

Abstract
Engineered zinc finger nucleases (ZFNs) are promising tools for genome manipulation and determining off-target cleavage sites of these enzymes is of great interest. We developed an in vitro selection method that interrogates 1011 DNA sequences for cleavage by active, dimeric ZFNs. The method revealed hundreds of thousands of DNA sequences, some present in the human genome, that can be cleaved in vitro by two ZFNs: CCR5-224 and VF2468, which target the endogenous human CCR5 and VEGF-A genes, respectively. Analysis of the identified sites in cultured human cells revealed CCR5-224-induced mutagenesis at nine off-target loci, though this remains to be tested in other relevant cell types. Similarly, we observed 31 off-target sites cleaved by VF2468 in cultured human cells. Our findings establish an energy compensation model of ZFN specificity in which excess binding energy contributes to off-target ZFN cleavage and suggest strategies for the improvement of future ZFN design.
Introduction
Zinc finger nucleases (ZFNs) are enzymes engineered to recognize and cleave desired target DNA sequences. A ZFN monomer consists of a zinc finger DNA-binding domain fused with a non-specific FokI restriction endonuclease cleavage domain1. Since the FokI nuclease domain must dimerize and bridge two DNA half-sites to cleave DNA2, ZFNs are designed to recognize two unique sequences flanking a spacer sequence of variable length and to cleave only when bound as a dimer to DNA. ZFNs have been used for genome engineering in a variety of organisms including mammals3–9 by stimulating either non-homologous end joining or homologous recombination. In addition to providing powerful research tools, ZFNs also have potential as gene therapy agents. Indeed, two ZFNs have recently entered clinical trials: one as part of an anti-HIV therapeutic approach (NCT00842634, NCT01044654, NCT01252641) and the other to modify cells used as anti-cancer therapeutics (NCT01082926).
DNA cleavage specificity is a crucial feature of ZFNs. The imperfect specificity of some engineered zinc fingers domains has been linked to cellular toxicity10 and therefore determining the specificities of ZFNs is of significant interest. ELISA assays11, microarrays12, a bacterial one-hybrid system13, SELEX and its variants14–16, and Rosetta-based computational predictions17 have all been used to characterize the DNA-binding specificity of monomeric zinc finger domains in isolation. However, the toxicity of ZFNs is believed to result from DNA cleavage, rather than binding alone18,19. As a result, information about the specificity of zinc finger nucleases to date has been based on the unproven assumptions that (i) dimeric zinc finger nucleases cleave DNA with the same sequence specificity with which isolated monomeric zinc finger domains bind DNA; and (ii) the binding of one zinc finger domain does not influence the binding of the other zinc finger domain in a given ZFN. The DNA-binding specificities of monomeric zinc finger domains have been used to predict potential off-target cleavage sites of dimeric ZFNs in genomes6,20, but to our knowledge no study to date has reported a method for determining the broad DNA cleavage specificity of active, dimeric zinc finger nucleases.
In this work we present an in vitro selection method to broadly examine the DNA cleavage specificity of active ZFNs. Our selection was coupled with high-throughput DNA sequencing technology to evaluate two obligate heterodimeric ZFNs, CCR5-2246, currently in clinical trials (NCT00842634, NCT01044654, NCT01252641), and VF24684, that targets the human VEGF-A promoter, for their abilities to cleave each of 1011 potential target sites. We identified 37 sites present in the human genome that can be cleaved in vitro by CCR5-224, 2,652 sites in the human genome that can be cleaved in vitro by VF2468, and hundreds of thousands of in vitro cleavable sites for both ZFNs that are not present in the human genome. We examined 34 or 90 sites for evidence of ZFN-induced mutagenesis in cultured human K562 cells expressing the CCR5-224 or VF2468 ZFNs, respectively. Ten of the CCR5-224 sites and 32 of the VF2468 sites we tested show DNA sequence changes consistent with ZFN-mediated cleavage in human cells, although we anticipate that cleavage is likely to be dependent on cell type and ZFN concentration. One CCR5-224 off-target site lies in a promoter of the malignancy-associated BTBD10 gene.
Our results, which could not have been obtained by determining binding specificities of monomeric zinc finger domains alone, indicate that excess DNA-binding energy results in increased off-target ZFN cleavage activity and suggest that ZFN specificity can be enhanced by designing ZFNs with decreased binding affinity, by lowering ZFN expression levels, and by choosing target sites that differ by at least three base pairs from their closest sequence relatives in the genome.
Results
In Vitro Selection for ZFN-Mediated DNA Cleavage
Libraries of potential cleavage sites were prepared as double-stranded DNA using synthetic primers and PCR (Supplementary Fig. S1). Each partially randomized position in the primer was synthesized by incorporating a mixture containing 79% wild-type phosphoramidite and 21% of an equimolar mixture of all three other phosphoramidites. Library sequences therefore differed from canonical ZFN cleavage sites by 21% on average, distributed binomially. We used a blunt ligation strategy to create a 1012-member minicircle library. Using rolling-circle amplification, >1011 members of this library were both amplified and concatenated into high molecular weight (>12 kb) DNA molecules. In theory, this library covers with at least 10-fold excess all DNA sequences that are seven or fewer mutations from the wild-type target sequences.
We incubated the CCR5-224 or VF2468 DNA cleavage site library at a total cleavage site concentration of 14 nM with two-fold dilutions, ranging from 0.5 nM to 4 nM, of crude in vitro-translated CCR5-224 or VF2468, respectively (Supplementary Fig. S2). Following digestion, we subjected the resulting DNA molecules (Supplementary Fig. S3) to in vitro selection for DNA cleavage and subsequent paired-end high-throughput DNA sequencing. Briefly, three selection steps (Fig. 1 and Supplementary Note 1) enabled the separation of sequences that were cleaved from those that were not. First, only sites that had been cleaved contained 5′ phosphates, which are necessary for the ligation of adapters required for sequencing. Second, after PCR, a gel purification step enriched the smaller, cleaved library members. Finally, a computational filter applied after sequencing only counted sequences that have filled-in, complementary 5′ overhangs on both ends, the hallmark for cleavage of a target site concatemer (Supplementary Table S1, Supplementary Note 2, and Supplementary Protocols 1–9). We prepared pre-selection library sequences for sequencing by cleaving the library at a PvuI restriction endonuclease recognition site adjacent to the library sequence and subjecting the digestion products to the same protocol as the ZFN-digested library sequences. High-throughput sequencing confirmed that the rolling-circle-amplified, pre-selection library contained the expected distribution of mutations (Supplementary Fig. S4).
Figure 1. In vitro selection for ZFN-mediated cleavage.
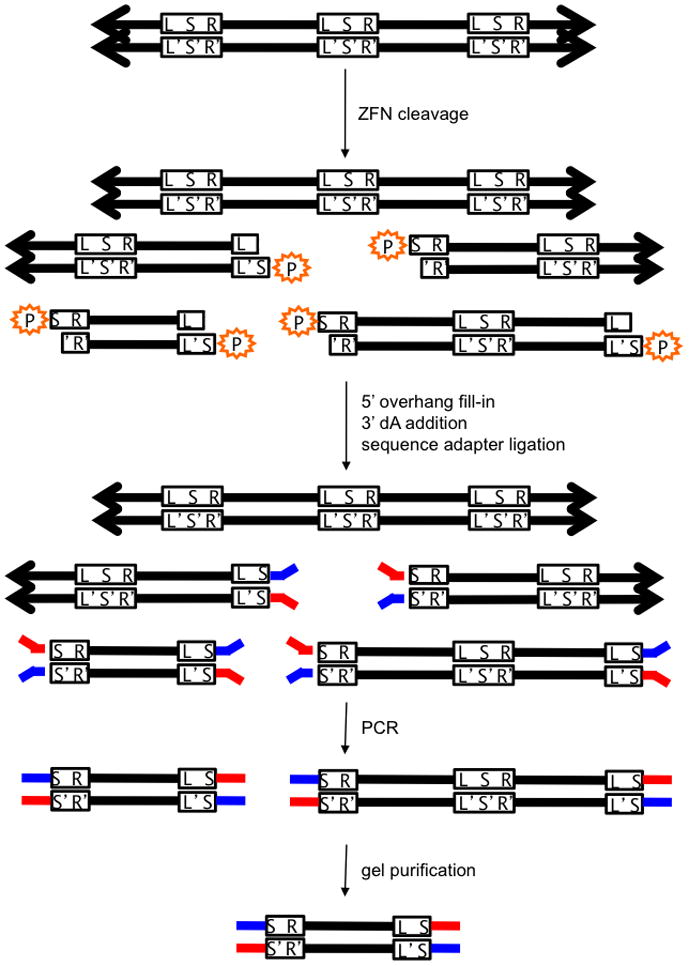
Pre-selection library members are concatemers (represented by arrows) of identical ZFN target sites lacking 5′ phosphates (orange). L = left half-site; R = right half-site, S = spacer; L′, S′, R′ = complementary sequences to L, S, R. ZFN cleavage reveals a 5′ phosphate, which is required for sequencing adapter (red and blue) ligation. The only sequences that can be amplified by PCR using primers complementary to the red and blue adapters are sequences that have been cleaved twice and have adapters on both ends. DNA cleaved at adjacent sites are purified by gel electrophoresis and sequenced. A computational screening step after sequencing ensures that the filled-in spacer sequences (S and S′) are complementary and therefore from the same molecule.
Off-Target Cleavage is Dependent on ZFN Concentration
As expected, only a subset of library members was cleaved by each enzyme. The pre-selection libraries for CCR5-224 and VF2468 had means of 4.56 and 3.45 mutations per complete target site (two half-sites), respectively, while post-selection libraries exposed to the highest concentrations of ZFN used (4 nM CCR5-224 and 4 nM VF2468) had means of 2.79 and 1.53 mutations per target site, respectively (Supplementary Fig. S4). We note that this selection strategy will most likely not recover all cleaved sequences (see Discussion for more details).
As ZFN concentration decreased, both ZFNs exhibited less tolerance for off-target sequences. At the lowest concentrations (0.5 nM CCR5-224 and 0.5 nM VF2468), cleaved sites contained an average of 1.84 and 1.10 mutations, respectively. We placed a small subset of the identified sites in a new DNA context and incubated in vitro with 2 nM CCR5-224 or 1 nM VF2468 for 4 hours at 37 °C (Supplementary Fig. S5). We observed cleavage for all tested sites and those sites emerging from the more stringent (low ZFN concentration) selections were cleaved more efficiently than those from the less stringent selections. Notably, all of the tested sequences contain several mutations, yet some were cleaved in vitro more efficiently than the designed target.
The DNA-cleavage specificity profile of the dimeric CCR5-224 ZFN (Fig. 2a and Supplementary Figs. S6a,b) was notably different than the DNA-binding specificity profiles of the CCR5-224 monomers previously determined by SELEX6. For example, some positions, such as (+)A5 and (+)T9, exhibited tolerance for off-target base pairs in our cleavage selection that were not predicted by the SELEX study. VF2468, which had not been previously characterized with respect to either DNA-binding or DNA-cleavage specificity, revealed two positions, (−)C5 and (+)A9, that exhibited limited sequence preference, suggesting that they were poorly recognized by the ZFNs (Fig. 2b and Supplementary Fig. S6c,d).
Figure 2. DNA cleavage sequence specificity profiles for CCR5-224 and VF2468 ZFNs.
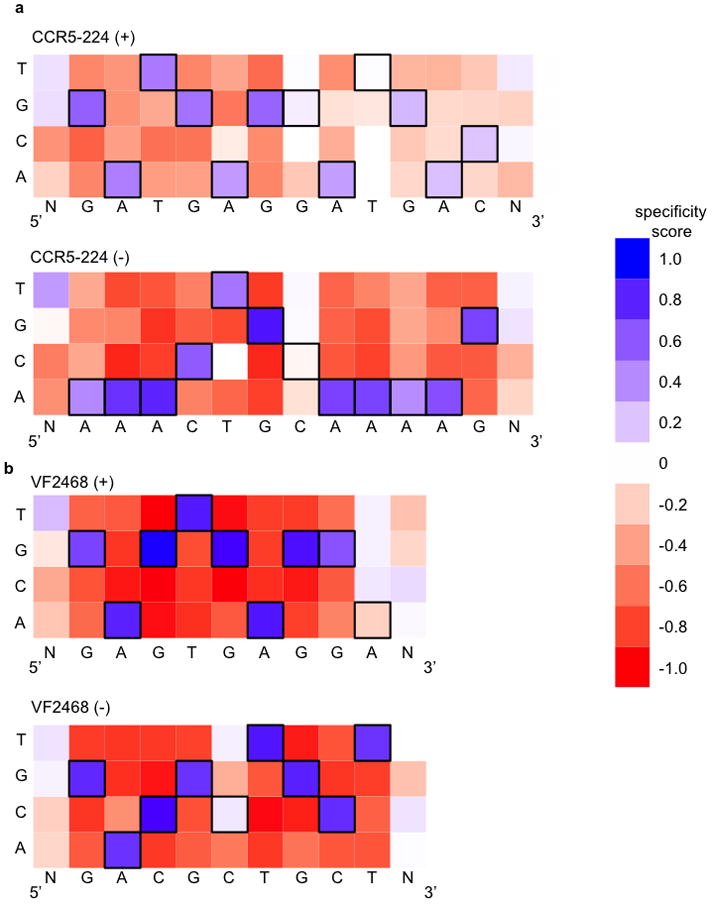
The heat maps show specificity scores compiled from all sequences identified in selections for cleavage of 14 nM of DNA library with (a) 2 nM CCR5-224 or (b) 1 nM VF2468. The target DNA sequence is shown below each half-site. Black boxes indicate target base pairs. Specificity scores were calculated by dividing the change in frequency of each base pair at each position in the post-selection DNA pool compared to the pre-selection pool by the maximal possible change in frequency from pre-selection library to post-selection library of each base pair at each position. Blue boxes indicate enrichment for a base pair at a given position, white boxes indicate no enrichment, and red boxes indicate enrichment against a base pair at a given position. The darkest blue shown in the legend corresponds to absolute preference for a given base pair (specificity score = 1.0), while the darkest red corresponds to an absolute preference against a given base pair (specificity score = −1.0).
Compensation Between Half-Sites Affects DNA Recognition
Our results reveal that ZFN substrates with mutations in one half-site are more likely to have additional mutations in nearby positions in the same half-site compared to the pre-selection library and less likely to have additional mutations in the other half-site. While this effect was found to be largest when the most strongly recognized base pairs were mutated (Supplementary Fig. S7), we observed this compensatory phenomenon for all specified half-site positions for both the CCR5 and VEGF-targeting ZFNs (Fig. 3 and Supplementary Fig. S8). For a minority of nucleotides in cleaved sites, such as VF2468 target site positions (+)G1, (−)G1, (−)A2, and (−)C3, mutation led to decreased tolerance of mutations in base pairs in the other half-site and also a slight decrease, rather than an increase, in mutational tolerance in the same half-site. When two of these mutations, (+)G1 and (−)G1, were enforced at the same time, mutational tolerance at all other positions decreased (Supplementary Fig. S9). Collectively, these results show that tolerance of mutations at one half-site is influenced by DNA recognition at the other half-site.
Figure 3. Evidence for a compensation model of ZFN target site recognition.
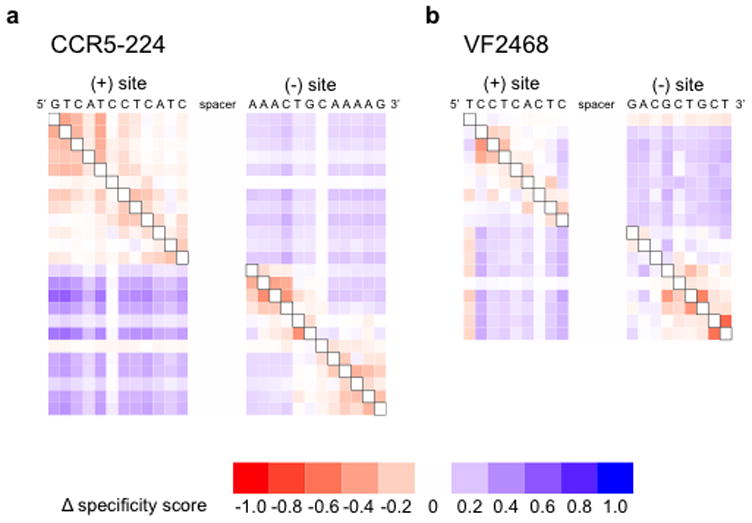
The heat maps show the changes in specificity score upon mutation at the black-boxed positions in selections with (a) 2 nM CCR5-224 or (b) 1 nM VF2468. Each row corresponds to a different mutant position (explained graphically in Supplementary Fig. S8). Sites are listed in their genomic orientation; the (+) half-site of CCR5-224 and the (+) half-site of VF2468 are therefore listed as reverse complements of the sequences found in Figure 2. Shades of blue indicate increased specificity score (more stringency) when the black boxed position is mutated and shades of red indicate decreased specificity score (less stringency).
This compensation model for ZFN site recognition applies not only to non-ideal half-sites, but also to spacers with non-ideal lengths. In general, the ZFNs cleaved at characteristic locations within the spacers (Supplementary Fig. S10), and five- and six-base pair spacers were preferred over four- and seven-base pair spacers (Supplementary Figs. S11 and S12). However, cleaved sites with five- or six-base pair spacers showed greater sequence tolerance at the flanking half-sites than sites with four- or seven-base pair spacers (Supplementary Fig. S13). Therefore, spacer imperfections, similar to half-site mutations, lead to more stringent in vitro recognition of other regions of the DNA substrate.
ZFNs Can Cleave Many Sequences With Up to Three Mutations
We calculated enrichment factors for all sequences containing three or fewer mutations by dividing each sequence’s frequency of occurrence in the post-selection libraries by its frequency of occurrence in the pre-selection libraries. Among sequences enriched by cleavage (enrichment factor > 1), CCR5-224 was capable of cleaving all unique single-mutant sequences, 93% of all unique double-mutant sequences, and half of all possible triple-mutant sequences (Fig. 4a and Supplementary Table S2a) at the highest enzyme concentration used. VF2468 was capable of cleaving 98% of all unique single-mutant sequences, half of all unique double-mutant sequences, and 17% of all triple-mutant sequences (Fig. 4b and Supplementary Table S2b).
Figure 4. ZFNs can cleave a large fraction of target sites with three or fewer mutations in vitro.
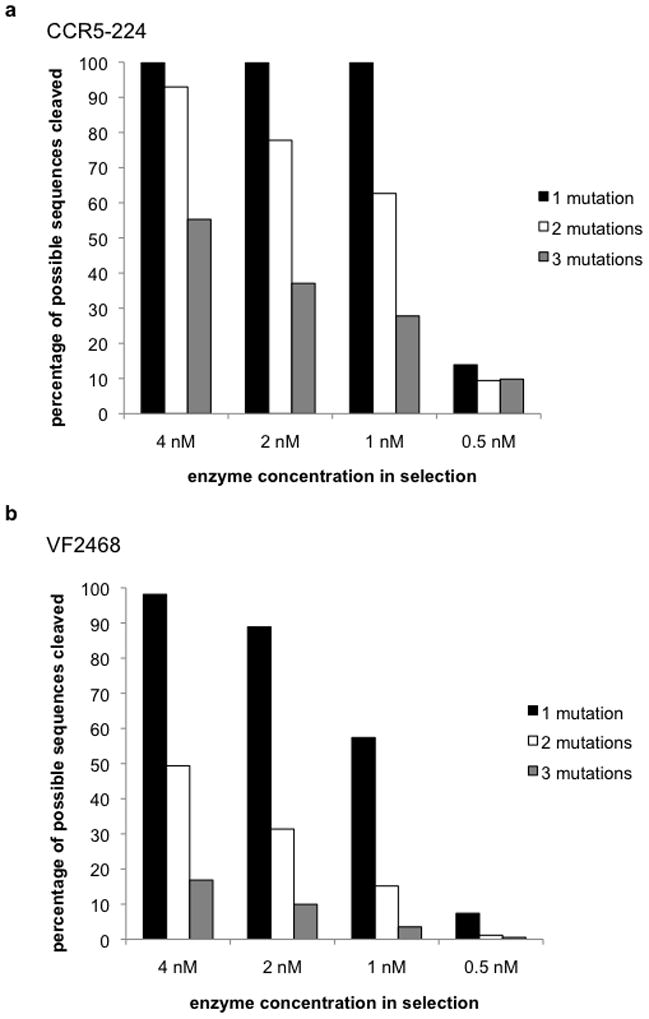
The percentages of the sequences with one, two, or three mutations that are enriched for in vitro cleavage (enrichment factor > 1) by the (a) CCR5-224 ZFN and (b) VF2468 ZFN are shown. Enrichment factors are calculated for each sequence identified in the selection by dividing the observed frequency of that sequence in the post-selection sequenced library by the frequency of that sequence in the pre-selection library.
Since our approach assays active ZFN dimers, it reveals the complete sequences of ZFN sites that can be cleaved. Ignoring the sequence of the spacer, the selection revealed 37 sites in the human genome with five- or six-base pair spacers that can be cleaved in vitro by CCR5-224 (Table 1 and Supplementary Table S3), and 2,652 sites in the human genome that can be cleaved by VF2468 (Supplementary Data). Among the genomic sites that were cleaved in vitro by VF2468, 1,428 sites had three or fewer mutations relative to the canonical target site (excluding the spacer sequence). Despite greater discrimination against single-, double-, and triple-mutant sequences by VF2468 compared to CCR5-224 (Fig. 4 and Supplementary Table S2), the larger number of in vitro-cleavable VF2468 sites reflects the difference in the number of sites in the human genome that are three or fewer mutations away from the VF2468 target site (3,450 sites) versus those that are three or fewer mutations away from the CCR5-224 target site (eight sites) (Supplementary Table S4).
Table 1. CCR5-224 off-target sites in the genome of human K562 cells.
Lower case letters indicate mutations compared to the target site. Sites marked with an ‘X’ were found in the corresponding in vitro selection dataset. ‘T’ refers to the total number of mutations in the site, and ‘(+)’ and ‘(−)’ to the number of mutations in the (+) and (−) half-sites, respectively. The sequences of the sites are listed as 5′ (+) half-site/spacer/(−) half-site 3′, therefore the (+) half-site is listed in the reverse sense as it is in the sequence profiles. K562 modification frequency is the frequency of observed sequences showing significant evidence of non-homologous end joining repair (see Online Methods) in cells expressing active ZFN compared to cells expressing empty vector. Sites that did not show statistically significant evidence of modifications are listed as not detected (n.d.), and K562 modification frequency is left blank for the three sites that were not analyzed due to non-specific PCR amplification from the genome. Supplementary Table S3 shows the sequence counts and P-values for the tested sites used to determine K562 modification frequency, and Supplementary Table S5 shows the modified sequences obtained for each site
| mutations | gene | in vitro selection stringency (nM) | K562 modification frequency | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| T | (+) | (−) | (+) half-site | spacer | (−) half-site | 4 | 2 | 1 | 0.5 | ||
| 0 | 0 | 0 | CCR5 (coding) | GTCATCCTCATC | CTGAT | AAACTGCAAAAG | X | X | X | X | 1: 2.3 |
| 2 | 1 | 1 | CCR2 (coding) | GTCgTCCTCATC | TTAAT | AAACTGCAAAAa | X | X | X | X | 1: 10 |
| 3 | 2 | 1 | BTBD10 (promoter) | GTttTCCTCATC | AAAGC | AAACTGCAAAAt | X | X | 1: 1,400 | ||
| 4 | 0 | 4 | GTCATCCTCATC | AGAGA | AAACTGgctAAt | X | X | n.d. | |||
| 4 | 3 | 1 | SLC4A8 | taaATCCTCATC | TCTATA | AAAaTGCAAAAG | X | X | n.d. | ||
| 3 | 2 | 1 | Z83955 RNA | GTCATCCcaATC | GAAGAA | AAACTGaAAAAG | X | X | n.d. | ||
| 3 | 1 | 2 | DGKK | cTCATCCTCATC | CATGC | AcAaTGCAAAAG | X | n.d. | |||
| 3 | 1 | 2 | GALNT13 | GTCATCCTCAgC | ATGGG | AAACaGCAgAAG | X | n.d. | |||
| 3 | 1 | 2 | GTCATCtTCATC | AAAAG | gAACTGCAAAAc | X | 1: 2,800 | ||||
| 4 | 0 | 4 | GTCATCCTCATC | CAATA | AAAgaaCAAAgG | X | n.d. | ||||
| 4 | 1 | 3 | TACR3 | GTCATCtTCATC | AGCAT | AAACTGtAAAgt | X | 1: 300 | |||
| 4 | 1 | 3 | PIWIL2 | GTCATCCTCATa | CATAA | AAACTGCcttAG | X | ||||
| 4 | 1 | 3 | aTCATCCTCATC | CATCC | AAtgTtCAAAAG | X | n.d. | ||||
| 4 | 3 | 1 | GTCcTgCTCAgC | AAAAG | AAACTGaAAAAG | X | 1: 4,000 | ||||
| 4 | 3 | 1 | KCNB2 | aTgtTCCTCATC | TCCCG | AAACTGCAAAtG | X | 1: 1,400 | |||
| 4 | 3 | 1 | GTCtTCCTgATg | CTACC | AAACTGgAAAAG | X | 1: 5,300 | ||||
| 4 | 3 | 1 | aaCATCCaCATC | ATGAA | AAACTGCAAAAa | X | n.d. | ||||
| 6 | 3 | 3 | aTCtTCCTCATt | ACAGG | AAAaTGtAAtAG | X | n.d. | ||||
| 6 | 4 | 2 | CUBN | GgCtTCCTgAcC | CACGG | AAACTGtAAAtG | X | ||||
| 6 | 5 | 1 | NID1 | GTttTgCaCATt | TCAAT | tAACTGCAAAAG | X | n.d. | |||
| 3 | 2 | 1 | GTCAaCCTCAaC | ACCTAC | AgACTGCAAAAG | X | 1: 1,700 | ||||
| 4 | 1 | 3 | WWOX | GTCATCCTCcTC | CAACTC | cAAtTGCtAAAG | X | n.d. | |||
| 4 | 2 | 2 | AMBRA1 | GTCtTCCTCcTC | TGCACA | tcACTGCAAAAG | X | n.d. | |||
| 4 | 2 | 2 | GTgATaCTCATC | ATCAGC | AAtCTGCAtAAG | X | n.d. | ||||
| 4 | 2 | 2 | WBSCR17 | GTtATCCTCAgC | AAACTA | AAACTGgAAcAG | X | 1: 860 | |||
| 4 | 2 | 2 | ITSN | cTCATgCTCATC | ATTTGT | tAACTGCAAAAt | X | n.d. | |||
| 4 | 4 | 0 | GcCAgtCTCAgC | ATGGTG | AAACTGCAAAAG | X | n.d. | ||||
| 4 | 4 | 0 | cTCATtCTgtTC | ATGAAA | AAACTGCAAAAG | X | n.d. | ||||
| 5 | 3 | 2 | GaagTCCTCATC | CCGAAG | AAACTGaAAgAG | X | n.d. | ||||
| 5 | 3 | 2 | ZNF462 | GTCtTCCTCtTt | CACATA | AAACcGCAAAtG | X | n.d. | |||
| 5 | 4 | 1 | aTaATCCTttTC | TGTTTA | AAACaGCAAAAG | X | n.d. | ||||
| 5 | 4 | 1 | GaCATCCaaATt | ACATGG | AAACTGaAAAAG | X | n.d. | ||||
| 5 | 5 | 0 | SDK1 | GTCtTgCTgtTg | CACCTC | AAACTGCAAAAG | X | n.d. | |||
| 4 | 1 | 3 | SPTB(coding) | GTCATCCgCATC | GCCCTG | gAACTGgAAAAa | X | n.d. | |||
| 4 | 2 | 2 | aTCATCCTCAaC | AAACTA | AAACaGgAAAAG | X | |||||
| 4 | 4 | 0 | KIAA1680 | GgaATgCcCATC | ACCACA | AAACTGCAAAAG | X | n.d. | |||
| 5 | 5 | 0 | GTttTgCTCcTg | TACTTC | AAACTGCAAAAG | X | n.d. | ||||
Identified Sites Are Cleaved by ZFNs in Human Cells
We tested whether CCR5-224 could cleave at sites identified by our selections in human cells by expressing CCR5-224 in K562 cells and examining 34 potential target sites within the human genome for evidence of ZFN-induced mutations using PCR and high-throughput DNA sequencing. We defined sites with evidence of ZFN-mediated cleavage as those with insertion or deletion mutations (indels) characteristic of non-homologous end joining (NHEJ) repair (Supplementary Table S5) that were significantly enriched (P < 0.05) in cells expressing active CCR5-224 compared to control cells containing an empty vector. We obtained 100,000 or more sequences for each site analyzed, which enabled us to detect that were modified at frequencies of approximately 1 in 10,000 or higher. Our analysis identified ten such sites: the intended target sequence in CCR5, a previously identified sequence in CCR2, and eight other off-target sequences (Table 1 and Supplementary Tables S3 and S5), one of which lies within the promoter of the BTBD10 gene. The eight newly identified off-target sites are modified at frequencies ranging from 1 in 300 to 1 in 5,300. We also expressed VF2468 in cultured K562 cells and performed the above analysis for 90 of the most highly cleaved sites identified by in vitro selection. Out of the 90 VF2468 sites analyzed, 32 showed indels consistent with ZFN-mediated targeting in K562 cells (Supplementary Table S6). We were unable to obtain site-specific PCR amplification products for three CCR5-224 sites and seven VF2468 sites and therefore could not analyze the occurrence of NHEJ at those loci. Taken together, these observations indicate that off-target sequences identified through the in vitro selection method include many DNA sequences that can be cleaved by ZFNs in human cells.
Discussion
The method presented here identified hundreds of thousands of sequences that can be cleaved by two active, dimeric ZFNs, including many that are present and can be cut in the genome of human cells. We note that the number of sequence reads obtained per selection (approximately one million) is likely insufficient to cover all cleaved sequences present in the post-selection libraries. It is therefore possible that additional off-target cleavage sites for CCR5-224 and VF2468 could be identified in the human genome as sequencing capabilities continue to improve. It is also possible that the data sets generated by this method could be used to develop computational models to predict ZFN cleavage sites in vitro and in cells.
One newly identified cleavage site for the CCR5-224 ZFN is within the promoter of the BTBD10 gene. When downregulated, BTBD10 has been associated with malignancy21 and with pancreatic beta cell apoptosis22. When upregulated, BTBD10 has been shown to enhance neuronal cell growth23 and pancreatic beta cell proliferation through phosphorylation of Akt family proteins22,23. This potentially important off-target cleavage site as well as seven others we observed in cells were not identified in a recent study6 that used in vitro monomer-binding data to predict potential CCR5-224 substrates.
We have previously shown that ZFNs that can cleave at sites in one cell line may not necessarily function in a different cell line4, most likely due to local differences in chromatin structure. Therefore, it is likely that a different subset of the in vitro-cleavable off-target sites would be modified by CCR5-224 or VF2468 when expressed in different cell lines. Purely cellular studies of endonuclease specificity, such as a recent study of homing endonuclease off-target cleavage24, may likewise be influenced by cell line choice. While our in vitro method does not account for some features of cellular DNA, it provides general, cell type-independent information about endonuclease specificity and off-target sites that can inform subsequent studies performed in cell types of interest.
Although both ZFNs we analyzed were engineered to a unique sequence in the human genome, both cleave a significant number of off-target sites in cells. This finding is particularly surprising for the four-finger CCR5-224 pair given that its theoretical specificity is 4,096-fold better than that of the three-finger VF2468 pair (CCR5-224 should recognize a 24-base pair site that is six base pairs longer than the 18-base pair VF2468 site). Examination of the CCR5-224 and VF2468 cleavage profiles (Fig. 2) and mutational tolerances of sequences with three or fewer mutations (Fig. 4) suggests different strategies may be required to engineer variants of these ZFNs with reduced off-target cleavage activities. The four-finger CCR5-224 ZFN showed a more diffuse range of positions with relaxed specificity and a higher tolerance of mutant sequences with three or fewer mutations than the three-finger VF2468 ZFN. For VF2468, re-optimization of only a subset of fingers may enable a substantial reduction in undesired cleavage events. For CCR5-224, in contrast, a more extensive re-optimization of many or all fingers may be required to eliminate off-target cleavage events. Analysis of a larger number of three-finger and four-finger ZFNs will be required to determine whether these patterns of off-target cleavage activities are a general property of these respective frameworks.
We note that not all four- and three-finger ZFNs will necessarily be as specific as the two ZFNs tested in this study. Both CCR5-224 and VF2468 were engineered using methods designed to optimize the binding activity of the ZFNs. Previous work has shown that for both three-finger and four-finger ZFNs, the specific methodology used to engineer the ZFN pair can have a tremendous impact on the quality and specificity of nucleases7,13,25,26. Therefore, it will be interesting and important to use a method such as the one described here to determine and compare the specificities of additional three-finger and four-finger ZFNs generated using various strategies.
Our findings have significant implications for the design and application of ZFNs with increased specificity. Half or more of all potential substrates with one or two site mutations could be cleaved by ZFNs, suggesting that binding affinity between ZFN and DNA substrate is sufficiently high for cleavage to occur even with suboptimal molecular interactions at mutant positions. We also observed that ZFNs presented with sites that have mutations in one half-site exhibited higher mutational tolerance at other positions within the mutated half-site and lower tolerance at positions in the other half-site. These results collectively suggest that in order to meet a minimum affinity threshold for cleavage, a shortage of binding energy from a half-site harboring an off-target base pair must be energetically compensated by excess zinc finger:DNA binding energy in the other half-site, which demands increased sequence recognition stringency at the non-mutated half-site (Supplementary Fig. S14). Conversely, the relaxed stringency at other positions in mutated half-sites can be explained by the decreased contribution of that mutant half-site to overall ZFN binding energy. This hypothesis is supported by a recent study showing that reducing the number of zinc fingers in a ZFN can actually increase, rather than decrease, activity27.
This model also explains our observation that sites with suboptimal spacer lengths, which presumably were bound less favorably by ZFNs, were recognized with higher stringency than sites with optimal spacer lengths. In vitro spacer preferences do not necessarily reflect spacer preferences in cells;28,29 however, our results suggest that the dimeric FokI cleavage domain can influence ZFN target-site recognition. Consistent with this model, Wolfe and co-workers recently observed differences in the frequency of off-target events in zebrafish of two ZFNs with identical zinc-finger domains but different FokI domain variants.20
Collectively, our findings suggest that (i) ZFN specificity can be increased by avoiding the design of ZFNs with excess DNA binding energy; (ii) off-target cleavage can be minimized by designing ZFNs to target sites that do not have relatives in the genome within three mutations; and (iii) ZFNs should be used at the lowest concentrations necessary to cleave the target sequence to the desired extent. While this study focused on ZFNs, our method should be applicable to all sequence-specific endonucleases that cleave DNA in vitro, including engineered homing endonucleases and engineered transcription activator-like effector (TALE) nucleases. This approach can provide important information when choosing target sites in genomes for sequence-specific endonucleases, and when engineering these enzymes, especially for therapeutic applications.
Methods
Oligonucleotides and Sequences
All oligonucleotides were purchased from Integrated DNA Technologies or Invitrogen and are listed in Supplementary Table S7. Primers with degenerate positions were synthesized by Integrated DNA Technologies using hand-mixed phosphoramidites containing 79% of the indicated base and 7% of each of the other standard DNA bases.
Library Construction
Libraries of target sites were incorporated into double-stranded DNA by PCR with Taq DNA Polymerase (NEB) on a pUC19 starting template with primers “N5-PvuI” and “CCR5-224-N4,” “CCR5-224-N5,” “CCR5-224-N6,” “CCR5-224-N7,” “VF2468-N4,” “VF2468-N5,” “VF2468-N6,” or “VF2468-N7,” yielding an approximately 545-bp product with a PvuI restriction site adjacent to the library sequence, and purified with the Qiagen PCR Purification Kit.
Library-encoding oligonucleotides were of the form 5′ backbone-PvuI site-NNNNNN-partially randomized half-site–N4–7–partially randomized half site-N-backbone 3′. The purified oligonucleotide mixture (approximately 10 μg) was blunted and phosphorylated with a mixture of 50 units of T4 Polynucleotide Kinase and 15 units of T4 DNA polymerase (NEBNext End Repair Enzyme Mix, NEB) in 1x NEBNext End Repair Reaction Buffer (50 mM Tris-HCl, 10 mM MgCl2, 10 mM dithiothreitol, 1 mM ATP, 0.4 mM dATP, 0.4 mM dCTP, 0.4 mM dGTP, 0.4 mM dTTP, pH 7.5) for 1.5 hours at room temperature. The blunt-ended and phosphorylated DNA was purified with the Qiagen PCR Purification Kit according to the manufacturer’s protocol, diluted to 10 ng/μL in NEB T4 DNA Ligase Buffer (50 mM Tris-HCl, 10 mM MgCl2, 10 mM dithiothreitol, 1 mM ATP, pH 7.5) and circularized by ligation with 200 units of T4 DNA ligase (NEB) for 15.5 hours at room temperature. Circular monomers were gel purified on 1% TAE-Agarose gels. 70 ng of circular monomer was used as a substrate for rolling-circle amplification at 30 °C for 20 hours in a 100 μL reaction using the Illustra TempliPhi 100 Amplification Kit (GE Healthcare). Reactions were stopped by incubation at 65 °C for 10 minutes. Target site libraries were quantified with the Quant-iT PicoGreen dsDNA Reagent (Invitrogen). Libraries with N4, N5, N6, and N7 spacer sequences between partially randomized half-sites were pooled in equimolar concentrations for both CCR5-224 and VF2468.
Zinc finger Nuclease Expression and Characterization
3xFLAG-tagged zinc finger proteins for CCR5-224 and VF2468 were expressed as fusions to FokI obligate heterodimers30 in mammalian expression vectors4 derived from pMLM290 and pMLM292. DNA and protein sequences are listed in Supplementary Figure S15. Complete vector sequences are available upon request. 2 μg of ZFN-encoding vector was transcribed and translated in vitro using the TNT Quick Coupled rabbit reticulocyte system (Promega). Zinc chloride (Sigma-Aldrich) was added at 500 μM and the transcription/translation reaction was performed for 2 hours at 30°C. Glycerol was added to a 50% final concentration. Western blots were used to visualize protein using the anti-FLAG M2 monoclonal antibody (Sigma-Aldrich). ZFN concentrations were determined by Western blot and comparison with a standard curve of N-terminal FLAG-tagged bacterial alkaline phosphatase (Sigma-Aldrich).
Test substrates for CCR5-224 and VF2468 were constructed by cloning into the HindIII/XbaI sites of pUC19. PCR with primers “test fwd” and “test rev” and Taq DNA polymerase yielded a linear 1 kb DNA that could be cleaved by the appropriate ZFN into two fragments of sizes ~300 bp and ~700 bp. Activity profiles for the zinc finger nucleases were obtained by modifying the in vitro cleavage protocols used by Miller et al.30 and Cradick et al.31. 1 μg of linear 1 kb DNA was digested with varying amounts of ZFN in 1x NEBuffer 4 (50 mM potassium acetate, 20 mM Tris-acetate, 10 mM magnesium acetate, 1 mM dithiothreitol, pH 7.9) for 4 hours at 37 °C. 100 μg of RNase A (Qiagen) was added to the reaction for 10 minutes at room temperature to remove RNA from the in vitro transcription/translation mixture that could interfere with purification and gel analysis. Reactions were purified with the Qiagen PCR Purification Kit and analyzed on 1% TAE-agarose gels.
In Vitro Selection
ZFNs of varying concentrations, an amount of TNT reaction mixture without any protein-encoding DNA template equivalent to the greatest amount of ZFN used (“lysate”), or 50 units PvuI (NEB) were incubated with 1 μg of rolling-circle amplified library for 4 hours at 37 °C in 1x NEBuffer 4 (50 mM potassium acetate, 20 mM Tris-acetate, 10 mM magnesium acetate, 1 mM dithiothreitol, pH 7.9). 100 μg of RNase A (Qiagen) was added to the reaction for 10 minutes at room temperature to remove RNA from the in vitro transcription/translation mixture that could interfere with purification and gel analysis. Reactions were purified with the Qiagen PCR Purification Kit. 1/10 of the reaction mixture was visualized by gel electrophoresis on a 1% TAE-agarose gel and staining with SYBR Gold Nucleic Acid Gel Stain (Invitrogen).
The purified DNA was blunted with 5 units DNA Polymerase I, Large (Klenow) Fragment (NEB) in 1x NEBuffer 2 (50 mM NaCl, 10 mM Tris-HCl, 10 mM MgCl2, 1 mM dithiothreitol, pH 7.9) with 500 μM dNTP mix (Bio-Rad) for 30 minutes at room temperature. The reaction mixture was purified with the Qiagen PCR Purification Kit and incubated with 5 units of Klenow Fragment (3′ exo−) (NEB) for 30 minutes at 37 °C in 1x NEBuffer 2 (50 mM NaCl, 10 mM Tris-HCl, 10 mM MgCl2, 1 mM dithiothreitol, pH 7.9) with 240 μM dATP (Promega) in a 50 μL final volume. 10 mM Tris-HCl, pH 8.5 was added to a volume of 90 μL and the reaction was incubated for 20 minutes at 75 °C to inactivate the enzyme before cooling to 12 °C. 300 fmol of “adapter1/2”, barcoded according to enzyme concentration, or 6 pmol of “adapter1/2” for the PvuI digest, were added to the reaction mixture, along with 10 ul 10x NEB T4 DNA Ligase Reaction Buffer (500 mM Tris-HCl, 100 mM MgCl2, 100 mM dithiothreitol, 10 mM ATP). Adapters were ligated onto the blunt DNA ends with 400 units of T4 DNA ligase at room temperature for 17.5 hours and ligated DNA was purified away from unligated adapters with Illustra Microspin S-400 HR sephacryl columns (GE Healthcare). DNA with ligated adapters were amplified by PCR with 2 units of Phusion Hot Start II DNA Polymerase (NEB) and 10 pmol each of primers “PE1” and “PE2” in 1x Phusion GC Buffer supplemented with 3% DMSO and 1.7 mM MgCl2. PCR conditions were 98 °C for 3 min, followed by cycles of 98 °C for 15 s, 60 °C for 15 s, and 72 °C for 15 s, and a final 5 min extension at 72 °C. The PCR was run for enough cycles (typically 20–30) to see a visible product on gel. The reactions were pooled in equimolar amounts and purified with the Qiagen PCR Purification Kit. The purified DNA was gel purified on a 1% TAE-agarose gel, and submitted to the Harvard Medical School Biopolymers Facility for Illumina 36-base paired-end sequencing.
Data Analysis
Illumina sequencing reads were analyzed using programs written in C++. Algorithms are described in the Supplementary Information section (Supplementary Protocols 1–9), and the source code is available on request. Sequences containing the same barcode on both paired sequences and no positions with a quality score of ‘B’ were binned by barcode. Half-site sequence, overhang and spacer sequences, and adjacent randomized positions were determined by positional relationship to constant sequences and searching for sequences similar to the designed CCR5-224 and VF2468 recognition sequences. These sequences were subjected to a computational selection step for complementary, filled-in overhang ends of at least 4 base pairs, corresponding to rolling-circle concatemers that had been cleaved at two adjacent and identical sites. Specificity scores were calculated with the formulae: positive specificity score = (frequency of base pair at position[post-selection] - frequency of base pair at position[pre-selection])/(1 - frequency of base pair at position[pre-selection]) and negative specificity score = (frequency of base pair at position[post-selection] - frequency of base pair at position[pre-selection])/(frequency of base pair at position[pre-selection]).
Positive specificity scores reflect base pairs that appear with greater frequency in the post-selection library than in the starting library at a given position; negative specificity scores reflect base pairs that are less frequent in the post-selection library than in the starting library at a given position. A score of +1 indicates an absolute preference, a score of −1 indicates an absolute intolerance, and a score of 0 indicates no preference.
Assay of Genome Modification at Cleavage Sites in Human Cells
CCR5-224 ZFNs were cloned into a CMV-driven mammalian expression vector in which both ZFN monomers were translated from the same mRNA transcript in stoichiometric quantities using a self-cleaving T2A peptide sequence similar to a previously described vector32. This vector also expresses enhanced green fluorescent protein (eGFP) from a PGK promoter downstream of the ZFN expression cassette. An empty vector expressing only eGFP was used as a negative control.
To deliver ZFN expression plasmids into cells, 15 μg of either active CCR5-224 ZFN DNA or empty vector DNA were used to Nucleofect 2×106 K562 cells in duplicate reactions following the manufacturer’s instructions for Cell Line Nucleofector Kit V (Lonza). GFP-positive cells were isolated by FACS 24 hours post-transfection, expanded, and harvested five days post-transfection with the QIAamp DNA Blood Mini Kit (Qiagen).
PCR for 37 potential CCR5-224 substrates and 97 potential VF2468 substrates was performed with Phusion DNA Polymerase (NEB) and primers “[ZFN] [#] fwd” and “[ZFN] [#] rev” (Supplementary Table S8) in 1x Phusion HF Buffer supplemented with 3% DMSO. Primers were designed using Primer333. The amplified DNA was purified with the Qiagen PCR Purification Kit, eluted with 10 mM Tris-HCl, pH 8.5, and quantified by 1K Chip on a LabChip GX instrument (Caliper Life Sciences) and combined into separate equimolar pools for the catalytically active and empty vector control samples. PCR products were not obtained for 3 CCR5 sites and 7 VF2468 sites, which excluded these samples from further analysis. Multiplexed Illumina library preparation was performed according to the manufacturer’s specifications, except that AMPure XP beads (Agencourt) were used for purification following adapter ligation and PCR enrichment steps. Illumina indices 11 (“GGCTAC”) and 12 (“CTTGTA”) were used for ZFN-treated libraries while indices 4 (“TGACCA”) and 6 (“GCCAAT”) were used for the empty vector controls. Library concentrations were quantified by KAPA Library Quantification Kit for Illumina Genome Analyzer Platform (Kapa Biosystems). Equal amounts of the barcoded libraries derived from active- and empty vector- treated cells were diluted to 10 nM and subjected to single read sequencing on an Illumina HiSeq 2000 at the Harvard University FAS Center for Systems Biology Core facility. Sequences were analyzed using Supplementary Protocol 9 for active ZFN samples and empty vector controls.
Statistical Analysis
In Supplementary Figure 4, P-values were calculated for a one-sided test of the difference in the means of the number of target site mutations in all possible pairwise comparisons among pre-selection, 0.5 nM post-selection, 1 nM post-selection, 2 nM post-selection, and 4 nM post-selection libraries for CCR5-224 or VF2468. The t-statistic was calculated as t = (x_bar1 - x_bar2)/sqrt(l × p_hat1× (1-p_hat1)/n1+ l × p_hat2× (1 - p_hat2)/n2), where x_bar1 and x_bar2 are the means of the distributions being compared, l is the target site length (24 for CCR5-224; 18 for VF2468), p_hat1 and p_hat2 are the calculated probabilities of mutation (x_bar/l) for each library, and n1 and n2 are the total number of sequences analyzed for each selection (Supplementary Table S1). All pre- and post-selection libraries were assumed to be binomially distributed.
In Supplementary Tables S3 and S6, P-values were calculated for a one-sided test of the difference in the proportions of sequences with insertions or deletions from the active ZFN sample and the empty vector control samples. The t-statistic was calculated as t = (p_hat1 - p_hat2)/sqrt((p_hat1× (1 - p_hat1)/n1)+ (p_hat2× (1 - p_hat2)/n2)), where p_hat1 and n1 are the proportion and total number, respectively, of sequences from the active sample and p_hat2 and n2 are the proportion and total number, respectively, of sequences from the empty vector control sample.
Plots
All heat maps were generated in the R software package with the following command: image([variable], zlim = c(−1,1), col = color Ramp Palette(c(“red”, “white”, “blue”), space= “Lab”)(2500)
Supplementary Material
Supplementary Figure S1. In vitro synthesis of target site library
Supplementary Figure S2. Expression and quantification of ZFNs
Supplementary Figure S3. Library cleavage with ZFNs
Supplementary Figure S4. ZFN off-target cleavage is dependent on enzyme concentration
Supplementary Figure S5. Cleavage efficiency of individual sequences is related to selection stringency
Supplementary Figure S6. Concentration-dependent sequence profiles for CCR5-224 and VF2468 ZFNs
Supplementary Figure S7. Stringency at the (+) half-site increases when CCR5- 224 cleaves sites with mutations at highly specified base pairs in the (−) half-site
Supplementary Figure S8. Data processing steps used to create mutation compensation difference maps
Supplementary Figure S9. Stringency at both half-sites increases when VF2468 cleaves sites with mutations at the first base pair of both half-sites
Supplementary Figure S10. ZFN cleavage occurs at characteristic locations in the DNA target site
Supplementary Figure S11. CCR5-224 preferentially cleaves five- and six-base pair spacers and cleaves five-base pair spacers to leave five-nucleotide overhangs
Supplementary Figure S12. VF2468 preferentially cleaves five- and six-base pair spacers, cleaves five-base pair spacers to leave five- nucleotide overhangs, and cleaves six-base pair spacers to leave four-nucleotide overhangs
Supplementary Figure S13. ZFNs show spacer length-dependent sequence preferences
Supplementary Figure S14. Model for ZFN tolerance of off-target sequences
Supplementary Figure S15. Sequences of ZFNs used in this study
Supplementary Table S1. Sequencing statistics
Supplementary Table S2. Both ZFNs tested have the ability to cleave a large fraction of target sites with three or fewer mutations
Supplementary Table S3. Potential CCR5-224 genomic off-target sites
Supplementary Table S4. There are many more potential genomic VF2468 target sites than CCR5-224 target sites
Supplementary Table S5. Sequences of CCR5-224-mediated genomic DNA modifications identified in cultured human K562 cells
Supplementary Table S6. Potential VF2468 genomic off-target sites
Supplementary Table S7. Oligonucleotides used in this study
Supplementary Note 1. Design of an In Vitro Selection for ZFN-Mediated DNA Cleavage
Supplementary Note 2. Analysis of CCR5-224 and VF2468 ZFNs Using the DNA Cleavage Selection
Supplementary Protocol 1. Quality score filtering and sequence binning
Supplementary Protocol 2. Filtering by ZFN
Supplementary Protocol 3. Library filtering
Supplementary Protocol 4. Sequence profiles
Supplementary Protocol 5. Genomic matches
Supplementary Protocol 6. Enrichment factors for sequences with 0, 1, 2, or 3 mutations
Supplementary Protocol 7. Filtered sequence profiles
Supplementary Protocol 8. Compensation difference map
Supplementary Protocol 9. NHEJ searching
Acknowledgments
This research was supported by NIH/NIGMS R01 GM065400 (D.R.L.), DARPA HR0011-11-2-0003 (D.R.L.), the Howard Hughes Medical Institute (D.R.L.), NIH/NIGMS R01 GM088040 (J.K.J.), NIH/OD DP1 OD006862 (J.K.J.), and the Jim and Ann Orr MGH Research Scholar Award (J.K.J). V.P. was supported by an NIH training grant to the Harvard University Training Program in Molecular, Cellular, and Chemical Biology (MCCB). C.L.R. was supported by a National Science Foundation Graduate Research Fellowship and a Ford Foundation Predoctoral Fellowship. The HMS Neuroscience core facility, supported by NIH/NINDS P30 NS045776, provided qPCR capabilities. We thank J. Carlson, B. Dorr, C. Pattanayak, D. Reyon, J. Sander, and D. Thompson for helpful discussions, M. Goodwin for technical assistance, and M. Maeder (Massachusetts General Hospital) for mammalian cell ZFN expression plasmids.
Footnotes
Author Contributions
V.P. performed the experiments, designed the research, analyzed the data, and wrote the manuscript. C.L.R. performed the experiments, designed the research, analyzed the data, and wrote the manuscript. J.K.J. designed the research, analyzed the data, and wrote the manuscript. D.R.L. designed the research, analyzed the data, and wrote the manuscript.
Competing Financial Interests
All authors declare no competing financial interests.
Supplementary Data. Potential VF2468 genomic off-target sites
References
- 1.Kim YG, Cha J, Chandrasegaran S. Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc Natl Acad Sci U S A. 1996;93:1156–60. doi: 10.1073/pnas.93.3.1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vanamee ES, Santagata S, Aggarwal AK. FokI requires two specific DNA sites for cleavage. J Mol Biol. 2001;309:69–78. doi: 10.1006/jmbi.2001.4635. [DOI] [PubMed] [Google Scholar]
- 3.Hockemeyer D, et al. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat Biotechnol. 2009;27:851–7. doi: 10.1038/nbt.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maeder ML, et al. Rapid “open-source” engineering of customized zinc-finger nucleases for highly efficient gene modification. Mol Cell. 2008;31:294–301. doi: 10.1016/j.molcel.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zou J, et al. Gene targeting of a disease-related gene in human induced pluripotent stem and embryonic stem cells. Cell Stem Cell. 2009;5:97–110. doi: 10.1016/j.stem.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perez EE, et al. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat Biotechnol. 2008;26:808–16. doi: 10.1038/nbt1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Urnov FD, et al. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature. 2005;435:646–51. doi: 10.1038/nature03556. [DOI] [PubMed] [Google Scholar]
- 8.Santiago Y, et al. Targeted gene knockout in mammalian cells by using engineered zinc-finger nucleases. Proc Natl Acad Sci U S A. 2008;105:5809–14. doi: 10.1073/pnas.0800940105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cui X, et al. Targeted integration in rat and mouse embryos with zinc-finger nucleases. Nat Biotechnol. 2011;29:64–7. doi: 10.1038/nbt.1731. [DOI] [PubMed] [Google Scholar]
- 10.Cornu TI, et al. DNA-binding specificity is a major determinant of the activity and toxicity of zinc-finger nucleases. Mol Ther. 2008;16:352–8. doi: 10.1038/sj.mt.6300357. [DOI] [PubMed] [Google Scholar]
- 11.Segal DJ, Dreier B, Beerli RR, Barbas CF., 3rd Toward controlling gene expression at will: selection and design of zinc finger domains recognizing each of the 5′-GNN-3′ DNA target sequences. Proc Natl Acad Sci U S A. 1999;96:2758–63. doi: 10.1073/pnas.96.6.2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bulyk ML, Huang X, Choo Y, Church GM. Exploring the DNA-binding specificities of zinc fingers with DNA microarrays. Proc Natl Acad Sci U S A. 2001;98:7158–63. doi: 10.1073/pnas.111163698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meng X, Thibodeau-Beganny S, Jiang T, Joung JK, Wolfe SA. Profiling the DNA-binding specificities of engineered Cys2His2 zinc finger domains using a rapid cell-based method. Nucleic Acids Res. 2007;35:e81. doi: 10.1093/nar/gkm385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wolfe SA, Greisman HA, Ramm EI, Pabo CO. Analysis of zinc fingers optimized via phage display: evaluating the utility of a recognition code. J Mol Biol. 1999;285:1917–34. doi: 10.1006/jmbi.1998.2421. [DOI] [PubMed] [Google Scholar]
- 15.Segal DJ, et al. Evaluation of a modular strategy for the construction of novel polydactyl zinc finger DNA-binding proteins. Biochemistry. 2003;42:2137–48. doi: 10.1021/bi026806o. [DOI] [PubMed] [Google Scholar]
- 16.Zykovich A, Korf I, Segal DJ. Bind-n-Seq: high-throughput analysis of in vitro protein-DNA interactions using massively parallel sequencing. Nucleic Acids Res. 2009;37:e151. doi: 10.1093/nar/gkp802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yanover C, Bradley P. Extensive protein and DNA backbone sampling improves structure-based specificity prediction for C2H2 zinc fingers. Nucleic Acids Res. 2011 doi: 10.1093/nar/gkr048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beumer K, Bhattacharyya G, Bibikova M, Trautman JK, Carroll D. Efficient gene targeting in Drosophila with zinc-finger nucleases. Genetics. 2006;172:2391–403. doi: 10.1534/genetics.105.052829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bibikova M, Golic M, Golic KG, Carroll D. Targeted chromosomal cleavage and mutagenesis in Drosophila using zinc-finger nucleases. Genetics. 2002;161:1169–75. doi: 10.1093/genetics/161.3.1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gupta A, Meng X, Zhu LJ, Lawson ND, Wolfe SA. Zinc finger protein-dependent and -independent contributions to the in vivo off-target activity of zinc finger nucleases. Nucleic Acids Res. 2011;39:381–92. doi: 10.1093/nar/gkq787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen J, et al. Molecular cloning and characterization of a novel human BTB domain-containing gene, BTBD10, which is down-regulated in glioma. Gene. 2004;340:61–9. doi: 10.1016/j.gene.2004.05.028. [DOI] [PubMed] [Google Scholar]
- 22.Wang X, et al. Glucose metabolism-related protein 1 (GMRP1) regulates pancreatic beta cell proliferation and apoptosis via activation of Akt signalling pathway in rats and mice. Diabetologia. 2011;54:852–63. doi: 10.1007/s00125-011-2048-1. [DOI] [PubMed] [Google Scholar]
- 23.Nawa M, Kanekura K, Hashimoto Y, Aiso S, Matsuoka M. A novel Akt/PKB-interacting protein promotes cell adhesion and inhibits familial amyotrophic lateral sclerosis-linked mutant SOD1-induced neuronal death via inhibition of PP2A-mediated dephosphorylation of Akt/PKB. Cell Signal. 2008;20:493–505. doi: 10.1016/j.cellsig.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 24.Petek LM, Russell DW, Miller DG. Frequent endonuclease cleavage at off-target locations in vivo. Mol Ther. 2010;18:983–6. doi: 10.1038/mt.2010.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hurt JA, Thibodeau SA, Hirsh AS, Pabo CO, Joung JK. Highly specific zinc finger proteins obtained by directed domain shuffling and cell-based selection. Proc Natl Acad Sci U S A. 2003;100:12271–6. doi: 10.1073/pnas.2135381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ramirez CL, et al. Unexpected failure rates for modular assembly of engineered zinc fingers. Nat Methods. 2008;5:374–5. doi: 10.1038/nmeth0508-374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shimizu Y, et al. Adding Fingers to an Engineered Zinc Finger Nuclease Can Reduce Activity. Biochemistry. 2011;50:5033–41. doi: 10.1021/bi200393g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bibikova M, et al. Stimulation of homologous recombination through targeted cleavage by chimeric nucleases. Mol Cell Biol. 2001;21:289–97. doi: 10.1128/MCB.21.1.289-297.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Handel EM, Alwin S, Cathomen T. Expanding or restricting the target site repertoire of zinc-finger nucleases: the inter-domain linker as a major determinant of target site selectivity. Mol Ther. 2009;17:104–11. doi: 10.1038/mt.2008.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Miller JC, et al. An improved zinc-finger nuclease architecture for highly specific genome editing. Nat Biotechnol. 2007;25:778–85. doi: 10.1038/nbt1319. [DOI] [PubMed] [Google Scholar]
- 31.Cradick TJ, Keck K, Bradshaw S, Jamieson AC, McCaffrey AP. Zinc-finger nucleases as a novel therapeutic strategy for targeting hepatitis B virus DNAs. Mol Ther. 2010;18:947–54. doi: 10.1038/mt.2010.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Doyon Y, et al. Heritable targeted gene disruption in zebrafish using designed zinc-finger nucleases. Nat Biotechnol. 2008;26:702–8. doi: 10.1038/nbt1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365–86. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1. In vitro synthesis of target site library
Supplementary Figure S2. Expression and quantification of ZFNs
Supplementary Figure S3. Library cleavage with ZFNs
Supplementary Figure S4. ZFN off-target cleavage is dependent on enzyme concentration
Supplementary Figure S5. Cleavage efficiency of individual sequences is related to selection stringency
Supplementary Figure S6. Concentration-dependent sequence profiles for CCR5-224 and VF2468 ZFNs
Supplementary Figure S7. Stringency at the (+) half-site increases when CCR5- 224 cleaves sites with mutations at highly specified base pairs in the (−) half-site
Supplementary Figure S8. Data processing steps used to create mutation compensation difference maps
Supplementary Figure S9. Stringency at both half-sites increases when VF2468 cleaves sites with mutations at the first base pair of both half-sites
Supplementary Figure S10. ZFN cleavage occurs at characteristic locations in the DNA target site
Supplementary Figure S11. CCR5-224 preferentially cleaves five- and six-base pair spacers and cleaves five-base pair spacers to leave five-nucleotide overhangs
Supplementary Figure S12. VF2468 preferentially cleaves five- and six-base pair spacers, cleaves five-base pair spacers to leave five- nucleotide overhangs, and cleaves six-base pair spacers to leave four-nucleotide overhangs
Supplementary Figure S13. ZFNs show spacer length-dependent sequence preferences
Supplementary Figure S14. Model for ZFN tolerance of off-target sequences
Supplementary Figure S15. Sequences of ZFNs used in this study
Supplementary Table S1. Sequencing statistics
Supplementary Table S2. Both ZFNs tested have the ability to cleave a large fraction of target sites with three or fewer mutations
Supplementary Table S3. Potential CCR5-224 genomic off-target sites
Supplementary Table S4. There are many more potential genomic VF2468 target sites than CCR5-224 target sites
Supplementary Table S5. Sequences of CCR5-224-mediated genomic DNA modifications identified in cultured human K562 cells
Supplementary Table S6. Potential VF2468 genomic off-target sites
Supplementary Table S7. Oligonucleotides used in this study
Supplementary Note 1. Design of an In Vitro Selection for ZFN-Mediated DNA Cleavage
Supplementary Note 2. Analysis of CCR5-224 and VF2468 ZFNs Using the DNA Cleavage Selection
Supplementary Protocol 1. Quality score filtering and sequence binning
Supplementary Protocol 2. Filtering by ZFN
Supplementary Protocol 3. Library filtering
Supplementary Protocol 4. Sequence profiles
Supplementary Protocol 5. Genomic matches
Supplementary Protocol 6. Enrichment factors for sequences with 0, 1, 2, or 3 mutations
Supplementary Protocol 7. Filtered sequence profiles
Supplementary Protocol 8. Compensation difference map
Supplementary Protocol 9. NHEJ searching