positive, negative, and hydrophobic) is comparable in the two active sites.
Figure 3.
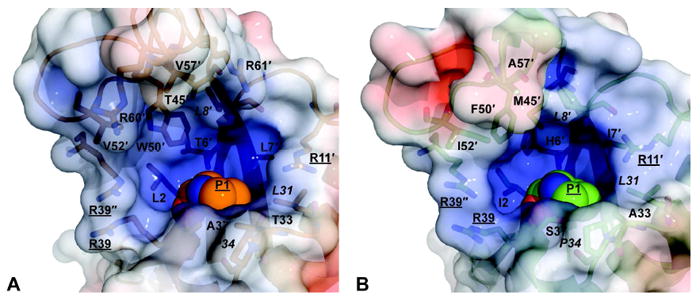
A detailed comparison of the TomN and 4-OT active sites with the electrostatic surface potentials shown. (A) Electrostatic surface potential of the TomN active site. Residues that define the active site pocket are represented as sticks and are labeled as in Figure 2. The italicized labels indicate identical residues between the two proteins. (B) Electrostatic surface potential of the 4-OT active site. In both panels, blue indicates positive charge, red indicates negative charge, and white indicates hydrophobic regions. Surfaces were generated as described for Figure 1.
DISCUSSION
Three 4-OTs (defined by their ability to convert 4 to 5) have now been characterized: the homohexamer 4-OT from P. putida mt-2, the heterohexamer 4-OT from C. aurantiacus J-10-fl, and the homohexamer TomN from Streptomyces achromogenes (4,6,7,15)5. Although they all catalyze the canonical 4-OT reaction with similar efficiencies, there are two distinctions. First, TomN probably has a different biological substrate. Second, the homo- and heterohexamer 4-OTs are found in catabolic pathways and TomN is found in a biosynthetic pathway that, at first glance, has a few parallel reactions to those found in the catabolic pathways (1,6,15).
The P. putida mt-2 4-OT is part of a so-called catechol meta-fission pathway, which is a bacterial catabolic pathway that transforms simple aromatic hydrocarbons such as benzene, toluene, and xylenes into useful cellular intermediates (6,7). The presence of this pathway enables the host bacteria to use these compounds as their sole sources of carbon and energy. The enzyme is a homohexamer with six active sites, which are made up of residues from three adjoining monomers (6,20).
A structural variation of the homohexamer 4-OT was discovered in the thermophilic bacteria C. aurantiacus J-10-fl (15). The hh4-OT consists of α- and β-subunits (forming a functional α,β-dimer), where the α-subunit, which does not have an amino-terminal proline, is annotated as a tautomerase-like protein (15). The β-subunit provides the catalytic amino-terminal proline. The hh4-OT is part of a meta-fission pathway based on its genomic context, but this has not been confirmed by biochemical characterization of the enzymes. Other than the quaternary structure, the factors conferring thermostability, and the absence of the low level dehalogenase activity, the mechanistic and kinetic properties of the hh4-OT largely resemble those of 4-OT (15). In both 4-OT and the hh4-OT, Pro-1 (β-Pro-1), Arg-11 (α-Arg-12), and Arg-39 (α-Arg-40) have been identified as key catalytic residues.
TomN is found in a biosynthetic pathway, but some of the initial reaction chemistry for the proposed ring C biosynthesis seemingly parallels that typically found in meta-fission degradative pathways (Scheme 5) (1). For example, the second step in the proposed five-step biosynthetic sequence involves oxidative extradiol aromatic ring cleavage of L-Dopa (42). The third step is an enzyme-catalyzed hydrolytic cleavage of the dienol ring-fission product (43). TomN is then proposed to tautomerize the product (i.e., 2). However, these assigned functions are tentative, and with the exception of TomN, the biochemical characterization of the enzymes is only in the very early stages. Moreover, the dioxygenase (TomH) and hydrolase (TomK) show no detectable sequence identity with the enzymes in various meta-fission pathways. Like 4-OT, TomN is a homohexamer, and Pro-1 and two arginine residues are critical for activity.
Scheme 5.
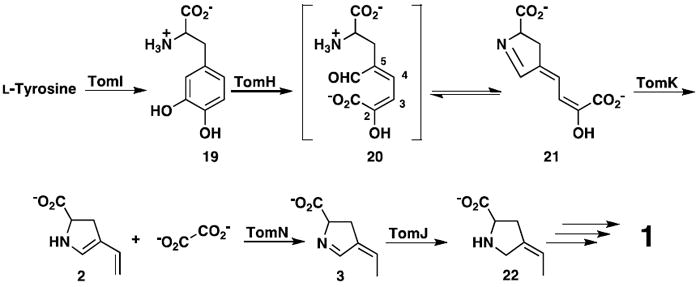
The proposed sequence of events converting L-tyrosine to the C ring of tomaymycin (1).
The mutagenesis experiments indicate that Pro-1, Arg-11, and Arg-39 in TomN play analogous roles to those proposed for these residues in the P. putida mt-2 4-OT and the hh4-OT (in the conversion of 4 to 5) (Table 2). Accordingly, replacing Pro-1 with an alanine affects kcat more significantly than Km, and this substantial decrease is the primary factor responsible for the decrease in kcat/Km. The decrease in kcat can be ascribed to an effect on the reaction chemistry, product release, or both. Alanine is less basic and more flexible than proline so that the decrease in basicity along with the sub-optimal positioning of the base could account for the reduced activity of the mutant (10). The decrease in Km could be due to the removal of the “bulky” ring of proline, making more room for the substrate. Similar results were obtained for the P1A mutant of 4-OT and the hh4-OT, except the decrease in kcat for TomN is ~10-fold greater (10,15). The results are consistent with the role of Pro-1 as a base in the reaction. Changing Arg-11 to an alanine impacts both Km and kcat, and follows the same trend observed for 4-OT and the hh4-OT. As would be anticipated for the removal of the group involved in C-6 carboxylate binding, the Km increases. Likewise, the kcat decreases because alanine cannot draw electron density away from C-5 and facilitate protonation. Finally, changing Arg-39 to an alanine has a profound effect on kcat and only a minimal effect on Km. The decrease in kcat is much more dramatic than that observed for the other 4-OTs, suggesting that Arg-39 plays a much more substantial role in catalysis (11,15).
In 4-OT, Pro-1 can function as a base because it has a pKa value of ~6.4, which is due in part to the presence of Phe-50 (13). The side chain of Phe-50 creates a nearby pocket of hydrophobicity. The inactivation of TomN by 2-oxo-3-pentynoate (17) by the covalent modification of the prolyl nitrogen suggests that the pKa of Pro-1 is comparable to that of 4-OT (also inactivated by 17). The crystal structure places Trp-50 in an identical position to Phe-50 in 4-OT, suggesting a similar role (Figures 2 and 3).
Two observations from the substrate specificity studies may provide clues about the properties of a biological substrate for TomN. First, 4 is an excellent substrate for TomN. However, with the exception of 4 and 6, TomN is not particularly efficient at carrying out 1,5-keto-enol tautomerization reactions (to produce the so-called conjugated ketones) with two other dienols (9 and 12) used in this study, whereas 4-OT is (11,16,19). This observation is somewhat surprising, but it could suggest that subtle differences in TomN preclude the efficient formation of conjugated ketones from dienols and that the biological function for TomN does not involve this type of reaction. Second, TomN, like 4-OT, processes mono- and diacid compounds, although for both enzymes the monoacids (9 and 15) are not processed as efficiently as the diacids (4 and 6) (15). A future stereochemical analysis will show if the substrates bind similarly in the two active sites (5,11,16,17).
The crystal structure of TomN shows that it largely resembles those of 4-OT and the hh4-OT, with main chain RMS deviations of 0.8 Å and 1.1 Å, respectively (7,15,20). The active sites are nearly identical and the key residues are positionally conserved (Figure 2). The structure of TomN and the conservation of essential catalytic residues suggest that similar catalytic and substrate recognition mechanisms are used in the three enzymes, and are fully consistent with the kinetic and mutagenic results.
On the basis of the kinetic, mechanistic, and structural results presented here, it is apparent that TomN functions very much like a canonical 4-OT, processing 4 to 5. Subtle differences in the active site of TomN could account for the different kinetic parameters obtained for the various substrates. These results have two implications. First, the striking similarities question whether the proposed substrate for TomN (i.e., 2), is, in fact, the biological substrate, or whether TomN processes a substrate that more closely resembles 4. Second, if 2 is not the substrate for TomN, then the proposed role for the TomN reaction in the biosynthetic pathway for the C ring of 1 may not be correct, and the proposed steps leading to ring formation may have to be re-examined.
In the currently proposed version, tyrosine is hydroxylated to yield L-Dopa (19, Scheme 5) (1)6. L-Dopa undergoes meta-fission (catalyzed by TomH) to produce (presumably) initially 20, which is proposed to cyclize to the dihydropyrrolidine species 21 (1). The subsequent TomK-catalyzed reaction yields oxalate and 2, the putative substrate for TomN. Tautomerization by TomN affords 3, which is reduced by the F420-dependent enzyme, TomJ, to generate 22. A series of reactions then incorporates 22 into the tricyclic ring system to yield 1 (1). It should be emphasized that these assignments are tentative, and are not based on extensive biochemical characterization of the individual enzymes.
The proposed conversion of 19 to 22 poses three potential problems (Scheme 5). First, the electron donating properties of the 2-hydroxy group (in 20) could attenuate the electrophilicity of the aldehyde carbonyl group and preclude a spontaneous ring closure (to 21) or make spontaneous ring closure a slow process (44). Second, dienols (such as 20 or 21) are likely in equilibrium with their 1,3- and 1,5-keto-enol tautomers (23 and 24, Scheme 6) (45). Hydrolytic cleavage (by TomK) most reasonably occurs via the 1,3 tautomer (i.e., 23), yet the reaction with 4 suggests that 24 might be favored. Third, compounds 21 and 3 could be in equilibrium with the acyclic counterparts such that the assignment of a substrate to an enzyme is compounded further. The reduction of 3 by TomJ might ultimately drive the pathway forward to converge on 22.
Scheme 6.
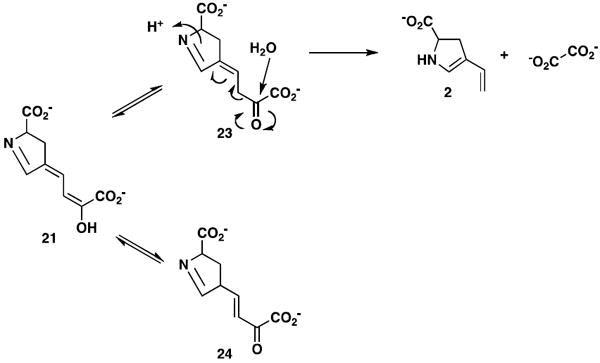
Tautomers of 21 and carbon-carbon bond fission of 23.
One possible alternative reaction sequence, which addresses some issues, is shown in Scheme 7. Ring opening again yields 20, which presumably exists in the s-trans form. TomN could then tautomerize 20 (which has the 2-hydroxymuconate framework) to 25 or 26. Although protonation can occur at C-3 (to yield 25) or C-5 (to yield 26), steric hindrance at C-5 (the aldehyde and amino acid moieties) might preclude C-5 protonation and favor formation of 25 (45). Moreover, TomN could favor one tautomer (i.e., 25) so that a wasteful non-enzymatic partitioning of the reactive 20 to both isomers is minimized. If the reaction proceeds via 25, TomK can then carry out a hydrolytic cleavage and produce 27 and oxalate, a known product of the pathway. A spontaneous ring closure of 27 to 28 is more likely (than that of 20 to 21), with 27 and 28 in equilibrium. If 3 is the substrate for TomJ (and 22 is the product), then a 1,3-allylic rearrangement of 28 to 3 is required at this stage. If and how this happens are not known. In one variation of this pathway, TomK could be responsible for the ketonization of 20 to 25, the hydrolytic cleavage of 25 to 27, and ring closure (43). In this scenario, TomN is available to convert 28 to 3. Clearly, the chemistry of the pathway intermediates is complex and must be sorted out before definitive roles can be assigned to the enzymes.
Scheme 7.
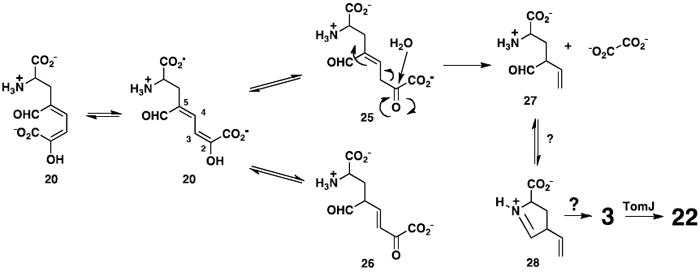
An alternative pathway for the conversion of 20 to 22.
It remains a possibility that TomN processes 2 to 3. The substrate specificity studies do not rule out the processing of monoacids. The appropriate experiment will be carried out when and if 2 can be generated (most likely, in situ from 19). In the absence of the genomic and pathway context, TomN might have been misannotated as a 4-OT based on its sequence similarity to the 4-OT family and its activity with 4. This raises the question of whether several enzymes annotated as “4-OTs” that lack an operon context actually carry out different biological reactions. This observation underscores the difficulty of functional annotation because the subtle sequence and structural features defining the individual reaction and substrate specificities are not yet known.
One reason for characterization of the tomaymycin biosynthetic cluster is to produce more potent tomaymycin analogues that lack the dose-limiting toxicity (1). If this is to become a reality, the actual sequence of events must be established for the pathway (including the steps resulting in C ring construction). In addition, a more detailed understanding of the chemistry must be obtained including a better understanding of how substituents affect the reactivity of 20 and 21 (if 21 is generated) and the equilibrium of acyclic and cyclic forms of proposed intermediates along the pathway (e.g., 20 and 21 or 27 and 28). Efforts are now underway to do this.
Acknowledgments
The protein mass spectrometry analysis was conducted in the Protein and Metabolite Analysis Facility. We thank Steve D. Sorey (Department of Chemistry, The University of Texas at Austin) for his expert assistance in the acquisition of the 1H NMR spectra.
Abbreviations
- CaaD and cis-CaaD
trans- and cis-3-chloroacrylic acid dehalogenase, respectively
- ESI
electrospray ion-trap
- HEPES
N-2-hydroxyethylpiperazine-N’-2-ethane sulfonate
- IPTG
isopropyl-β-d-thiogalactoside
- Kn
kanamycin
- LB
Luria-Bertani
- MALDI
matrix-assisted laser desorption/ionization
- MSAD
malonate semialdehyde decarboxylase
- MR
molecular replacement
- NMR
nuclear magnetic resonance
- 4-OT
4-oxalocrotonate tautomerase
- PEG
polyethylene glycol
- PCR
polymerase chain reaction
- PBD
pyrrolo[1,4]benzodiazepine
- RMS
root-mean-square
- SDS-PAGE
sodium dodecyl sulfate-polyacrylamide gel electrophoresis
Footnotes
This research was supported by the National Institutes of Health Grants GM-41239 (CPW) and GM-084473 (BG), a fellowship award (F32 GM089083) from the National Institute of General Medical Sciences (GKS), and the Robert A. Welch Foundation Grant F-1334 (CPW).
The proposed substrate for TomN (i.e., 2) is not available (1).
The sequence for the tomaymycin gene cluster is deposited in the GenBank and the gene for TomN annotated (1).
The primed residues refer to different subunits in TomN.
The 4-OT isozyme from P. sp CF600 is not considered in this discussion because it is so similar to the P. putida mt-2 4-OT (7).
Conner, K.L. and Gerratana, B., unpublished data, 2011.
The atomic coordinates and structure factors have been deposited with the Brookhaven Protein Data Bank (PDB code 3ry0).
References
- 1.Li W, Chou SC, Khullar A, Gerratana B. Cloning and characterization of the biosynthetic cluster for tomaymycin, an SJG-136 monomeric analog. Appl Environ Microbiol. 2009;75:2958–2963. doi: 10.1128/AEM.02325-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Li W, Khullar A, Chou SC, Sacramo A, Gerratana B. Biosynthesis of sibiromycin, a potent antitumor antibiotic. Appl Environ Microbiol. 2009;75:2869–2878. doi: 10.1128/AEM.02326-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gerratana B. Biosynthesis, synthesis, and biological activities of pyrrolobenzodiazepines. Med Res Rev. 2010 doi: 10.1002/med.20212. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Whitman CP, Aird BA, Gillespie WR, Stolowich NJ. Chemical and enzymatic ketonization of 2-hydroxymuconate, a conjugated enol. J Am Chem Soc. 1991;113:3154–3162. [Google Scholar]
- 5.Wang SC, Johnson WH, Jr, Czerwinski RM, Stamps SL, Whitman CP. Kinetic and stereochemical analysis of YwhB, a 4-oxalocrotonate tautomerase homologue in Bacillus subtilis: mechanistic implications for the YwhB-and 4-oxalocrotonate tautomerase-catalyzed reactions. Biochemistry. 2007;46:11919–11929. doi: 10.1021/bi701231a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harayama S, Rekik M, Ngai K-L, Ornston LN. Physically associated enzymes produce and metabolize 2-hydroxy-2,4-dienoate, a chemically unstable intermediate formed in catechol metabolism via meta cleavage in Pseudomonas putida. J Bacteriol. 1989;171:6251–6258. doi: 10.1128/jb.171.11.6251-6258.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Subramanya HS, Roper DI, Dauter Z, Dodson EJ, Davies GJ, Wilson KS, Wigley DB. Enzymatic ketonization of 2-hydroxymuconate: specificity and mechanism investigated by the crystal structures of two isomerases. Biochemistry. 1996;35:792–802. doi: 10.1021/bi951732k. [DOI] [PubMed] [Google Scholar]
- 8.Stivers JT, Abeygunawardana C, Mildvan AS, Hajipour G, Whitman CP, Chen LH. Catalytic role of the amino-terminal proline in 4-oxalocrotonate tautomerase: affinity labeling and heteronuclear NMR studies. Biochemistry. 1996;35:803–813. doi: 10.1021/bi951077g. [DOI] [PubMed] [Google Scholar]
- 9.Stivers JT, Abeygunawardana, Mildvan AS, Hajipour G, Whitman CP. 4-Oxalocrotonate tautomerase: pH dependences of catalysis and pKa values of active site residues. Biochemistry. 1996;35:814–823. doi: 10.1021/bi9510789. [DOI] [PubMed] [Google Scholar]
- 10.Czerwinski RM, Johnson WH, Jr, Whitman CP, Harris TK, Abeygunawardana C, Mildvan AS. Kinetic and structural effects of mutations of the catalytic amino-terminal proline in 4-oxalocrotonate tautomerase. Biochemistry. 1997;36:14551–14560. doi: 10.1021/bi971545h. [DOI] [PubMed] [Google Scholar]
- 11.Harris TK, Czerwinski RM, Johnson WH, Jr, Legler PM, Abeygunawardana C, Massiah MA, Stivers JT, Whitman CP, Mildvan AS. Kinetic, stereochemical, and structural effects of mutations of the active site arginine residues in 4-oxalocrotonate tautomerase. Biochemistry. 1999;38:12343–12357. doi: 10.1021/bi991116e. [DOI] [PubMed] [Google Scholar]
- 12.Czerwinski RM, Harris TK, Johnson WH, Jr, Legler PM, Stivers JT, Mildvan AS, Whitman CP. Effects of mutations of the active site arginine residues in 4-oxalocrotonate tautomerase on the pKa values of active site residues and on the pH dependence of catalysis. Biochemistry. 1999;38:12358–12366. doi: 10.1021/bi9911177. [DOI] [PubMed] [Google Scholar]
- 13.Czerwinski RM, Harris TK, Massiah MA, Mildvan AS, Whitman CP. The structural basis for the perturbed pKa of the catalytic base in 4-oxalocrotonate tautomerase: kinetic and structural effects of mutations of Phe-50. Biochemistry. 2001;40:1984–1995. doi: 10.1021/bi0024714. [DOI] [PubMed] [Google Scholar]
- 14.Wang SC, Johnson WH, Jr, Whitman CP. The 4-oxalocrotonate tautomerase- and YwhB-catalyzed hydration of 3E-haloacrylates: implications for evolution of new enzymatic activities. J Am Chem Soc. 2003;125:14282–14283. doi: 10.1021/ja0370948. [DOI] [PubMed] [Google Scholar]
- 15.Burks EA, Fleming CD, Mesecar AD, Whitman CP, Pegan SD. Kinetic and structural characterization of a heterohexamer 4-oxalocrotonate tautomerase from Chloroflexus aurantiacus J-10-fl: implications for functional and structural diversity in the tautomerase superfamily. Biochemistry. 2010;49:5016–5027. doi: 10.1021/bi100502z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lian H, Czerwinski RM, Stanley TM, Johnson WH, Jr, Watson RJ, Whitman CP. The contribution of the substrate’s carboxylate group to the mechanism of 4-oxalocrotonate tautomerase. Bioorg Chem. 1998;26:141–156. [Google Scholar]
- 17.Hajipour G, Johnson WH, Jr, Dauben PD, Stolowich NJ, Whitman CP. Chemical and enzymatic ketonization of 5-(carboxymethyl)-2-hydroxymuconate. J Am Chem Soc. 1993;115:3533–3542. [Google Scholar]
- 18.Stanley TM, Johnson WH, Jr, Burks EA, Whitman CP, Hwang C-C, Cook PF. Expression and stereochemical and isotope effect studies of active 4-oxalocrotonate decarboxylase. Biochemistry. 2000;39:718–726. doi: 10.1021/bi9918902. [DOI] [PubMed] [Google Scholar]
- 19.Burks EA, Johnson WH, Jr, Whitman CP. Stereochemical and isotopic labeling studies of 2-oxo-hept-4-ene-1,7-dioate hydratase: evidence for an enzyme-catalyzed ketonization step in the hydration reaction. J Am Chem Soc. 1998;120:7665–7675. [Google Scholar]
- 20.Taylor AB, Czerwinski RM, Johnson WH, Jr, Whitman CP, Hackert ML. Crystal structure of 4-oxalocrotonate tautomerase inactivated by 2-oxo-3-pentynoate at 2.4 Å resolution: analysis and implications for the mechanism of inactivation and catalysis. Biochemistry. 1998;37:14692–14700. doi: 10.1021/bi981607j. [DOI] [PubMed] [Google Scholar]
- 21.Poelarends GJ, Almrud JJ, Serrano H, Darty JE, Johnson WH, Jr, Hackert ML, Whitman CP. Evolution of enzymatic activity in the tautomerase superfamily: mechanistic and structural consequences of the L8R mutation in 4-oxalocrotonate tautomerase. Biochemistry. 2006;45:7700–7708. doi: 10.1021/bi0600603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1989. [Google Scholar]
- 23.Wang SC, Person MD, Johnson WH, Jr, Whitman CP. Reactions of trans-3-chloroacrylic acid dehalogenase with acetylene substrates: consequences of and evidence for a hydration reaction. Biochemistry. 2003;42:8762–8773. doi: 10.1021/bi034598+. [DOI] [PubMed] [Google Scholar]
- 24.Waddell WJ. A simple ultraviolet spectrophotometric method for the determination of protein. J Lab Clin Med. 1956;48:311–314. [PubMed] [Google Scholar]
- 25.Schagger H, von Jagow G. Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range of 1 to 100 kDa. Anal Biochem. 1987;166:368–379. doi: 10.1016/0003-2697(87)90587-2. [DOI] [PubMed] [Google Scholar]
- 26.Ho SN, Hunt HD, Horton RM, Pullen JK, Pease LR. Site-directed mutagenesis by overlap extension using the polymerase chain reaction. Gene. 1989;77:51–59. doi: 10.1016/0378-1119(89)90358-2. [DOI] [PubMed] [Google Scholar]
- 27.Taylor AB, Johnson WH, Jr, Czerwinski RM, Li H-S, Hackert ML, Whitman CP. Crystal structure of macrophage migration inhibitory factor complexed with (E)-2-fluoro-p-hydroxycinnamate at 1.8 Å resolution: implications for enzymatic catalysis and inhibition. Biochemistry. 1999;38:7444–7452. doi: 10.1021/bi9904048. [DOI] [PubMed] [Google Scholar]
- 28.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 29.Otwinowski Z, Minor W. Processing of x-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 30.Collaborative Computational Project, Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr Sect D: Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 31.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Crystallogr. 1993;26:283–291. [Google Scholar]
- 32.DeLano WL. The PyMol Molecular Graphics System. DeLano Scientific; San Carlos, CA: 2002. [Google Scholar]
- 33.Medema MH, Trefzer A, Kovalchuk A, van den Berg M, Müller U, Heijne W, Wu L, Alam MT, Ronning CM, Nierman WC, Bovenberg RA, Breitling R, Takano E. The sequence of a 1.8-mb bacterial linear plasmid reveals a rich evolutionary reservoir of secondary metabolic pathways. Genome Biol Evol. 2010;2:212–224. doi: 10.1093/gbe/evq013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wilkinson P, Waterfield NR, Crossman L, Corton C, Sanchez-Contreras M, Vlisidou I, Barron A, Bignell A, Clark L, Ormond D, Mayho M, Bason N, Smith F, Simmonds M, Churcher C, Harris D, Thompson NR, Quail M, Parkhill J, Ffrench-Constant RH. Comparative genomics of the emerging human pathogen Photorhabdus asymbiotica with the insect pathogen Photorhabdus luminescens. BMC Genomics. 2009;10:302. doi: 10.1186/1471-2164-10-302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Poelarends GJ, Saunier R, Janssen DB. trans-3-Chloroacrylic acid dehalogenase from Pseudomonas pavonaceae 170 shares structural and mechanistic similarities with 4-oxalocrotonate tautomerase. J Bacteriol. 2001;183:4269–4277. doi: 10.1128/JB.183.14.4269-4277.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Poelarends GJ, Serrano H, Johnson WH, Jr, Hoffman DW, Whitman CP. The hydratase activity of malonate semialdehyde decarboxylase: mechanistic and evolutionary implications. J Am Chem Soc. 2004;126:15658–15659. doi: 10.1021/ja044304n. [DOI] [PubMed] [Google Scholar]
- 37.Poelarends GJ, Veetil VP, Whitman CP. The chemical versatility of the β-α-β fold: catalytic promiscuity and divergent evolution in the tautomerase superfamily. Cell Mol Life Sci. 65:3606–3618. doi: 10.1007/s00018-008-8285-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Whitman CP. The 4-oxalocrotonate tautomerase family of enzymes: how nature makes new enzymes using a β-α-β structural motif. Arch Biochem Biophys. 2002;402:1–13. doi: 10.1016/S0003-9861(02)00052-8. [DOI] [PubMed] [Google Scholar]
- 39.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: application to microtubules and the ribosome. Proc Natl Acad Sci USA. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dolinsky TJ, Czodrowski P, Li H, Nielsen JE, Jensen JH, Klebe G, Baker NA. PDB2PQR: Expanding and upgrading automated preparation of biomolecular structures for molecular simulations. Nucleic Acids Res. 2007;35:W522–525. doi: 10.1093/nar/gkm276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dolinsky TJ, Nielsen JE, McCammon JA, Baker NA. PDB2PQR: an automated pipeline for the setup, execution, and analysis of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004;32:W665–W667. doi: 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bugg TD, Ramaswamy S. Non-heme iron-dependent dioxygenases: unravelling catalytic mechanisms for complex enzymatic oxidations. Curr Opin Chem Biol. 2008;12:134–140. doi: 10.1016/j.cbpa.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 43.Li C, Montgomery MG, Mohammed F, Li JJ, Wood SP, Bugg TD. Catalytic mechanism of C-C hydrolase MhpC from Escherichia coli: kinetic analysis of His263 and Ser110 site-directed mutants. J Mol Biol. 2005;346:241–251. doi: 10.1016/j.jmb.2004.11.032. [DOI] [PubMed] [Google Scholar]
- 44.Colabroy KL, Begley TP. The pyridine ring of NAD is formed by a nonenzymatic pericyclic reaction. J Am Chem Soc. 2005;127:840–841. doi: 10.1021/ja0446395. [DOI] [PubMed] [Google Scholar]
- 45.Whitman CP. Keto-enol tautomerization in enzymatic reactions. In: Barton D, Nakanishi K, Poulter CD, editors. Comprehensive Natural Products Chemistry. Vol. 5. Elsevier Science Ltd; Oxford: 1999. pp. 31–50. [Google Scholar]