

Abstract
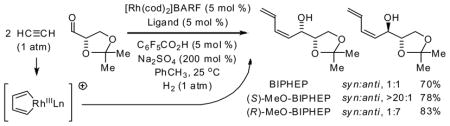
Hydrogenative coupling of acetylene to α-chiral aldehydes 1a–4a using enantiomeric rhodium catalysts ligated by (S)-MeO-BIPHEP and (R)-MeO-BIPHEP delivers the diastereomeric products of carbonyl-(Z)-butadienylation 1b–4b and 1c–4c, respectively, with good to excellent levels of catalyst directed diastereofacial selectivity. Diastereomeric L-glyceraldehyde acetonide adducts 1b and 1c were converted to the four isomeric enoates 6b, 8b, 6c, and 8c, representing a formal synthesis of all eight L-hexoses.
The broad role of carbohydrates in diverse biological processes evokes a persistent need for efficient synthetic strategies toward natural and unnatural monosaccharides.1 Beginning with the synthesis of glucose, fructose and mannose from glyceraldehyde reported by Emil Fischer (1890); 2 numerous protocols for the synthesis and interconversion of monosaccharides have appeared.1 However, nearly a century elapsed before the first enantioselective de novo synthesis of a monosaccharide was reported by Sharpless and Masamune (1983), who prepared all eight L-hexoses through asymmetric epoxidation.3 Subsequently, elegant syntheses of various hexose stereoisomers were disclosed based upon catalytic enantioselective alkene dihydroxylation, 4 catalytic enantioselective Payne rearrangement, 5 and catalytic enantioselective aldol addition. 6
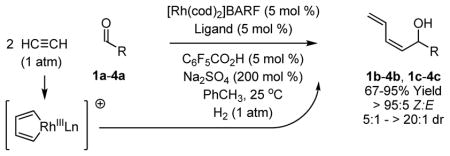
Here, using catalytic enantioselective hydrogenative C-C couplings of acetylene recently developed in our laboratory,7,8 we report a concise formal synthesis of all eight L-hexoses through serial catalyst-directed diastereofacial selection, the sequential use of transformations wherein the stereochemical bias of an enantiomeric catalyst overrides the diastereofacial bias of a chiral nonracemic substrate. 9 Additionally, catalyst-directed diastereofacial selection in hydrogenative couplings of acetylene to α-chiral aldehydes 1a–4a is described. In each case, the stereochemical bias of the catalyst was found to override the inherent diastereofacial bias of the α-chiral aldehyde.
Initial studies focused on catalyst-directed stereoinduction in the hydrogenative coupling of acetylene to L-glyceraldehyde 1a. Under previously disclosed conditions using the achiral ligand BIPHEP,7 an equimolar distribution of diastereomers 1b and 1c is formed. This absence of substrate-directed diastereofacial selectivity suggested the feasibility of catalyst-directed diastereofacial selection. Indeed, employing a chiral rhodium catalyst ligated by (S)-MeO-BIPHEP, a ≥ 20:1 diastereomeric ratio of adducts 1b and 1c is obtained, as determined by 1H NMR. Using the enantiomeric rhodium catalyst ligated by (R)-MeO-BIPHEP, a 1:7 diastereomeric ratio of adducts 1b and 1c is obtained, representing an inversion in diastereofacial selectivity (Table 1, entry 1).
Table 1.
Catalyst-directed diastereofacial selection in hydrogenative couplings of acetylene to α-chiral aldehydes.
 | ||||
|---|---|---|---|---|
| entry | substrate | ligand | diastereomeric products, dr | yielda |
| 1 |
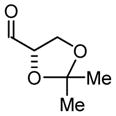 1a |
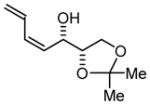 1b |
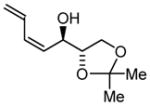 1c |
|
| BIPHEP (S)-MeO-BIPHEP (R)-MeO-BIPHEP |
1b:1c, 1:1 1b:1c, >20:1 1b:1c, 1:7 |
70% 78% 83%b |
||
| 2 |
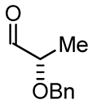 2a |
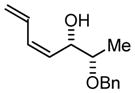 2b |
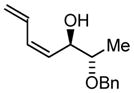 2c |
|
| BIPHEP (S)-MeO-BIPHEP (R)-MeO-BIPHEP |
2b:2c, 1.5:1 2b:2c, 11:1 2b:2c, 1:5 |
76% 95% 92% |
||
| 3 |
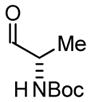 3a |
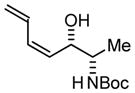 3b |
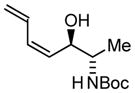 3c |
|
| BIPHEP (S)-MeO-BIPHEP (R)-MeO-BIPHEP |
3b:3c, 2:1 3b:3c, 16:1 3b:3c, 1:5 |
73% 75% 67% |
||
| 4 |
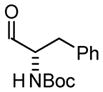 4a |
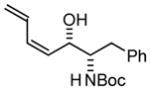 4b |
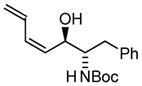 4c |
|
| BIPHEP (S)-MeO-BIPHEP (R)-MeO-BIPHEP |
4b:4c, 1:1 4b:4c, 12:1 4b:4c, 1:12 |
70% 80% 73% |
||
Cited yields are of isolated material. Best results are obtained using an apparatus in which mixtures of hydrogen and acetylene are delivered from a gas bag via cannula. See Supporting Information for detailed experimental procedures.
Reaction was performed at 4 °C.
Based on these results, catalyst-directed diastereofacial selection was explored in hydrogenative couplings of acetylene to aldehydes 2a–4a using enantiomeric rhodium catalysts ligated by (S)-MeO-BIPHEP and (R)-MeO-BIPHEP. For each aldehyde, good to excellent levels of catalyst-directed stereoinduction are observed in both the matched and mismatched cases. For α-alkoxy aldehydes 1a and 2a and N-Boc-L-alaninal 3a, anti-Felkin-Anh addition represents the matched mode of C-C coupling. In the case of N-Boc-L-phenylalaninal 4a, equivalent levels of diastereofacial selectivity are observed in additions employing enantiomeric rhodium catalysts. To corroborate the relative stereochemical assignment of adducts 1b, 2c, 3b and 4b, the diene side chain of these materials was exhaustively hydrogenated under the conditions of iridium catalysis10 to furnish the corresponding n-butyl adducts, which were correlated to authentic samples. 11
To showcase the utility of this methodology, the L-glyceraldehyde acetonide adducts 1b and 1c were transformed to cis-enoates 6b and 6c and trans-enoates 8b and 8c, representing a formal synthesis of all eight L-hexoses (Scheme 1). Oxidative cleavage of diene terminus of 1b and 1c using the Johnson-Lemieux protocol 12 delivers cis-enal 5b and 5c, respectively. Under the oxidative cleavage conditions, olefin isomerization to form the corresponding trans-enals was not detected by 1H NMR. Exposure of cis-enals 5b and 5c to manganese oxide in the presence of sodium cyanide in methanol provides the methyl cis-enoates 6b and 6c, respectively. The stereochemical integrity of cis-olefin moieties of 6b and 6c is retained in the presence of cyanide, a nucleophilic catalyst. The corresponding ethyl trans-enoates 8b and 8c were prepared in a similar fashion. Exposure of cis-enals 5b and 5c to manganese oxide in the presence of sodium cyanide in ethanol provides the ethyl cis-enoates 7b and 7c, respectively. Exposure of 7b and 7c to trimethylphosphine in dilute butanol results in formation of the corresponding ethyl trans-enoates 8b and 8c.
Scheme 1.

Conversion of D-glyceraldehyde adducts 1b and 1c to isomeric enoates 6b, 8c and 8b, 8c representing a formal synthesis of all eight L-hexoses via serial catalyst-directed diastereofacial selection.a
aCited yields are of isolated material.
As reported by Sasaki,4e Sharpless asymmetric dihydroxylation of the diastereomeric methyl cis-enoates 6b and 6c delivers diols 9a, 9b, 9e and 9f, which have been transformed to L-talose, L-gulose, L-mannose and L-allose, respectively. Sharpless asymmetric dihydroxylation of the diastereomeric ethyl trans-enoates 8b and 8c delivers diols 9c, 9d, 9g and 9h, which have been transformed to L-idose, L-galactose, L-altrose and L-glucose, respectively. Diastereofacial selectivities obtained using the indicated pseudo-enantiomeric osmium-based catalysts are indicated explicitly for the convenience of the reader.
In summary, we report catalyst-directed diastereoselectivity in the hydrogenative coupling of acetylene to aldehydes 1a–4a. Further, through sequential catalyst-directed diastereoselective hydrogenative carbonyl-(Z)-butadienylation-olefin asymmetric dihydroxylation, a concise formal synthesis of all eight L-hexoses is achieved from L-glyceraldehyde acetonide 1a. These studies demonstrate the utility of serial catalyst-directed diastereofacial selection as a means for the controlled preparation of contiguous stereochemical arrays.
Supplementary Material
Acknowledgments
Acknowledgment is made to the Welch Foundation, the NIH-NIGMS (RO1-GM069445) and the ACS-GCI Pharmaceutical Roundtable for partial support of this research. Oliver Briel of Umicore is thanked for the kind donation of [Rh(cod)2]BARF.
Footnotes
Supporting Information Available. Experimental procedures and tabulated spectral data and scanned images of 1H and 13C NMR spectra for all new compounds. This material is available free of charge via the internet at http://pubs.acs.org.
References
- 1.For selected reviews encompassing de novo synthetic approaches to monosaccharides, see: Zamoiski A, Banaszek A, Grynkiewicz G. Adv Carbohydr Chem Biochem. 1982;40:1.Hudlicky T, Entwistle DA, Pitzer KK, Thorpe AJ. Chem Rev. 1996;96:1195. doi: 10.1021/cr9403300.Gijsen HJM, Qiao L, Fitz W, Wong CH. Chem Rev. 1996;96:443. doi: 10.1021/cr950031q.Vogel P. In: Glycoscience. Fraser-Reid BO, Tatsuta K, Thiem J, editors. II Chapter 4.4. Springer-Verlag; Berlin: 2001. p. 1023.
- 2.(a) Fischer E. Ber Dtsch Chem Ges. 1890;23:370. [Google Scholar]; (b) Fischer E. Ber Dtsch Chem Ges. 1890;23:799. [Google Scholar]
- 3.For monosaccharide synthesis employing alkene enantioselective epoxidation, see: Ko SY, Lee AWM, Masamune S, Reed LA, III, Sharpless KB, Walker FJ. Science. 1983:949. doi: 10.1126/science.220.4600.949.Also see reference 9a.
- 4.For monosaccharide synthesis employing enantioselective alkene dihydroxylation, see: Harris JM, Keranen MD, O’Doherty GA. J Org Chem. 1999;64:2982. doi: 10.1021/jo990410+.Takeuchi M, Taniguchi T, Ogasawara K. Synthesis. 1999:341.Harris JM, Keranen MD, Nguyen H, Young VG, O’Doherty GA. Carbohydr Res. 2000;328:17. doi: 10.1016/s0008-6215(00)00031-8.Ahmed MdM, Berry BP, Hunter TJ, Tomcik DJ, O’Doherty GA. Org Lett. 2005;7:745. doi: 10.1021/ol050044i.Ermolenka L, Sasaki NA. J Org Chem. 2006;71:693. doi: 10.1021/jo0521192.
- 5.For monosaccharide synthesis employing enantioselective Payne rearrangement, see: Covell DJ, Vermeulen NA, Labenz NA, White MC. Angew Chem Int Ed. 2006;45:8217. doi: 10.1002/anie.200603321.
- 6.For monosaccharide synthesis employing enantioselective organocatalyzed aldol addition, see: Northrup AB, MacMillan DWC. Science. 2004;305:1752. doi: 10.1126/science.1101710.Enders D, Grondal C. Angew Chem Int Ed. 2005;44:1210. doi: 10.1002/anie.200462428.
- 7.For hydrogen-mediated couplings of acetylene to carbonyl compounds and imines, see: Kong JR, Krische MJ. J Am Chem Soc. 2006;128:16040. doi: 10.1021/ja0664786.Skucas E, Kong JR, Krische MJ. J Am Chem Soc. 2007;129:7242. doi: 10.1021/ja0715896.
- 8.For selected reviews of hydrogenative C-C coupling, see: Ngai MY, Kong JR, Krische MJ. J Org Chem. 2007;72:1063. doi: 10.1021/jo061895m.Iida H, Krische MJ. Top Curr Chem. 2007;279:77.Skucas E, Ngai MY, Komanduri V, Krische MJ. Acc Chem Res. 2007;40:1394. doi: 10.1021/ar7001123.
- 9.For selected examples of catalyst directed diastereofacial selection, see: Minami N, Ko SS, Kishi Y. J Am Chem Soc. 1982;104:1109.Kobayashi S, Ohtsubo A, Mukaiyama T. Chem Lett. 1991:831.Hammadi A, Nuzillard JM, Poulin JC, Kagan HB. Tetrahedron: Asymmetry. 1992;3:1247.Doyle MP, Kalinin AV, Ene DG. J Am Chem Soc. 1996;118:8837.Trost BM, Calkins TL, Oertelt C, Zambrano J. Tetrahedron Lett. 1998;39:1713.Balskus EEP, Jacobsen EN. Science. 2007;317:1736. doi: 10.1126/science.1146939.Also, see reference 3.
- 10.For exhaustive hydrogenation of conjugated dienes catalyzed by iridium, see: Cui X, Burgess K. J Am Chem Soc. 2003;125:14212. doi: 10.1021/ja037653a.and references therein.
- 11.O-Benzyl derivative of adduct 1b, Ito M, Kibayashi C. Tetrahedron. 1991;45:9329.Adduct 2c, Fujita M, Hiyama T. J Org Chem. 1988;53:5415.Adduct 3b, Reetz MT, Rolfing K, Greibenow N. Tetrahedron Lett. 1994;35:1969.Adduct 4b, Barrow JC, Coburn CA, Nantermet PG, Selnick HG, Stachel SJ, Stanton MG, Stauffer SR, Zhuang L, Davis JR. WO 2005/065195. International Patent. 2005
- 12.For Johnson-Lemieux reaction of conjugated dienes, see: Sakya SM, Suarez-Contreras M, Dirlam JP, O’Connell TN, Hayashi SF, Santoro SL, Kamicker BJ, George DM, Ziegler CB. Bioorg Med Chem Lett. 2001;11:2751. doi: 10.1016/s0960-894x(01)00567-4.Cho CW, Krische MJ. Org Lett. 2006;8:891. doi: 10.1021/ol052976s.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.