

Abstract
The innate immune system responds within minutes of infection to produce type I interferons and pro-inflammatory cytokines. Interferons induce the synthesis of cell proteins with antiviral activity, and also shape the adaptive immune response by priming T cells. Despite the discovery of interferons over 50 years ago, only recently have we begun to understand how cells sense the presence of a virus infection. Two families of pattern recognition receptors have been shown to distinguish unique molecules present in pathogens, such as bacterial and fungal cell wall components, viral RNA and DNA, and lipoproteins. The first family includes the membrane-bound toll-like receptors (TLRs). Studies of the signaling pathways that lead from pattern recognition to cytokine induction have revealed extensive and overlapping cascades that involve protein-protein interactions and phosphorylation, and culminate in activation of transcription proteins that control the transcription of genes encoding interferons and other cytokines. A second family of pattern recognition receptors has recently been identified, which comprises the cytoplasmic sensors of viral nucleic acids, including MDA-5, RIG-I, and LGP2. In this review we summarize the discovery of these cytoplasmic sensors, how they recognize nucleic acids, the signaling pathways leading to cytokine synthesis, and viral countermeasures that have evolved to antagonize the functions of these proteins. We also consider the function of these cytoplasmic sensors in apoptosis, development and differentiation, and diabetes.
Keywords: Antiviral innate immunity, MDA-5, RIG-I, domain grafting, cell signaling, apoptosis, viral pathogenesis
1. Introduction
The number of pathogens that we encounter daily is astronomical. Most are halted by an efficient defense system that has evolved over millions of years in the face of microbial infections. An important component of this defense arsenal is the innate immune system, which responds within minutes of infection to produce type I interferons (Isaacs & Lindenmann, 1957; Rubinstein, et al., 1979; Friesen, et al., 1981; Pestka & Baron, 1981). These antiviral proteins are produced by infected cells and lead to the synthesis of cell proteins, which halt viral replication, and also shape the adaptive immune response by priming T cells. Additionally, interferons also modulate a plethora of other important cellular functions including cell growth and differentiation, histocompatibility and tumor antigen expression, gene expression and anti-tumor effects (Fisher, et al., 1983; Fisher & Grant, 1984; Giacomini, et al., 1984; Grant, et al., 1985; Greiner, et al., 1984; Huang, et al., 1999a; Huang, et al., 1999b; Jiang, et al., 2000; Moulton, et al., 1992; Sen & Sarkar, 2007; Su, et al., 2008).
Despite the discovery of interferons over 50 years ago, only recently have we begun to understand how cells sense the presence of a virus infection and initiate cytokine synthesis. First insights came from the discovery of the toll-like receptors (TLRs) during a study of genes essential for establishment of the dorsal-ventral axis in Drosophila (Nusslein-Volhard & Wieschaus, 1980). The TLRs were subsequently shown to be transmembrane proteins that distinguish unique molecules present in pathogens, such as bacterial and fungal cell wall components, viral RNA and DNA, and lipoproteins (reviewed in Mezhitov, 2007). We now understand that pathogens are recognized as foreign by a family of host pattern-recognition receptors. Examination of the signaling pathways that lead from pattern recognition to cytokine induction have revealed extensive and overlapping cascades that involve protein-protein interactions and phosphorylation. These culminate in activation of transcription proteins that control the transcription of genes encoding interferons and other cytokines (reviewed in Kawai & Akira 2008).
A second family of pattern recognition receptors has recently been identified, which comprises the cytoplasmic sensors of viral nucleic acids, including MDA-5, RIG-I, and LGP2. Here we review the discovery of these proteins, how they recognize nucleic acids, the signaling pathways leading to cytokine synthesis, and viral countermeasures that have evolved to antagonize the functions of these proteins. We also consider the role of these cytoplasmic sensors in apoptosis, development and differentiation, and diabetes.
2. Characterization of RIG-I and MDA-5
2.1 Identification of RIG-I and MDA-5
Retinoic acid-inducible gene I (RIG-I, also known as DDX58) and melanoma differentiation associated gene-5 (MDA-5, also known as Helicard or IFIH1) are virus sensors expressed ubiquitously in the cytoplasm. RIG-I was initially identified as a gene induced in acute promyelocytic leukemia cells after treatment with retinoic acid (Sun, 1997). A few years later its role as an antiviral protein was reported. In fact, screening an expression cDNA library obtained from IFN-β-treated cells lead to the isolation of RIG-I (Yoneyama, et al., 2004). An IRF-reporter gene was introduced in L929 cells together with the cDNA library, and clones were selected for their ability to induce the promoter after transfection of poly (I:C) (Yoneyama, et al., 2004). Recently a splice variant of RIG-I has been identified (Gack, et al., 2008) which carries a deletion (residues 36-80) within the first caspase activation and recruitment domain (CARD) (Fig.1).
Fig. 1.

Schematic representation of the primary structure and functional domains of MDA-5, RIG-I, RIG-I-SV (splice variant) and LGP2. CARD: caspase activating recruitment domain. RD: repressor domain. CTD: C terminal domain.
MDA-5 was identified in a differentiation induction subtraction hybridization (DISH) screen (Huang et al., 1999a; Huang et al., 1999b) that was designed to define genes regulated as a function of induction of terminal differentiation in human HO-1 melanoma cells (Jiang and Fisher, 1993). These DISH genes, many of which represented unique sequences not reported in the 1999 gene database, were named melanoma differentiation associated (mda) genes. mda-5 was one such novel upregulated DISH gene in HO-1 human melanoma cells induced to irreversibly lose growth potential and terminally differentiate by treatment with IFN-β and mezerein, a protein kinase C-activating compound (Kang, et al., 2002).
2.2 Functional domains
RIG-I and MDA-5 are DExD/H RNA helicases, which possess two (CARD) domains at their amino terminus (Fig.1). Together with LGP2, laboratory of genetics and physiology 2, they form the RIG-I-like receptors (RLRs) family. LGP2 possesses only the helicase domain and lacks the CARD domain (Takeuchi & Akira, 2008; Yoneyama & Fujita, 2008). RIG-I and MDA-5 share approximately 25% homology within the CARD domain regions and 40% within the helicase domain.
RIG-I and LGP2 have a similar repressor domain (RD) localized at the carboxyl terminus (Saito, et al., 2007). Evidence suggests an interaction between the RIG-I repressor region (aa723-925) and the CARD and helicase domains (helicase linker region aa 420-627). Interestingly, over-expression of the repressor domain blocks RIG-I-mediated signaling.
Recently, a C-terminal domain (CTD) has been described, which partly overlaps with the previously identified RD. The structure of the CTD of RIG-I has been determined by X-ray crystallography (Cui, et al., 2008) and nuclear magnetic resonance (NMR) (Takahasi, et al., 2008). It consists of a basic concave surface and the opposite side contains acidic residues. A positive cleft is the binding site for dsRNA and 5′ ppp-RNA and also encompasses the biological activity of signal repression. The crystal structure of RD reveals a zinc-binding domain coordinated by four cysteines (C810, C813, C864 and C869) conserved also in MDA-5 and LGP2. Mutational studies have shown that the zinc coordination site is a key structural motif and is essential for RIG-I signaling.
The CARD is the effector domain, that transduces the signal when the molecule is activated. Overexpression of CARD induces constitutive signaling independent of viral infection (Yoneyama, et al., 2004). It has also been shown that the CARDs negatively regulate the ATPase activity of RIG-I preventing its signaling in the absence of viral RNA (Gee, et al., 2008).
Based on structural and functional studies, a model has been proposed for RIG-I activation. In the absence of its ligand, non-self RNA generated during viral infection, RIG-I is in an inactivated form. Binding of dsRNA or 5′ ppp-RNA to the basic cleft in the CTD induces a conformational change, that in the presence of ATP causes uncovering of the CARDs, that are then able to interact with the adaptor protein IPS-1 and transduce the signal (Takahasi, et al., 2008), (Fig. 2).
Fig. 2. Model for activation of RIG-I and MDA-5 by RNA.
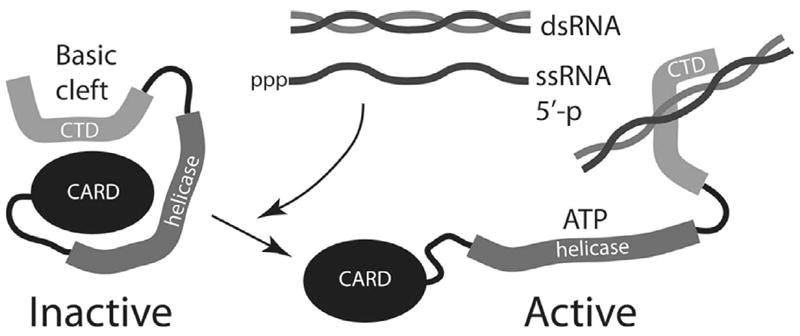
In the absence of RNA, the sensor molecule is folded in an inactive state, with the CARD domain occluded by the CTD. The latter also contains the binding sites for RNA. When RNAs are produced in virus-infected cells (either dsRNA or ssRNA with 5’-phosphates), these bind the CTD and cause a conformational change that exposes the CARD domain. ATP is required for the conformational change. The CARD domain then interacts with downstream signaling molecules, leading to IFN transcription.
2.3 Subcellular distribution of RIG-I and MDA-5
Recent evidence suggests that RIG-I localizes to membrane ruffles in non-polarized epithelial cells where it associates with the F-actin cytoskeleton (Mukherjee et. al., 2009). MDA-5, however, localizes to the cytoplasm with no appreciable co-localization with F-actin. The specific distribution of RIG-I to the membrane ruffles is dependent on RacGTPase activity (Mukherjee et al., 2009). Deletional analysis has shown that association of RIG-I with the cytoskeleton was dependent on CARDs.
In further studies it has been shown that RIG-I is able to induce cellular migration through its actin association (Mukherjee et. al., 2009). RIG-I, unlike MDA-5, is also localized to apical junction complexes in polarized epithelial and endothelial cells. The biological consequences of the interaction of RIG-I with F-actin are significant. It appears that actin depolymerization results in RIG-I redistribution and activation of IRF3 and NFκB and IFNβ promoter activity (Mukherjee et. al., 2009).
3. rig-I and mda-5 promoter regulation
rig-I was first cloned as a retinoic acid (RA)-inducible gene (Sun, 1997). However, the molecular mechanism by which RA regulates rig-I expression remains to be determined. The promoter region of rig-I has not been mapped for RA-responsive elements and with the accrual of information on the role of rig-I in RA signaling, efforts have not been expended in analyzing RA-regulation of rig-I. On the other hand, both rig-I and mda-5 are type I IFN-inducible genes and analysis of the ~2-kb promoter region of human rig-I revealed interesting findings (Su, et al., 2007). The promoter was more active in diverse normal cells compared to their malignant counterparts that correlated with higher rig-I mRNA expression in normal cells compared to cancer cells. In an experimental mouse brain tumor model, rig-I expression was abundantly detected in the surrounding normal brain while it was hardly detectable in the tumor tissue. In both normal and tumor cells rig-I promoter activity could be induced by IFN-β or by poly(I).poly(C). Serial deletion and mutation analysis identified an interferon regulatory factor-1 (IRF-1)-binding site just proximal to the transcription start site that conferred the basal, IFN-β- and dsRNA-mediated induction of the rig-I promoter. As a corollary to rig-I, IRF-1 expression was also higher in normal cells than that in cancer cells and IRF-1 induction by IFN-β followed a similar kinetics to that of rig-I. IRF-1 is a tumor suppressor and the expression profile of rig-I together with its regulation by IRF-1 indicates that rig-I might also have tumor suppressor properties. Although mda-5 was cloned as a type I IFN-inducible gene and mda-5 induction by type I IFN has been studied extensively (Kang, et al., 2004; Kang, et al., 2002), as yet no studies have been published analyzing in detail the promoter region of mda-5. It would be useful to have a comparative study between rig-I and mda-5 promoters, to check if the mda-5 promoter also displays normal cell specificity and whether IFN regulation of the mda-5 promoter is controlled by the classical IFN-stimulated regulatory elements (ISRE) or by other factors, such as IRF-1. More importantly, these promoters might be screened for identification of small molecules with potential anti-viral properties.
4. The RIG-I and MDA-5 signaling pathway
4.1 Recognition of RNA
A role for RIG-I as a cytoplasmic sensor of viral RNA was first discovered during a screen for molecules involved in intracellular dsRNA-induced expression of IFN (Yoneyama, et al., 2004). Overexpression of RIG-I in mouse L929 cells lead to increased activation of the IFN-β promoter after introduction of dsRNA into cells. Activation of this promoter was shown to be dependent upon the RIG-I helicase domain. RIG-I was found to bind poly(I:C) linked to agarose beads, but not single stranded RNA (poly(A)) or dsDNA. Synthesis of RIG-I also augmented the IFN response of cells infected with Newcastle disease virus, and reduced the viral yield of vesicular stomatitis virus, a rhabdovirus, and encephalomyocarditis virus (EMCV), a picornavirus. Based on these observations, it was suggested that RIG-I specifically recognizes dsRNA produced during the replication of RNA viruses (Yoneyama, et al., 2004). However, the interaction of RIG-I with its RNA ligand was clearly not sufficient for induction of IFN. This conclusion came from the observation that an amino acid change in the ATP binding domain of RIG-I blocked induction of IFN, but did not affect the ability of the protein to bind dsRNA (Yoneyama, et al., 2004).
The importance of RIG-I in sensing RNA virus infections was emphasized by the results of experiments with mice lacking the rig-I gene (Kato, et al., 2005). The production of IFN in response to infection with Sendai and Newcastle disease viruses (two paramyxoviruses), and VSV was abrogated in cDCs from rig-I-/- mice. In contrast, IFN production by pDCs was not dependent upon RIG-I, but upon toll-like receptors. These observations indicated that sensing of viral RNA by RIG-I occurs in a cell-type dependent manner.
MDA-5 was originally implicated in the recognition of poly(I:C) and was shown to bind this synthetic nucleic acid, although with less affinity than RIG-I (Kang, et al., 2002; Yoneyama, et al., 2005). Overproduction of MDA-5 inhibited the growth of EMCV and VSV, as was also observed for RIG-I (Yoneyama, et al., 2005). In another study, it was shown that MDA-5, RIG-I and TLR3 are activated during measles virus infection (Berghall, et al., 2006) and overexpression of MDA-5 but not RIG-I or TLR3 leads to enhanced IFN-β promoter activity in MV-infected A549 cells, suggesting that MDA-5 is involved in MV-induced expression of antiviral cytokines in human epithelial cells (Berghall, et al., 2006). It has also been demonstrated that RIG-I and MDA-5 expression, but not TLR3, resulted in the activation of the IFN-β promoter in influenza A virus-infected epithelial cells (Siren, el al., 2006), suggesting that RIG-I and MDA-5 mediate influenza A virus-induced IFN-β synthesis in epithelial cells (Siren, el al., 2006).
Gene disruptions provided evidence for differences in RNA sensing (Gitlin, et al., 2006; Kato, et al., 2006). Administration of poly(I:C) to wild type or rig-I-/- mice lead to similar levels of cytokines, including IFNs, in sera. In contrast, mda-5-/- mice did not produce IFN or interleukins in response to poly(I:C) (Kato, et al., 2006). Cultured bone-marrow-derived DCs isolated from rig-I-/- or wild type mice produced similar levels of IFN after delivery of poly(I:C) into the cytoplasm. In contrast, after stimulation with poly(I:C), production of IFN-α/β by bone-marrow DCs, macrophages, or fibroblasts derived from mda-5-/- mice was severely impaired. Fibroblasts from mda-5-/- mice responded normally to dsRNAs transcribed in vitro, while cells from rig-I-/- mice did not. These dsRNAs were produced by in vitro transcription of the mouse lamin A/C gene to produce (+) and (-) strand RNAs, which were subsequently annealed. Therefore, it was concluded that MDA-5 recognizes poly(I:C) in vivo and in vitro, while RIG-I can recognize poly(I:C) in vitro, but not in vivo; the latter protein principally recognizes RNA transcripts.
IFN production after infection of cultured cells with paramyxoviruses, VSV, influenza virus, and Japanese encephalitis virus (a flavivirus) was impaired in MEFs from rig-I-/- mice. In contrast, RIG-I was dispensable for production of IFN in cells infected with EMCV. GMCSF-DCs from rig-I-/- mice failed to produce IFN after transfection with VSV RNA, but not after transfection of EMCV RNA. The authors concluded that in mice, RIG-I recognizes dsRNA produced during RNA virus replication, and not poly(I:C) (Kato, et al., 2006).
The conclusion that the ligand for RIG-I is dsRNA was not consistent with the fact that many negative-strand RNA viruses, such as members of the paramyxovirus and orthomyxovirus families, do not produce dsRNA during their replicative cycles. It was subsequently discovered that RIG-I also recognizes RNA molecules with a 5′ ppp (Hornung, et al., 2006; Pichlmair, et al., 2006; Plumet, et al., 2007). The triphosphate is found at the 5′-end of in vitro transcribed RNAs, explaining previous results indicating that these molecules are sensed by RIG-I. In cells infected with measles virus, the leader RNA, a small, uncapped RNA with 5′ ppp, activates RIG-I (Hornung, et al., 2006). Similarly, the influenza virus single stranded genomic RNA, which bears 5′-phosphates, is also a ligand for RIG-I (Pichlmair, et al., 2006). Removal of the 5′-phosphates does not affect RNA binding to RIG-I, but abrogates IFN induction. Cellular RNAs do not display a 5′-triphosphate: this structure is either removed or replaced with a 5′-cap structure. Therefore, ‘self’ RNAs do not activate RIG-I.
Infection of mice lacking the gene encoding MDA-5 revealed that this protein is critical for detection of infection with picornaviruses, but not flaviviruses, influenza viruses, or paramyxoviruses (Gitlin, et al., 2006; Kato, et al., 2006). How this selective recognition was achieved was explained by the finding that RIG-I, but not MDA-5, recognizes RNA with 5′-ppp. The 5′-ends of the RNA genomes and RNA replication intermediates of picornaviruses are not phosphorylated, but are covalently linked to a small protein, VPg. Such RNA molecules therefore would not be recognized by RIG-I. Picornavirus infections are sensed by MDA-5 in mice lacking RIG-I. However, in the absence of MDA-5, picornavirus genomes are not sensed. It is not known why dsRNAs produced during the replicative cycles of picornaviruses are not recognized by RIG-I – perhaps they are too long (see below). Although poly(I:C) and EMCV viral RNA can activate MDA-5, the actual ligand in virus-infected cells has not been identified. Candidates include the highly structured regions of RNA in the 5′- and 3′-noncoding regions of the viral genome.
Although RIG-I contains a domain with homology to RNA helicases, it does not have an RNA-binding motif, and therefore how it interacts with RNA was initially not understood. The C-terminal domain (amino acids 792-925) of recombinant RIG-I binds dsRNA (including short RNAs, ~25 bp) and 5′ ppp RNA within this RNA (Takahasi, et al., 2008). The amino acid sequence of RIG-I indicates that it is a ligand-dependent ATPase (Cui, et al., 2008; Gee, et al., 2008; Takahasi, et al., 2008). When the ATP binding site at amino acid 270 is altered, RIG-I cannot induce IFN synthesis, but it can still bind dsRNA and 5′ ppp RNA (Takahasi, et al., 2008). Although RIG-I has RNA helicase activity, it is not required for triggering IFN synthesis – consistent with the fact that single-stranded 5′ ppp RNA is a functional ligand for RIG-I. The precise role of ATP in RIG-I activation is not known; it has been suggested to play a role in the structural change of the RNA-RIG-I complex to an active form (Yoneyama & Fujita, 2008).
The C-terminal domain (CTD) of RIG-I contains a repressor domain (aa 723-925), which can bind the CARD and helicase domain and inhibit RIG-I mediated signaling (Saito, et al., 2007). The repressor domain is believed to act in cis by masking the CARD domain. A region that overlaps with the repressor domain (aa 792-925) was identified by limited protease digestion of RIG-I bound to RNA (Takahasi, et al., 2008). This fragment is sufficient to bind dsRNA or 5′ -ppp RNA. The atomic structure of the CTD reveals similarity to the mammalian suppressor of Sec4 (Mss4), a guanine nucleotide exchange factor that is not an RNA-binding protein (Cui, et al., 2008; Takahasi, et al., 2008). A positively-charged cleft on one side of the molecule has been suggested to be the RNA recognition surface. Alteration of amino acids within this cleft reduces RNA binding. Based on the observation that the CTD binds 5′ -ppp RNA as a dimer, it has been proposed that one dsRNA molecule can bind multiple CTDs. Precise understanding of the RIG-I – ligand interaction awaits structural determination of the complex.
Amino acid changes in the basic cleft that inactivate RNA recognition do not produce constitutively active RIG-I, suggesting that the surfaces that bind RNA and repressor functions do not overlap. This finding, together with the observation that RNA recognition does not require ATP binding or hydrolysis, has lead to a model in which the repressor domain maintains RIG-I in a closed, inactive conformation (Figure 2). Upon binding the RNA ligand on the concave surface of the CTD, a conformational change (possibly requiring ATP) occurs, exposing the CARD domain, and allowing the CARD domain to initiate a signal transduction pathway leading to IFN synthesis.
The mechanism for the discrimination of RNA ligands by RIG-I and MDA-5 is not known. Both proteins have a CTD with common residues, although there are differences in surface charge that may influence substrate recognition (Yoneyama & Fujita, 2008). Commercially available poly(I:C), which has been frequently used to study RNA sensing, is at least 3-kb in length. By producing poly (I:C) of various lengths it was determined that short segments of the polymer (~300-bp) do not activate MDA-5, but are potent ligands for RIG-I (Kato, et al., 2008). In contrast, longer segments of poly(I:C) preferentially activate MDA-5. Consistent with these findings is the observation that the short dsRNA segments of the reovirus genome (1.2-1.4-kbp) selectively activate RIG-I, while the longer RNA segments (3.4-kbp) activate MDA-5. The mechanism underlying the discrimination between long and short dsRNA of MDA-5 and RIG-I remains to be elucidated. One possibility is that long segments of dsRNA bind RIG-I but induce a conformation of the protein that cannot engage in downstream signaling (Takahasi, et al., 2008). This hypothesis is supported by the observation that binding of short dsRNA and 5′ ppp RNA to RIG-I leads to the formation of 17- and 30-kD protease resistant fragments; in contrast, binding of long dsRNA causes formation of a 60-kD protease resistant fragment (Takahasi, et al., 2008).
It is likely that there are exceptions to the simple RNA recognition rules described above for MDA-5 and RIG-I. For example, it has been reported that IFN production occurs during infection with dengue and West Nile viruses in the absence of either RIG-I or MDA-5 (Fredericksen, et al., 2008; Kato, et al., 2006; Loo, et al., 2008; Sumpter, et al., 2005). These results suggest that perhaps other sensors of viral RNA remain to be discovered.
Collectively, studies to date indicate that RIG-I and MDA-5 can recognize different types of viruses, although there is significant evidence to also indicate possible redundant roles for these two helicases, probably due to the fact that some viruses are able to inadvertently produce both long and short types of dsRNA species following infection. Both RIG-I and MDA-5 require IPS-1 to direct innate immune defenses and are cooperatively essential for mounting an effective response to positive- and negative-stranded virus infection.
4.2 Induction of cytokine signaling and the role of adaptors FADD/RIP1, IPS-1 and a new player STING
4.2.1 FADD/RIP1
While it was apparent that RIG-I and MDA-5 were important for intracellular recognition of viral RNAs, it was less obvious which molecules resided downstream of these helicases and facilitated their ability to activate IRF3, IRF7 and NF-κB, required for efficient type I IFN transcription. At approximately the same time as RIG-I was reported to be important in TLR-independent signaling, another study also reported the discovery of a TLR-independent innate immune pathway that involved the death domain containing proteins FADD and RIP1 (Balachandran, et al., 2004). FADD (Fas associated death domain) was previously isolated by its ability to interact with the death receptor Fas, and to mediate caspase-8 dependent cell death in response to FasL (Chinnaiyan, et al., 1995). In contrast, RIP1 (receptor-interacting serine-threonine kinase 1) has been reported to play a key role in facilitating tumor necrosis factor-alpha (TNF-α) signaling and is recruited by TRADD (TNFR1-associated death domain protein) to the TNF receptor to induce NF-κB activation (Hsu, et al., 1996). Both FADD and RIP1 can interact through death domain association, with TRADD to mediate TNF-α-induced apoptosis (Kelliher, et al., 1998; Micheau & Tschopp, 2003). In the absence of FADD or RIP1, cells were also found to be defective in their ability to produce type I IFN (Balachandran, et al., 2004; Balachandran, et al., 2007). Normal MEFs lacking these death domain-containing molecules were extremely sensitive to RNA virus infection and also exhibited defective IFN-mediated antiviral gene activity through, amongst other things, a lack of IRF7 production. Subsequent studies indicated that FADD and RIP1 are essential for RIG-I- and MDA-5-mediated signaling and likely formed an ‘innateosome’ complex with these helicases to facilitate the activity of NF-κB and IRF3 and IRF7 (Balachandran, et al., 2004; Balachandran, et al., 2007). Of interest is that insect homologues, referred to as IMD (for Rip1) and dFADD (for FADD) were also shown to play a critical role in TLR-independent signaling in Drosophila and were essential for protecting flies against gram-negative bacteria (Ferrandon, et al., 2004; Georgel, et al., 2001; Lemaitre, et al., 1995; Leulier, et al., 2002; Naitza, et al., 2002). Insects lacking IMD or dFADD fail to activate the Drosophila IKK complex which is essential for the production of appropriate stress responses to anti-bacterial genes (Aggarwal & Silverman, 2008). Thus, FADD and RIP1 may be evolutionarily conserved death domain containing molecules important in host defense against pathogen infection.
4.2.2 IFN-β promoter stimulator 1 (IPS-1)
In addition to the studies described above related to FADD/RIP1, a number of groups in 2005 reported the isolation of a new adaptor molecule that also appeared to mediate RIG-I/MDA-5 function. One group utilized an expression cloning strategy to identify molecules that activated the IFN-β promoter and isolated a gene that they referred to as IFN-β promoter stimulator 1 (IPS-1) (Kawai et al., 2005). A second group isolated the same protein by screening uncharacterized molecules known to be potent inducers of NF-κB. They termed the protein VISA (for virus-induced signaling adaptor) (Xu el al., 2005). In contrast two other groups performed profile searches in human protein databases to identify new CARD-domain-containing proteins, similar to domains evident in RIG-I/MDA-5 and called the products Cardif (CARD adaptor inducing IFN-β) and MAVS (mitochondrial antiviral signaling) (Meylan, el al., 2005; Seth et al., 2005). Herein we will term the protein IPS-1.
IPS-1 appears to be expressed in a variety of tissues and is 540 amino acids in length, predicting a protein of 65-kDa. IPS-1 was found to activate type I IFN transcription, the NF-κB promoter as well IP-10, RANTES, and ISRE promoters, that are also regulated by the IRF family. IPS-1 was also found to associate with RIG-I and MDA-5, through CARD domain interactions, as well as with FADD and RIP1, which as mentioned earlier, are also required to facilitate RIG-I/MDA-5 activity.
RNAi knockdown of IPS-1 reduced the ability of poly(IC) and RIG-I/MDA-5 to induce type I IFN, but not TRIF, indicating that IPS-1 did not play a significant role in the TLR-pathway (although one group did report that IPS-1 associated with TRIF and TRAF6 to mediate bifurcation of the TLR3-triggered NF-κB and IRF-3 activation pathways) (Xu, et al., 2005). IPS-1 was unable to activate the IFN-α and IFN-β promoters in cells deficient in both TBK1 and IKKi, indicating that it lies upstream of these kinases (Kawai, et al., 2005; Meylan, et al., 2005; Q. Sun, et al., 2006; Xu, et al., 2005). Interestingly, IPS-1 was found to localize in the mitochondrial membrane, although little is known about protein partners that may associate with IPS-1 in this organelle (Sun, et al., 2006). It also remains to be determined how IPS-1 is able to associate with RIG-I/MDA-5 in the cell following virus infection.
Biochemical analysis has indicated that the CARD domain of IPS-1 is essential for signaling activity, although a transmembrane domain was reported as being required for IPS-1 dimerization, which was also deemed essential for activity. It was further found that NS3-4A, a multifunctional protein of hepatitis C virus (HCV) that has serine protease activity essential for the production of mature viral proteins, targeted IPS-1 to subvert interferon mediated antiviral activity (Meylan, et al., 2005). Thus, HCV-mediated resistance to IFN action may involve IPS-1 deregulation (Meylan, et al., 2005). IPS-1-/- mice appear viable and a variety of cells derived from these animals, such as MEFs, macrophages and conventional dendritic cells had impaired type I IFN responses to both RIG-I-recognized RNA viruses such as SeV, NDV, and VSV, as well as MDA-5-recognized viruses such as EMCV (Kumar, et al., 2006; Sun, et al., 2006). However plasmacytoid dendritic cells did not appear affected by the loss of IPS-1 and were able to produce IFN in response to SeV infection.
IPS-1 deficient animals were severely impaired in their ability to produce type I IFN as well as other cytokines such as IL-6 and IP-10, following infection with EMCV. Loss of IPS-1 was not found to affect TLR signaling, for example in response to LPS or exogenous poly(IC). Accordingly, IPS-1 deficient animals were extremely sensitive to lethal VSV infection and slightly more sensitive to EMCV infection, indicating the importance of this molecule in both RIG-I and MDA-5-mediated innate immune signaling (Kumar, et al., 2006; Sun, et al., 2006). While one group indicated that IPS-1 was partially required to facilitate DNA-virus mediated signaling (vaccinia), other reports indicated that IPS-1 was not required for IFN induction by cytosolic DNA or Listeria monocytogenes (Kumar, et al., 2006; Sun, et al., 2006).
In summary, the experimental data indicate that IPS-1 is a potent inducer of type I IFN following oligermerization and largely resides in the membrane of mitochondria. IPS-1 facilitates both RIG-I and MDA-5 signaling through CARD domain association and is essential for in vivo innate immunity to RNA, but not DNA virus infection. While the exact signaling mechanisms remain to be determined, it has recently been reported that TRADD may be recruited to IPS-1 to orchestrate ‘innateosome’ complex formation with the E3 ubiquitin ligase TRAF3 and TANK, and with FADD and RIP1, which culminates in the activation of IRF3 and NF-κB (Michallet et al., 2008; Hacker et al., 2006; Oganesyan et al., 2006). Further work will undoubtedly elucidate this complex series of events.
4.2.3 Stimulator of IFN genes (STING)
While significant progress has been made in unraveling the molecular components of TLR-independent innate signaling in response to RNA viruses, less was known about innate signaling in response to DNA virus infection. While plasmacytoid dendritic cells were known to utilize TLR9 in the recognition of CpG-containing DNA, it was apparent that important TLR-independent pathways also existed to recognize DNA in alternative tissues, the mechanisms of action of which remain to be determined (Takeuchi & Akira, 2007). However, in 2008, a molecule referred to as STING (Stimulator of IFN Genes) was isolated using an expression cloning strategy to identify molecules that activated the IFN-β promoter (Ishikawa & Barber, 2008). This molecule was predicted to be 379-aa in length and comprised several putative transmembrane regions in the amino terminal region. Two other groups also isolated STING around this time (referred to as MITA and MYPS) (Jin, et al., 2008; Zhong, et al., 2008).
Confocal analysis indicated that STING was ubiquitously expressed, resided in the endoplasmic reticulum (ER) and following overexpression potently induced the activation of both NF-κB as well as IRF3 to stimulate type I IFN production (Ishikawa & Barber, 2008). STING deficient animals were generated and were viable and MEFs lacking STING were found to be sensitive to VSV and SeV infection. However, loss of STING did not appear to dramatically effect poly(IC) signaling. Indeed, co-immunoprecipitation experiments indicated that STING appeared to associate with RIG-I, rather than MDA-5, which may explain sensitivity to VSV but not poly(IC), which are dependent on RIG-I and MDA-5, respectively. IPS-1 was also found to associate with STING in co-immunoprecipitation experiments but it was not clear if this was a direct or indirect (complex) association. Of further interest was that STING was found by two hybrid screening to associate with SSR2/TRAP, a member of the TRAP complex comprising four subunits (α – Δ) that facilitates translocation of proteins into the ER following translation (Hartmann, et al., 1993; Ishikawa & Barber, 2008; Lipschutz, et al., 2003; Menetret, et al., 2005).
The TRAP complex is known to associate with the ‘translocon’, a protein complex that forms an aqueous pore spanning the ER membrane, that consists of three copies of the SEC61 heterotrimer. Thus, STING is predominantly an ER resident protein that may link RIG-I mediated intracellular innate signaling to the ‘translocon’. Speculatively, RIG-I may detect translating viral RNAs at the intersection of ribosome/ER ‘translocon’ association and require STING to exert effective innate immune function. The mitochondrial network is also known to associate with the ER and such mitochondria-associated membranes (MAM) may provide direct physical contact between the ER and mitochondria, thus facilitating a role for IPS-1 in the ‘innateosome’ complex (Bozidis, et al., 2007; Hartmann, et al., 1993; Ishikawa & Barber, 2008; Lipschutz, et al., 2003; Menetret, et al., 2005). Alternatively, STING may participate in mediating ER stress response pathways, but this remains to be verified.
Although it is not clear how signaling from the ‘translocon’ to IRF3/NF-kB occurs, it has recently been established that the ‘translocon’ physically associates with the ‘exocyst’—the octameric Sec6–Sec8 complex that also associates with the ER and tethers secretory vesicles to membranes, and facilitates protein synthesis and secretion (Guo & Novick, 2004; Lipschutz, et al., 2003). Recently, the ‘exocyst’ complex was found to recruit and activate TBK1 and play a role in type I IFN-β induction (Chien, et al., 2006). Preliminary analysis indicated that STING co-immunoprecipitated with TBK1, and that RNAi ablation of Sec5 also rendered cells defective in the STING function.
Importantly, the generation of STING deficient MEFs and macrophages yielded the surprising finding that STING was essential for recognizing non CpG DNA species, such as short double-stranded DNA oligonucleotides that are able to potently induce IFN (ISD), as well as bacterial DNA and poly d(AdT). Thus, STING may facilitate the detection of intracellular viral RNA and DNA species as well as B-form types of DNA in general, indicating convergence of these intracellular pathogen-associated molecular pattern (PAMP) recognition pathways.
4.3 RNase L and the antiviral response
The activation of signaling pathways by double stranded RNA molecules leads to the expression of IFNs and these, in turn, initiate expression of IFN-induced genes (ISGs). Double-stranded RNA is sensed by RIG-I and MDA-5. It had generally been considered that the RNA molecules arose as a by-product of viral infection; however, recent evidence suggests that cellular generated small RNAs can amplify the antiviral response (Malathi, et al., 2007). Specifically, it has been shown that the antiviral endoribonuclease, RNaseL, is activated by 2′, 5′ oligoadenylate, and produces small RNA fragments from cellular RNA which contribute to IFN production and this involves RIG-I, MDA-5 and IFN stimulator protein 1 (IPS-1) and IRF-3 (Malathi, et al., 2007). Using normal and RNaseL deficient mice, viral infection produces appreciably more IFN-β if RNaseL is expressed (Malathi, et al., 2007). Similarly the introduction of 2′-5′ oligoadenylate into normal mice, but not RNaseL null mice, caused a marked increase in levels of circulating IFN. It has previously been demonstrated that activation of RNaseL by 2′-5′ oligoadenylate increases expression of a number of IFN-stimulated genes (Malathi, et al., 2005). Although RNaseL is stimulated by cellular (self) RNAs, it is interesting to note that viral dsRNAs activate 2′-5′ oligoadenylate synthetase, which causes production of 2′-5′ oligoadenylate, activating RNaseL. RNaseL cleaves single-stranded RNA resulting in double-stranded (duplex) structures, which can then activate signaling by RIG-I and MDA-5, producing IFN.
5. Negative regulation of the signaling pathway
Limitation of IFN production is an essential physiological requisite necessary for the overall well-being of the organism. Following initiation of antiviral and antiproliferative responses or the activation of innate immunity, restriction of production of excess IFN must occur. The fact that there exist multiple mechanisms to control IFN levels confirms the importance of counteracting deleterious effects of IFN, that include chronic cellular toxicity and the initiation of inflammatory or autoimmune diseases.
A number of mechanisms contribute to the regulation of the RIG-I signaling pathway to limit its action under steady-state conditions. In particular, a number of negative regulators of RIG-I signaling have been characterized, although significantly fewer have been identified for MDA-5. Fundamentally, some of these mechanisms contribute to a negative feedback loop whereas others regulate steady-state levels.
5.1 Inhibition of RIG-I/MDA-5 by LGP2
Of the recently characterized novel regulators of RIG-I and MDA-5 signaling, most appear to function to maintain a tight control of virus-initiated IFN production. One such negative regulator is a member of the RLH family, LGP2, unlike RIG-I and MDA-5, lacks the N-terminal CARD required for activating IPS-1-dependent signaling events (see below). It has also been observed that RIG-I-mediated production of IFN can lead to an increase in RIG-I transcription, initiating an IFN amplification loop.
Under normal physiological conditions, RIG-I exists in an autorepressed state determined by its repressor domain (RD) located at the end of the C-terminus (Saito, et al., 2007). Besides this autoinhibition, which occurs in the absence of stimuli and controls the basal activity of RIG-I, several proteins have been found to have an inhibitory role in the RIG-I-mediated signaling pathway. LGP2 was the first protein characterized as an inhibitor of the RIG-I/MDA-5 signaling pathways. It exerts this function in different ways. One mechanism is by sequestering ligands from RIG-I/MDA-5 recognition. In fact LGP2 is also a DExD/H RNA helicase, which has a high homology with RIG-I and MDA-5 in the C-terminal region, but is completely devoid of the N-terminal region containing the effector CARD domain. Therefore, it can bind to RNA through its CTD, competing with RIG-I and MDA-5, but, lacking the N-terminal CARD, cannot interact with the adaptor protein IPS-1, losing the ability to transmit the signal (Rothenfusser, et al., 2005; Yoneyama, et al., 2004). Another way in which LGP2 exerts an inhibitory role on RIG-I is mediated by the presence of a RD located in the C-terminal region, analogous to that present in RIG-I. This domain associates with the CARD domain and with a linker region between the CARD and the RNA helicase domain and causes RIG-I dimerization that is necessary for its activation and binding with the downstream protein IPS-1. It was found that, through this analogous domain, LGP2 associates with RIG-I, acting in trans preventing RIG-I dimerization necessary for its activation (Saito, et al., 2007). LGP2 was also found to interact with the C-terminal and the transmembrane regions of IPS-1 competing for binding of the downstream kinase Ikki/Ikke essential for IRF3 activation (Komuro & Horvath, 2006). Therefore, LGP2 acts at two levels in the signaling pathway attenuating the production of IFN. Interestingly LGP2 is also an IFN-inducible gene and it is induced by viral infection playing a negative feedback role in the antiviral signaling loop.
Interestingly LGP2-/- mice are more resistant to VSV infection than wt animals, but are more susceptible to encephalomyocarditis (EMCV) infection (Venkataraman, et al., 2007). VSV produces RNA intermediates during replication, which are specifically recognized by RIG-I, whereas EMCV infection causes the activation of MDA-5. It might therefore be postulated that LGP2 is a positive regulator of MDA-5 mediated signaling pathways.
5.2 Inhibition by RNF125
The RIG-I and MDA-5 signaling pathway is also negatively regulated by the RING finger E3 ubiquitin ligase RNF125 (Arimoto, et al., 2007). This protein is IFN-inducible and by conjugating ubiquitin to RIG-I and MDA-5 causes their proteosomal degradation thereby decreasing their cellular levels. By mutational analysis it was shown that the N-terminal region of RIG-I, which contains the CARD domain, is the main substrate for the RNF125 protein, with minimal ubiquitination occurring in the C-terminal region. Overexpression of RNF125 in virus-infected or dsRNA-stimulated cells decreases IFN-β production, whereas its reduction, mediated by si-RNA knock-down, augments IFN-β expression. RNF125 also acts downstream of RIG-I and MDA-5, by targeting, for proteosomal degradation, the adaptor protein IPS-1. It is now clear that ubiquitination is not only a mechanism for targeting proteins for proteosomal degradation (usually through Lys 48-linked polyubiquitination) but can also play a role in promoting protein interactions or catalytic activation, regulating their functions (through Lys 63-linked ubiquitination) (Liu, et al., 2005). Ubiquitination is a reversible process in which the action of ubiquitin-conjugating enzymes is counteracted by de-ubiquitinating enzymes (DUB). In the case of RIG-I besides being ubiquitinated by RNF125 for proteosomal degradation, it is also ubiquitinated by TRIM25 causing activation.
5.3 Inhibition by de-ubiquitinating enzymes
Several de-ubiquitinating (DUB) enzymes have also been shown to regulate the level of ubiquitinated RIG-I, consequently acting as negative regulators of the RIG-I signaling pathway. The anti-apoptotic protein A20, a known inhibitor of NF-κB, blocks RIG-I-mediated signaling (Lin, et al., 2006). A20 is a peculiar protein that contains an ovarian tumor (OTU) domain at its N-terminus that has a de-ubiquitinating function and a C-terminal ubiquitin ligase domain. Both of these regions are necessary for inhibition of NF-κB (Wertz, et al., 2004), but only the C-terminal region is responsible for inactivation of RIG-I signaling. Stable transfection of the constitutively active domain of RIG-I confers more resistance to VSV replication and this effect is reversed by the addition of A20. In other studies, it has been shown that the de-ubiquitinazing enzyme A (DUBA) removes the lysine-63-linked polyubiquitin chain from the TNF receptor-associated factor 3 (TRAF3), resulting in the dissociation of TRAF3 from TANK-binding kinase 1 (TBK1) and consequently disrupting the signal (Kayagaki, et al., 2007). DUBA is another member of the ovarian tumor (OTU) family of de-ubiquitinating enzymes and was identified in a siRNA-based screen as an enzyme able to negatively regulate IFN production in the antiviral pathway. Knocking-down DUBA by siRNA leads to more IFN production and its overexpression has the opposite effect. The tumor suppressor CYLD (cylindromatosis), another OTU de-ubiquitinating enzyme that removes Lys 63-linked polyubiquitin chains, was also recently shown to negatively regulate RIG-I (Friedman, et al., 2008; M. Zhang, et al., 2008). CYLD negatively regulates the signaling function of RIG-I by direct interaction with RIG-I causing its de-ubiquitination. It also acts as a de-ubiquitinating enzyme of TBK1, the kinase that phosphorylates IRF3, leading to inactivation of IRF3 signaling pathway and IFN production. RIG-I is ubiquitinated in its CARD domain by the E3 ubiquitin ligase TRIM25, and this is necessary to render RIG-I active. CYLD acts as a de-conjugating enzyme, removing the K63 ubiquitin chain and inactivating the signal.
5.4 Inhibition by ISG15
Another protein that has an inhibitory effect on RIG-I signaling pathway is the ubiquitin-like IFN-stimulated gene 15 (ISG15). ISG15 was found to interact with RIG-I (Zhao, et al., 2005) and later it was shown that RIG-I ISGylation negatively regulates RIG-I (Kim, et al., 2008). ISG15 is a ubiquitin-like protein, which is strongly induced by IFNs during viral infection (Narasimhan, et al., 2005).
5.5 Inhibiton of MDA-5 by DAK
Using a yeast two-hybrid screen the dihydroxyacetone kinase (DAK) was shown to interact with MDA-5 and to have a specific inhibitory effect on MDA-5-mediated IFN signaling (Diao, et al., 2007). It was observed that overexpression of DAK inhibits activation of IRF3 and the IFN-β promoter, but does not inhibit the NF-κB activation pathway. Although MDA-5 and RIG-I are very similar in domain structure and function, DAK does not interact with RIG-I and does not inhibit the RIG-I-mediated signaling pathway. The interaction of DAK with MDA-5 is disrupted during viral infection, suggesting that DAK is a physiological inhibitor of MDA-5.
5.6 Inhibition by the autophagy complex Atg12-Atg5
The RIG-I/MDA-5 signaling pathway is also inhibited by Atg12-Atg5, a protein conjugate, which plays a key role in regulating autophagy (Jounai, et al., 2007). Specifically, Atg-deficient embryonic mouse fibroblasts (MEFs) are more resistant to VSV infection and produce more IFN than wt MEFs. Regulation of IFN production by the Atg12-Atg5 complex depends on binding to the CARD of RIG-I, MDA-5 and IPS-1. In an analogous fashion, Atg7-/- MEFs are also resistant to VSV infection, indicating that Atg12-Atg5 complex formation, which requires Atg7, is essential for inhibition of signaling.
5.7 Inhibition by NLRX1
NLRX1 is a member of the nucleotide-binding domain and leucine-rich repeat containing (NLR) protein family (Moore, et al., 2008). NLRX1 is a widely expressed protein that is generally localized to the outer mitochondrial membrane and has recently been shown to bind to IPS-1. The CARD domain on IPS-1 determines this interaction through a proposed nucleotide-binding domain in NLRX1 and results in marked inhibition of RIG-I and MDA-5 mediated IFN release. Thus, NLRX1 can be viewed as a negative regulator of RIG-I and MDA-5 activity such that depletion of NLRX1 with siRNAs results in increased virus-induced IFN production (Moore, et al., 2008). NLRX1 contains a mitochondrial targeting sequence, and it is notable that deletion of the leucine-rich repeat results in loss of the IPS-1 inhibitory activity. It appears that NLXR1 competes with RIG-I for IPS-1 possibly through interaction between the CARDs. As the level of NLXR1 does not change in response to IFN treatment or MDA-5 and RIG-I expression it appears that it acts to regulate steady-state antiviral responses.
5.8 Inhibition by caspase-1
In a recent study it was demonstrated that caspase-1, which functions as an inflammatory caspase and whose expression is activated during viral infection, negatively regulates RIG-I signaling activity by promoting RIG-I secretion, consequently controlling its intracellular levels (Kim & Yoo, 2008). It is not clear how RIG-I is secreted, but since RIG-I interacts with caspase-1 and it was found in the supernatant together with caspase-1, one hypothesis is that RIG-I secretion involves the ‘inflammasome’ complex. However, the physiological significance of RIG-I secretion is not yet clear.
Another recent report describes a RIG-I splicing variant (RIG-I SV) which was found to be induced during viral infection or upon IFN stimulation and shown to suppress RIG-I signaling (Gack, et al., 2008). The splicing variant lacks residues 36 to 80 located in the first CARD, therefore it cannot interact with TRIM25 or be ubiquitinated in the second CARD, losing its ability to bind the adaptor protein IPS-1 and consequently to carry out signal transduction. RIG-I-SV acts as a dominant inhibitor of the RIG-I–mediated antiviral response. In fact, RIG-I SV interacts with RIG-I WT inhibiting its multimerization and consequent interaction with IPS-1. This inhibition of virus-induced IFN signal transduction could potentially serve as a negative feedback mechanism.
6. Cell type specificity of RIG-I and MDA-5
In vivo evaluation of the importance of RIG-I and MDA5 in recognizing virus infection came through the generation of viable knockout mice that lacked these genes. rig-I-/- MEFs did not significantly produce type I IFN in response to a variety of negative-stranded RNA viruses, such as paramyxoviruses, rhabdoviruses and orthomyxoviruses, since the major mechanism of 5′-triphosphate viral RNA recognition was absent (Kato el al., 2005). Conventional dendritic cells (cDC’s) lacking rig-I (but not MyD88-/-TRIF-/- double knockout cDC’s), induced from bone marrow in the presence of GM-CSF, were also defective in their ability to manufacture type I IFN following exposure to the paramyxovirus, Newcastle disease virus (NDV). However, plasmacytoid dendritic cells (pDC’s) lacking RIG-I were able to normally generate type I IFN following NDV infection through activation of the TLR pathway since MyD88-/-TRIF-/- defective pDC’s had severely impaired type I IFN responses following similar virus exposure (Kato et al., 2005; Kato et al., 2006). Despite this, rig-I -/- mice remained sensitive to lethal infection by the above viruses. In contrast, macrophages, cDC’s and MEFs lacking MDA-5 were impaired in their ability to produce type I IFN in response to the positive-stranded virus EMCV (but not the above negative-stranded viruses) as well as poly(IC) (Kato et al., 2006; Gitlin et al., 2006). pDC’s from MDA-5 deficient animals exhibited normal ability to induce type I IFN following exposure to EMCV, in contrast to pDC’s lacking MyD88. Nevertheless, mice lacking MDA-5 remained highly susceptible to picornavirus infection. These data largely confirmed concomitant in vitro RNA binding analysis performed on these helicase proteins (as described earlier) (Takeuchi and Akira, 2008; Takeuchi and Akira 2007). For example, EMCV generates longer RNA species that are replicate intermediates compared to negative-stranded viruses, which are recognized by MDA-5 rather than RIG-I. However, it is worth noting that some viruses, such as the flaviviruses, dengue virus and Japanese encephalitis virus, were able to activate both the MDA-5 and RIG-I pathways in MEFs, probably because these viruses generated both long and short viral dsRNA structures following infection (Loo et al., 2008). These studies indicate that RIG-I and MDA-5 play a pivotal role in recognizing virus infection in most cell types, except in pDC’s, where the TLR (predominantly TLR 7) pathway appears to be the important mechanism in manifesting anti-viral host defense, for reasons that remain unknown.
7. Viral countermeasures
The various defense mechanisms that operate in healthy hosts have evolved for millions of years, yet are imperfect because viral genomes encode gene products that block every step of host defense. The RNA sensing pathways described in this review are no exception. Viral gene products may antagonize nearly every step of the sensing of RNA by RIG-I and MDA-5 (Figure 4). Understanding such viral countermeasures not only improves our understanding of innate sensing pathways, but may also suggest avenues for therapeutic intervention.
Fig. 4. Viral countermeasures in the RNA sensing and signaling pathway.
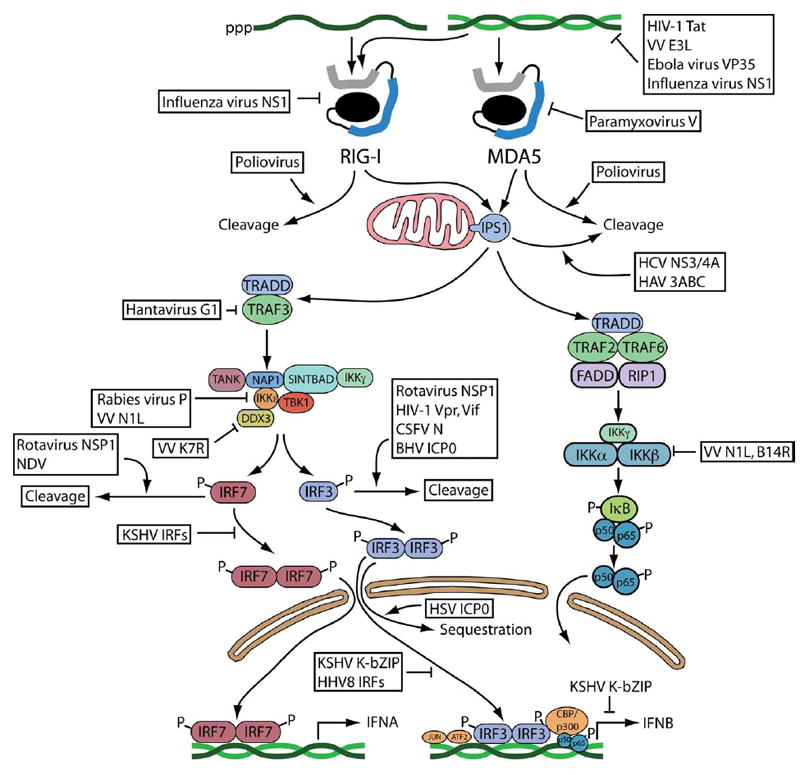
The steps leading from detection of RNA by RIG-I and MDA-5 through signaling and transcription of IFN genes are depicted. Viral gene products that interfere at different steps of this pathway are listed. An arrow indicates stimulation, and a bar indicates inhibition of the pathways
7.1 Interference with RNA sensing
As described previously, host RNAs are not detected by RIG-I or MDA-5 because they do not contain 5′-phosphates. Many viral RNAs are also capped, either by cellular or viral capping enzymes, or, in the case of influenza viruses, when the viral polymerase primes the synthesis of mRNAs with capped fragments derived from the 5′-ends of host mRNAs (Plotch, et al., 1981). A phosphatase is produced during infection with certain negative strand RNA viruses (Borna disease virus, Crimean-Congo Hemorrhagic fever virus, Hantaan virus) that converts the 5′ ppp on viral mRNAs to a monophosphate (Habjan, et al., 2008), thereby preventing detection by RIG-I. The genomic RNA of picornaviruses is covalently linked to a viral protein, VPg (Flanegan, et al., 1977; Lee, et al., 1977), which also prevents activation of RIG-I. These modifications of viral RNAs can all be viewed as strategies to avoid recognition by RIG-I.
Some viral genomes encode proteins that bind dsRNA and prevent their detection by RIG-I and MDA-5 in the cytoplasm. These include the vaccinia virus E3L protein (Smith, et al., 2001; Xiang, et al., 2002), Ebola virus VP35 (Haasnoot, et al., 2007), and human cytomegalovirus proteins TRS1 and m142/m143 (Cassady, 2005; Child, et al., 2004; Child, et al., 2006; Hakki & Geballe, 2005; Valchanova, et al., 2006).
Infection of cells with members of the picornavirus family leads to degradation of MDA-5 and RIG-I. Cleavage of MDA-5 is carried out by cellular caspases, which are activated by virus infection (Barral, et al., 2007; Rebsamen, et al., 2008). Cleavage of MDA-5 can also be accomplished in the absence of virus infection by inducing apoptosis pharmacologically (Barral, et al., 2007; Kovacsovics, et al., 2002). In contrast, the viral 3Cpro protease cleaves RIG-I directly in cells infected with poliovirus, rhinovirus, and other picornaviruses (Barral et al., submitted). It is not known if sensor cleavage plays a role in attenuating IFN response during picornavirus infection. Cleavage of RIG-I is a somewhat puzzling event because it is assumed that picornavirus infections are sensed by MDA-5. Perhaps cleavage of RIG-I/MDA-5 antagonizes other functions of the sensor proteins.
Some viral proteins have been shown to bind RIG-I or MDA-5, thereby blocking induction of IFN. The V proteins of paramyxoviruses bind MDA-5 and block IFN induction (Andrejeva, et al., 2004; Childs, et al., 2007). The C-terminal domain of V proteins bind a specific region of MDA-5, but not RIG-I; such binding prevents dsRNA binding, dimerization, and activation of MDA-5 (Childs, et al., 2008). However, the significance of such antagonism is not known, because mice lacking the gene encoding mda-5 are as susceptible to paramyxovirus infection as wild type mice, and mount similar IFN responses (Gitlin, et al., 2006; Kato, et al., 2006), and RIG-I appears to be the sensor for paramyxovirus infections. Perhaps the conserved MDA-5 binding ability of paramyxovirus V proteins targets a different activity of the protein. The NS1 protein of influenza A viruses binds RIG-I (Mibayashi, et al., 2007; Pichlmair, et al., 2006) and inhibits signaling and IFN induction (Guo, et al., 2007; Opitz, et al., 2007). As expected, viruses that do not produce the NS1 protein produce high levels of IFN in infected cells (Talon, et al., 2000). The G transmembrane glycoprotein of human metapneumovirus, a paramyxovirus, also binds RIG-I and blocks IFN-β induction (Bao, et al., 2008). There is some evidence that these viral proteins might not directly interact with RIG-I, and the mechanism of interference has not been elucidated.
7.2 Interference with signal transduction
The downstream adaptor protein IPS1 is cleaved in cells infected with a variety of viruses. As a consequence, IRF-3 phosphorylation does not occur and IFN transcription is attenuated. The 3Cpro proteinase of the picornavirus hepatitis A virus degrades IPS-1 only when part of the hydrophobic precursor protein 3ABC, which targets the proteinase to the mitochondrial membrane (Yang, et al., 2007). The NS3/4A protease of hepatitis C virus cleaves within the transmembrane domain of IPS1, causing its release from the mitochondrion (Lin, et al., 2006; Loo, et al., 2006; Meylan, et al., 2005). These examples demonstrated that IPS1 must be present in the mitochondrial membrane to function in the signaling pathway leading to IFN synthesis (Seth, et al., 2005).
Members of the signaling pathway downstream of IPS1 are also targets of viral antagonism. The cascades that begin with ligand-dependent activation of RIG-I and MDA-5 converge at the IKK family of proteins (Figure 4) whose role is to phosphorylate NF-kB, IRF3, and IRF7. Members of the IKK protein family are targets of viral antagonism. The G1 protein of hantavirus disrupts the interaction of TRAF3 with TBK1, thereby interrupting signaling (Alff, et al., 2008). The NS3/4A proteinase of hepatitis C virus and the N1L protein of vaccinia virus interact with TBK1, blocking phosphorylation of downstream transcription proteins (DiPerna, et al., 2004; Otsuka, et al., 2005). The consequence of inhibiting TBK1 activity is failure to phosphorylate IRF3, which cannot enter the nucleus to stimulate IFN transcription. The vaccinia virus K7R protein also prevents the activation of IRF3 and IRF7 by the TBK1-IKKε (Schroder, et al., 2008). The K7R protein binds the human DEAD box helicase DDX3, which is part of the TBK1-IKKε complex and is required for activation of the IFN-β promoter (Soulat, et al., 2008).
7.3 Inhibition of IRFs
The transcription proteins IRF3 and IRF7 are prime targets for inhibition by a variety of viral proteins. The papain-like protease of SARS virus binds IRF3 and prevents dimerization, thereby inhibiting nuclear translocation (Devaraj, et al., 2007). Other viral proteins act as IRF mimics. For example, the V proteins of paramyxoviruses bind TBK1-IKKε and compete with IRF3, thereby inhibiting its phosphorylation (Lu, et al., 2008). The genome of Kaposi’s sarcoma-associated herpesvirus (KSHV) forms dimers with IRF7, blocking its ability to bind DNA (Joo, et al., 2007). Several human herpesvirus 8 proteins are dominant-negative inhibitors of IRF3, preventing its association with the co-activators CBP and p300 (Lin, et al., 2001). The transcription protein K-bZIP of KSHV blocks activation of the IFN-β promoter by competing with IRF3 for binding sites in the promoter (Lefort, et al., 2007), while ICP0 of HSV binds IRF3 and localizes the protein in nuclear bodies where it cannot activate the IFN-β promoter (Melroe, et al., 2007).
Another strategy to prevent IRF mediated transcriptional activation is proteolytic degradation. The ICP0 protein from bovine herpesvirus degrades IRF3 instead of sequestering it like the HSV orthologue (Saira, et al. 2007). Degradation of IRF3 is also brought about by the Vpr and Vif proteins of HIV-1, the N protease of flaviviruses, and the NSP1 protein of rotaviruses; the latter also induces degradation of IRF7 (Barro & Patton, 2007; Bauhofer, et al., 2007; Okumura, et al., 2008). Of great interest is the observation that the vaccinia virus E3L protein disables the cellular ISG15 protein (Guerra, et al., 2008). ISG15 is an ISG that is believed to counter virus-induced proteolysis of IRF3; for example, it can prevent degradation of IRF3 by paramyxovirus V protein (Lu, et al., 2006). The vaccinia virus E3L protein therefore disables the cell’s attempt to block a viral antagonism of the innate immune response.
NF-κB function is also modulated during virus infection. This complex is not only involved in the production of IFN-β and chemokines, but also inhibits apoptosis and causes cell proliferation, which may benefit viral replication. Consequently, in some viral infections, NF-κB activity is blocked early in the replicative cycle, to prevent IFN-mediated clearing of infection. The A238L protein of African swine fever virus acts as a homolog of IκBα; it binds NF-κB and prevents its entry into the nucleus (Tait, et al., 2000). Later in infection, the viral A224L protein activates NF-κB, causing inhibition of apoptosis and cellular proliferation. Why cleavage of the p65-RelA subunit of NF-κB by poliovirus proteinase 3Cpro at a late stage in infection would be beneficial to virus replication is not clear (Neznanov, et al., 2005). Perhaps the concomitant stimulation of apoptosis helps stimulate virus release from cells (Jackson, et al., 2005).
Other viral proteins regulate NF-κB by disrupting IKK. The B14R protein of vaccinia virus binds IKKβ and prevents its phosphorylation and therefore activation (Chen, et al., 2008), while the K13 protein of KSHV binds the IKKα-IKKβ complex in a way that causes activation of NF-κB (Matta, et al., 2007). Curiously, cleavage of IκBα by the 3Cpro proteinase of the picornavirus coxsackvirus B3 limits NF-κB mediated signaling, but leads to reduced viral replication. The cleaved IκBα binds NF-κB, which enters the nucleus but cannot transactivate; therefore apoptosis increases, limiting viral replication (Zaragoza, et al., 2006). The authors suggest that IκBα acts as a sensor for viral replication.
8. Apoptosis and growth suppression
mda-5 was cloned as a transcript induced during induction of terminal differentiation of human melanoma cells and initiates an irreversible growth arrest program in these cancer cells (Kang, et al., 2002). Indeed, the first report on cloning and characterization of mda-5 revealed its growth suppressor properties (Kang, et al., 2002). Follow-up studies demonstrated that adenovirus-mediated delivery of mda-5 induced apoptosis that could be inhibited by active Ras/Raf pathway (Kang, et al., 2004; Lin, et al., 2006). In genetically modified rodent fibroblasts as well as in human pancreatic and colorectal cancer cell lines, constitutively active Ras/Raf/MEK/ERK pathway precluded mda-5-induced apoptosis that could be reversed by antisense inhibition of Ras or chemical inhibition of the ERK pathway (Lin, et al., 2006). Deletion analysis revealed that compared to the full-length mda-5, deletion of either the CARD or the helicase domain significantly abrogated the growth suppressor properties of mda-5 (Kang, et al., 2004). This loss-of-function was more pronounced when the CARD domain was deleted compared to the deletion of the helicase domain indicating that CARD domain is the active domain of the molecule, an observation later confirmed for RIG-I.
The cloning of human mda-5 was subsequently followed by cloning of the murine mda-5 gene, named helicard (Kovacsovics, et al., 2002). Apoptotic stimuli, such as FasL, resulted in cleavage of MDA-5/Helicard by caspases with subsequent nuclear translocation of the helicase domain, the consequences of which remain to be determined. MDA-5/Helicard augmented FasL-mediated DNA degradation while a mutant, resistant to caspase-cleavage, and a second mutant, with loss of helicase activity, lost this activity. However, MDA-5/Helicard alone did not induce apoptosis in 293T cells. Caspase-mediated cleavage of human MDA-5 has also been observed in poliovirus-infected cells (Barral, et al., 2007).
RH116, having 99.5% identity to mda-5, was cloned as a gene downregulated by Murabutide, an HIV-suppressive immunomodulator (Cocude, et al., 2003). Overexpression of RH116 inhibited growth, but did not induce apoptosis and the mechanism of this growth inhibition was not analyzed. Interestingly, counterintuitive to the anti-viral function of MDA-5, RH116 actually increased HIV-1 replication. HIV-1 treatment itself induced RH116 expression, which was localized predominantly in the nucleus. Thus, although it is accepted that MDA-5 inhibits growth, there is debate about induction of apoptosis by MDA-5 and possible differential effects of the human vs. the murine version of this gene. The lack of follow-up studies on Helicard or RH116 precludes resolution of this issue at the present time.
Similar to MDA-5, RIG-I also displays apoptosis-inducing properties. Cytosolic poly(I:C) or influenza A virus infection activates caspase 1 and 3 that results in pro-IL-18 processing (Rintahaka, et al., 2008). Overexpression of RIG-I or its active form (only the CARD domains) or IPS-1 activated caspases, processed pro-IL-18 and induced apoptosis in HaCaT keratinocytes cells. The reverse experiment, i.e., inhibition of rig-I to block these phenomena, was not carried out to strengthen the direct link of RIG-I in virus-induced apoptosis. The apoptosis induction by a synthetic RA (CD437) was shown to be inhibited by the hepatitis C virus (HCV) non-structural (NS)3/4A protease, which is know to cleave RIG-I, thereby indirectly showing an involvement of RIG-I in apoptosis induction (Pan, et al., 2008). Infection of cells with Sendai virus activates RIG-I and that leads to the activation of IRF-3. IRF-3 induces the expression of virus stress-inducible genes and causes host cell apoptosis by activating caspase-8 (Peters et al., 2008). A dominant-negative inhibitor of RIG-I, containing only the helicase domain, inhibits IRF-3 activation, prevents apoptosis and allows persistent virus infection. A novel strategy was used to capitalize on specific properties of RIG-I as an anti-cancer therapy (Poeck, et al., 2008). RIG-I, not MDA-5, is activated by 5′-triphosphates. An siRNA, with 5′-triphosphate ends, was generated against Bcl-2. The recognition of 5′-triphosphate by RIG-I activated innate immune cells, such as dendritic cells and directly induced expression of IFNs and apoptosis in tumor cells. These RIG-I-mediated effects synergized with Bcl-2 siRNA to induce apoptosis of tumor cells in lung metastasis in animal models. The observation that systemic administration of this bifunctional RNA molecule resulted in tumor-specific apoptosis offers promise of potential therapeutic use of this approach.
9. Role of RIG-I in development and differentiation
As has been discussed above, the best-characterized role of RIG-I is in viral RNA-induced type 1 IFN generation. However, recent evidence has suggested that it can act, in the absence of viral infection, to regulate myeloid cell differentiation (Zhang, et al., 2008). Initial experiments suggested that treatment of retinoic acid-sensitive myeloid leukaemia cell lines with all-trans-retinoic acid (ATRA) resulted in increased expression of RIG-I. As this is considered partially to mimic normal myelopoiesis it has been concluded that RIG-I is developmentally regulated during myeloid differentiation (Zhang, et al., 2008). A potential role for MDA-5 in differentiation was not investigated and is an area worth further exploration
It had been shown that most rig-I-/- mice die before birth (Kato, et al., 2005). However, in a later study, in which a different region of the RIG-I gene was deleted, mice grew to maturity. These animals developed granulocytosis, indicative of defects in myeloid development. In the same study it was noted that the granulocytosis in the rig-I-/- mice is attributable to reduction in expression of IFN consensus sequence-binding protein (Icsbp) which plays a central role in regulating myeloid differentiation (Zhang, et al., 2008).
10. MDA-5 and type 1 diabetes
In a large screen of single-nucleotide polymorphisms (SNPs) associated with type 1 diabetes, a number of mutations were observed in the IFN induced with helicase C domain 1 (IFIH1/MDA-5) linkage disequilibrium (LD) block on chromosome 2q. Indeed there was a strong correlation between SNPs in the MDA-5 gene itself and in the 3′ intergenic region and susceptibility to type 1 diabetes (Liu, et al., 2008; Smyth, et al., 2006; Todd, et al., 2007). Furthermore, after screening a large panel of controls and type 1 diabetes patients for MDA-5 expression it was suggested that SNPs leading to increased expression of MDA-5 may be associated with increased risk of developing the disease and that there might be an increase in MDA-5 expression overall in diabetes although this latter observation did not appear to be statistically significant in this cohort (Liu, et al., 2008). These studies suggest an intriguing possibility that blocking MDA-5 expression could provide a protective mechanism in the context of diabetes.
11. Evolution
The origin of the MDA-5 and RIG-I proteins has been examined from a phylogenetic perspective (Sarkar et al., 2008). What at first might seem a simple task is complicated by the presence of four protein domains in MDA-5 and RIG-I (Figure 5 A). Because the proteins are made up of these four domains that are themselves members of their own large domain families, it is possible that the MDA-5 and RIG-I proteins have a diverse and chimeric evolutionary history. In addition, the proteins that are closely related to MDA-5 and RIG-I can be determined using phylogenetic approaches.
Fig. 5.
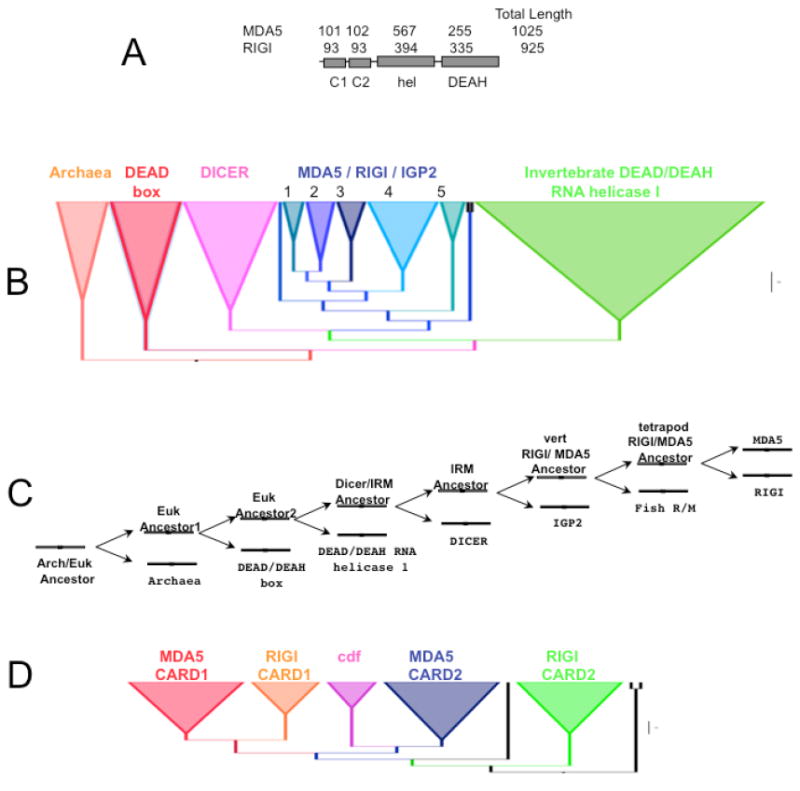
A. Schematic diagram showing the domain structure of RIG-I and MDA-5. The numbers above the cartoon indicate the size of the domains. Abbrevitions: C1= CARD domain 1; C2 = CARD domain 2; hel = helicase and DEAH= DEAH box domain. B. Phylogentic tree showing the overall topology of the helicase/DEAH domains. C. Stepwise diagram showing the duplication events involved in the evolution of the helicase/DEAH linked domains leading to RIGI and MDA5. Each double-headed arrow represents a gene duplication event. D. Phylogenetic tree showing the topology for the CARD domains.
Three steps were used to unravel the evolutionary history of these proteins. The first step was to examine if any of the domains had ancient linkage to each other or to other domains in other proteins. The second step was to identify whether any of the four domains had similar evolutionary histories, thus supporting the hypothesis that fusion events of previously independent domains occurred. The third step was to examine the evolutionary patterns of these singular and/or linked domains.
It is well known that helicase and DEAH domains are found linked in the genomes of species from the organismal superdomain Archaea as well as throughout the genomes of Eukaryotes (Abdelhaleem et al., 2003; Story et al., 2001). There are no known linked helicase DEAH domains in the organismal superdomain Bacteria. Given the well accepted topology of the tree of life with respect to the three organismal superdomains (Bacteria, Eukarya, Archaea), these observations suggest that the helicase DEAH linkage occurred in the common ancestor of Archaea and Eukarya. However, the helicase DEAH domain linkage could be a more recent phenomenon due to secondary fusion of these domains. If the latter scenario is correct then the evolutionary histories of the two domains will be divergent and if the former scenario is correct the evolutionary histories of the two domains will be congruent. When we examined the congruence of helicase and DEAH domains with respect to their evolutionary histories using the Incongruence Length Difference (ILD; 47–49) test, we found that both have statistically significant congruent histories (P<0.05; (Sarkar, et al., 2008) allowing us to reject the hypothesis that these two domains have fused after the divergence of Archaea. This result strongly implies that the helicase DEAH domain linkage in RIG-I and MDA-5 is an ancient relationship that arose in the common ancestor of the Archaea and Eukarya.
Understanding the ancient linkage of the CARD domains is more complex. These domains, most often associated with caspases, are found in a wide variety of proteins from a broad array of vertebrates (Inohara et al., 2005; Kersse et al., 2007). Because CARD domains appear in a large number of protein families and some of these protein families are not related, we suggest that the CARD domains may have experienced different evolutionary histories to the helicase DEAH domains. Most caspases (cysteine-dependent aspartate-specific proteases) have a single CARD domain, with the exceptions being MDA-5, RIG-I, and Nacht proteins. Because the Nacht CARD domains have diverged greatly from the MDA-5 and RIG-I CARD domains (see below), we can rule out an ancient linkage involving Nacht CARD domains. Furthermore, because the linked CARD1/CARD2 state in MDA-5 is found in all vertebrates, we conclude that the linkage of the two CARD domains in this protein occurred in the common ancestor of vertebrates.
The second step involved understanding the CARD1/CARD2 linked arrangement in RIG-I and MDA-5. The linkage of the CARD1/CARD2 domains could have arisen by two very different processes in the vertebrates. First, a single CARD domain could have simply duplicated to produce two linked domains. Second, two independently derived CARD domains could have fused to produce the linked state. Again the former scenario would result in the two CARD domains having congruent evolutionary history and in the latter the two domains would have incongruent histories. The domain comparisons for CARD1 and CARD2 revealed strong incongruence of CARD1 and CARD2 using the ILD test (p=0.68). In addition, when the congruence of CARD1 is tested relative to the helicase and DEAH domains there is also strong incongruence (p=0.45 and p=0.91 respectively). On the other hand, the CARD2 domain shows strong congruence with both the helicase and DEAH domains (both p<0.01). The ILD tests indicate that the evolutionary history of the CARD1 domain is divergent from the other three domains and suggests that the CARD1 domain was grafted onto an existing CARD2-helicase-DEAD/DEAH structure more than likely in the common ancestor of vertebrates.
The congruence analysis discussed above using the ILD test suggests that there are two independently evolving domain linkages that make up these proteins. Consequently, we constructed phylogenies for the domains in these proteins to examine the evolutionary history of each. We first examined the linked helicase/DEAD-DEAH domains and second, the genealogical relationships of the two CARD domains.
Figure B shows a phylogenetic analysis of a large representation of helicase, DEAH domains proteins in the database. This phylogenetic tree can be directly translated into the diagram shown in Figure 5 C. This scenario suggests that the helicase DEAH proteins have undergone at least seven duplication events to produce new families of helicase proteins in the evolution of eukaryotes. The final two duplications produced the LGP-2 helicases and then the MDA-5-RIG-I proteins. Because LGP2, MDA-5, and RIG-I are found only in vertebrates and DICER proteins are broadly distributed phylogenetically, we propose that the duplication event of MDA-5 and RIG-I occurred in the common ancestor of vertebrates.
The CARD domain phylogenetic analysis is consistent with our observations based on the ILD tests (Figure 5 D). The phylogenetic and incongruence results indicate that the CARD2 boxes were the first N-terminal elements to be grafted to the anciently duplicated helicase/DEAD domains. Next, the MDA-5 CARD1 domain was grafted to the CARD2-helicase/DEAD domains. Because RIG-I and MDA-5 genes are only found in vertebrates we propose that the CARD1 fusion occurred in the common ancestor of vertebrates.
The evolutionary history of the MDA-5 and RIG-I proteins that we propose here, demonstrates an intriguing and circuitous pathway that is consistent with intense selection pressure for the existence and maintenance of these genes. Gene duplication is generally considered the easiest way to construct proteins with similar domain structure. In other words, the most parsimonious route to RIG-I and MDA-5 proteins would have been to make one and then duplicate the entire assemblage. However, according to the phylogenetic analyses, there was independent fusion of CARD2 to the RIG-I or MDA-5 helicase/DEAD domains and then another independent fusion of CARD1 to the CARD2/helicase/ DEAD domain. This pathway rules out simple duplications as the only route to the eventual structure of the RIG-I and MDA-5 proteins. We suggest, from these data, that the MDA-5 and RIG-I domain structures are a case where the most parsimonious evolutionary path has not been taken, because the scenario the trees support have nearly twice as many steps in them than required to perform simple duplication events (see Sarkar, et al., 2008).
12. Perspectives
In just 6 years we have progressed from discovery of the cytoplasmic RNA sensors of the innate immune response, to developing a detailed understanding of RNA recognition and the signaling pathways that lead to interferon induction. The molecular mechanisms by which MDA-5 and RIG-I recognize specific RNA ligands remains to be determined; we expect that additional structural work will be required to answer this question. Specifically, it will be necessary to solve the atomic structures of full-length RIG-I and MDA-5 with and without an RNA ligand to provide a detailed understanding of the recognition process. The requirement for the RNA helicase activity is also enigmatic. Similarly, RNA unwinding is not needed for RNA recognition, although this activity may be important for the conformational change that leads to exposure of the CARD domains.
A relatively uncharted area of innate immunity is the recognition of viral DNA. There have been several reports of cytoplasmic DNA sensors, but precisely how these recognize nucleic acid and induce cytokine synthesis remains to be determined (Stetson, 2006; Ishii, 2006; Bürckstümmer, 2008). STING is essential for interferon production in response to cytoplasmic dsDNA, but it is not a nucleic acid sensor (Ishikawa & Barber, 2008). It will be of interest to determine whether DNA sensing pathways overlap with those described here for detection of RNA.
Viral genomes encode proteins that block every step of the RNA sensing pathways described in this review. Viral gene products may also antagonize nearly every step of the sensing of RNA by RIG-I and MDA-5, including RNA recognition, signaling, and induction of transcription. Understanding such viral countermeasures not only improves our understanding of innate sensing pathways, but may also suggest avenues for therapeutic intervention.
An important question is whether we can use our understanding of RNA sensing pathways for health benefit. We know that polymorphisms in genes encoding TLRs may influence outcome of virus infection (Awomoyi, 2007). Therefore it would be useful to identify changes in MDA-5 and RIG-I that might alter susceptibility to infection. Other approaches include induction of MDA-5 or RIG-I transcription with specific promoter activators, or restoring defective innate responses that occur during chronic diseases such as hepatitis C virus infection. It might also be possible to suppress the inappropriate activation of innate responses that take place during autoimmune and inflammatory disease. One approach to such therapy would be to design inhibitors of key components that are targeted by viral antagonism. Such approaches could bring unexpected health benefits beyond the prevention of infectious diseases.
Fig. 3. The RIG-I and MDA-5 signaling pathway.
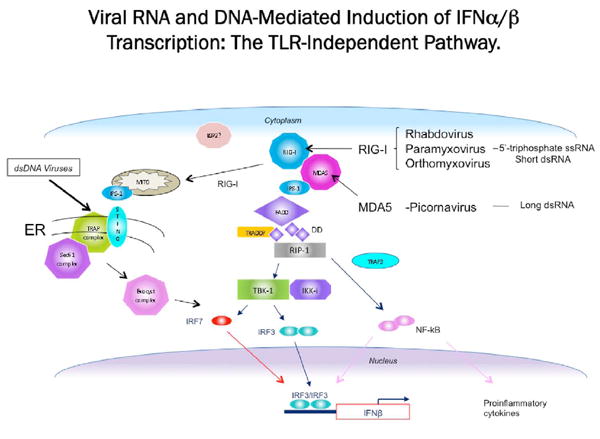
Short dsRNA’s or 5’-triphosphate ssRNA’s activate RIG-I which associates with mitochondrial IPS-1 to trigger FADD/RIP1 (innateosome) controlled activation of the type I interferon genes. Longer RNA’s generated from picornaviruses trigger MDA5-mediated activation of type I IFN. The RIG-I/IPS-1 pathway also requires the assistance of STING to activate type I IFN, which resides in the endoplasmic reticulum (ER). STING may regulate the translocon composed of the TRAP complex and Sec61, which in turn may regulate exocyst-mediated stimulation of the TBK-1 pathway. STING also appears pivotal in regulating DNA-mediated triggering of type I IFN production.
Acknowledgments
The present studies were supported in part by National Institutes of Health grants GM068448 (PBF), AI079336 (GNB) and AI50754 and AI068017 (VRR). Support was also provided by the Samuel Waxman Cancer Research Foundation (SWCRF) (PBF). RD thanks the Sackler Institute for Comparative Genomics, the Lewis and Dorothy B Cullman Program in Molecular Systematics and the Korein Family Foundation all at the American Museum of Natural History for their continued support. Support from the VCU Institute of Molecular Medicine to DS and PBF is acknowledged. DS is the Harrison Scholar in Cancer Research at the VCU Massey Cancer Center, VCU School of Medicine. PBF holds the Thelma Newmeyer Corman Chair in Cancer Research at the VCU Massey Cancer Center, VCU School of Medicine and is a SWCRF Investigator.
List of abbreviations
- CARD
caspase activation and recruitment domain
- cDCs
conventional dendritic cells
- CTD
C-terminal domain
- DISH
differentiation induction subtraction hybridization
- EMCV
encephalomyocarditis virus
- ER
endoplasmic reticulum
- FADD
fas associated death domain
- IFN
interferon
- GMCSF
granulocyte macrophage colony stimulating factor
- IKK
I kappa B kinase
- IRF
interferon regulatory factor
- IKKi
IkappaB-kinase-epsilon
- IPS-1
interferon β promoter stimulator 1
- LGP2
laboratory of genetics and physiology 2
- MDA-5
melanoma differentiation associated gene-5
- MEF
mouse embryo fibroblast
- Mss4
mammalian suppressor of Sec4
- PAMP
pathogen-associated molecular pattern
- pDCs
plasmacytoid dendritic cells
- Poly(I:C)
polyinosinic:polycytidylic acid
- RA
retinoic acid
- RIG-I
retinoic acid-inducible gene-I
- RIP1
receptor-interacting serine-threonine kinase 1
- RLH
RIG-like helicases
- SNP
single-nucleotide polymorphism
- STING
stimulator of IFN genes
- TBK1
TANK-binding kinase 1
- TLR
toll-like receptor
- TRADD
TNFR1-associated death domain protein
- TRAF3
TNF receptor associated factor 3
- TRAP
translocon-associated protein
- VSV
vesicular stomatitis virus
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- Abdelhaleem M, Maltais L, Wain H. The human DDX and DHX gene families of putative RNA helicases. Genomics. 2003;81(6):618–622. doi: 10.1016/s0888-7543(03)00049-1. [DOI] [PubMed] [Google Scholar]
- Aggarwal K, Silverman N. Positive and negative regulation of the Drosophila immune response. BMB Rep. 2008;41(4):267–277. doi: 10.5483/bmbrep.2008.41.4.267. [DOI] [PubMed] [Google Scholar]
- Alff PJ, Sen N, Gorbunova E, Gavrilovskaya IN, Mackow ER. The NY-1 hantavirus Gn cytoplasmic tail coprecipitates TRAF3 and inhibits cellular interferon responses by disrupting TBK1-TRAF3 complex formation. J Virol. 2008;82(18):9115–9122. doi: 10.1128/JVI.00290-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrejeva J, Childs KS, Young DF, Carlos TS, Stock N, Goodbourn S, et al. The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-beta promoter. Proc Natl Acad Sci U S A. 2004;101(49):17264–17269. doi: 10.1073/pnas.0407639101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arimoto K, Takahashi H, Hishiki T, Konishi H, Fujita T, Shimotohno K. Negative regulation of the RIG-I signaling by the ubiquitin ligase RNF125. Proc Natl Acad Sci U S A. 2007;104(18):7500–7505. doi: 10.1073/pnas.0611551104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Awomoyi AA, Rallabhandi P, Pollin TI, Lorenz E, Sztein MB, Boukhvalova MS, et al. Association of TLR4 polymorphisms with symptomatic respiratorysyncytial virus infection in high-risk infants and young children. J Immunol. 2007;179(5):3171–3177. doi: 10.4049/jimmunol.179.5.3171. [DOI] [PubMed] [Google Scholar]
- Balachandran S, Thomas E, Barber GN. A FADD-dependent innate immune mechanism in mammalian cells. Nature. 2004;432(7015):401–405. doi: 10.1038/nature03124. [DOI] [PubMed] [Google Scholar]
- Balachandran S, Venkataraman T, Fisher PB, Barber GN. Fas-associated death domain-containing protein-mediated antiviral innate immune signaling involves the regulation of Irf7. J Immunol. 2007;178(4):2429–2439. doi: 10.4049/jimmunol.178.4.2429. [DOI] [PubMed] [Google Scholar]
- Bao X, Liu T, Shan Y, Li K, Garofalo RP, Casola A. Human metapneumovirus glycoprotein G inhibits innate immune responses. PLoS Pathog. 2008;4(5):e1000077. doi: 10.1371/journal.ppat.1000077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barral PM, Morrison JM, Drahos J, Gupta P, Sarkar D, Fisher PB, et al. MDA-5 is cleaved in poliovirus-infected cells. J Virol. 2007;81(8):3677–3684. doi: 10.1128/JVI.01360-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barro M, Patton JT. Rotavirus NSP1 inhibits expression of type I interferon by antagonizing the function of interferon regulatory factors IRF3, IRF5, and IRF7. J Virol. 2007;81(9):4473–4481. doi: 10.1128/JVI.02498-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauhofer O, Summerfield A, Sakoda Y, Tratschin JD, Hofmann MA, Ruggli N. Classical swine fever virus Npro interacts with interferon regulatory factor 3 and induces its proteasomal degradation. J Virol. 2007;81(7):3087–3096. doi: 10.1128/JVI.02032-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berghäll H, Sirén J, Sarkar D, Julkunen I, Fisher PB, Vainionpää R, Matikainen S. The interferon-inducible RNA helicase, mda-5, is involved in measles virus-induced expression of antiviral cytokines. Microbes and Infection. 2006;8(8):2138–2144. doi: 10.1016/j.micinf.2006.04.005. [DOI] [PubMed] [Google Scholar]
- Bozidis P, Williamson CD, Colberg-Poley AM. Isolation of endoplasmic reticulum, mitochondria, and mitochondria-associated membrane fractions from transfected cells and from human cytomegalovirus-infected primary fibroblasts. Curr Protoc Cell Biol. 2007;Chapter 3(Unit 3):27. doi: 10.1002/0471143030.cb0327s37. [DOI] [PubMed] [Google Scholar]
- Burckstummer T, Baumann C, Bluml S, Dixit E, Durnberger G, Jahn H, et al. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat Immunol. 2009;10(3):266–272. doi: 10.1038/ni.1702. [DOI] [PubMed] [Google Scholar]
- Cassady KA. Human cytomegalovirus TRS1 and IRS1 gene products block the double-stranded-RNA-activated host protein shutoff response induced by herpes simplex virus type 1 infection. J Virol. 2005;79(14):8707–8715. doi: 10.1128/JVI.79.14.8707-8715.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen RA, Ryzhakov G, Cooray S, Randow F, Smith GL. Inhibition of IkappaB kinase by vaccinia virus virulence factor B14. PLoS Pathog. 2008;4(2):e22. doi: 10.1371/journal.ppat.0040022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien Y, Kim S, Bumeister R, Loo YM, Kwon SW, Johnson CL, et al. RalB GTPase-mediated activation of the IkappaB family kinase TBK1 couples innate immune signaling to tumor cell survival. Cell. 2006;127(1):157–170. doi: 10.1016/j.cell.2006.08.034. [DOI] [PubMed] [Google Scholar]
- Child SJ, Hakki M, De Niro KL, Geballe AP. Evasion of cellular antiviral responses by human cytomegalovirus TRS1 and IRS1. J Virol. 2004;78(1):197–205. doi: 10.1128/JVI.78.1.197-205.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Child SJ, Hanson LK, Brown CE, Janzen DM, Geballe AP. Double-stranded RNA binding by a heterodimeric complex of murine cytomegalovirus m142 and m143 proteins. J Virol. 2006;80(20):10173–10180. doi: 10.1128/JVI.00905-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childs K, Stock N, Ross C, Andrejeva J, Hilton L, Skinner M, et al. mda-5, but not RIG-I, is a common target for paramyxovirus V proteins. Virology. 2007;359(1):190–200. doi: 10.1016/j.virol.2006.09.023. [DOI] [PubMed] [Google Scholar]
- Childs KS, Andrejeva J, Randall RE, Goodbourn S. Mechanism of mda-5 inhibition by paramyxovirus V proteins. J Virol. 2008;83(3):1465–1473. doi: 10.1128/JVI.01768-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinnaiyan AM, O’Rourke K, Tewari M, Dixit VM. FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis. Cell. 1995;81(4):505–512. doi: 10.1016/0092-8674(95)90071-3. [DOI] [PubMed] [Google Scholar]
- Cocude C, Truong MJ, Billaut-Mulot O, Delsart V, Darcissac E, Capron A, et al. A novel cellular RNA helicase, RH116, differentially regulates cell growth, programmed cell death and human immunodeficiency virus type 1 replication. J Gen Virol. 2003;84(Pt 12):3215–3225. doi: 10.1099/vir.0.19300-0. [DOI] [PubMed] [Google Scholar]
- Cui S, Eisenacher K, Kirchhofer A, Brzozka K, Lammens A, Lammens K, et al. The C-terminal regulatory domain is the RNA 5’-triphosphate sensor of RIG-I. Mol Cell. 2008;29(2):169–179. doi: 10.1016/j.molcel.2007.10.032. [DOI] [PubMed] [Google Scholar]
- Devaraj SG, Wang N, Chen Z, Tseng M, Barretto N, Lin R, et al. Regulation of IRF-3-dependent innate immunity by the papain-like protease domain of the severe acute respiratory syndrome coronavirus. J Biol Chem. 2007;282(44):32208–32221. doi: 10.1074/jbc.M704870200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diao F, Li S, Tian Y, Zhang M, Xu LG, Zhang Y, et al. Negative regulation of MDA5- but not RIG-I-mediated innate antiviral signaling by the dihydroxyacetone kinase. Proc Natl Acad Sci U S A. 2007;104(28):11706–11711. doi: 10.1073/pnas.0700544104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiPerna G, Stack J, Bowie AG, Boyd A, Kotwal G, Zhang Z, et al. Poxvirus protein N1L targets the I-kappaB kinase complex, inhibits signaling to NF-kappaB by the tumor necrosis factor superfamily of receptors, and inhibits NF-kappaB and IRF3 signaling by toll-like receptors. J Biol Chem. 2004;279(35):36570–36578. doi: 10.1074/jbc.M400567200. [DOI] [PubMed] [Google Scholar]
- Ferrandon D, Imler JL, Hoffmann JA. Sensing infection in Drosophila: Toll and beyond. Semin Immunol. 2004;16(1):43–53. doi: 10.1016/j.smim.2003.10.008. [DOI] [PubMed] [Google Scholar]
- Fisher PB, Grant S. Effects of interferon on differentiation in normal and tumor cells. Pharmacol & Therapeut. 1984;27:143–166. doi: 10.1016/0163-7258(85)90067-1. [DOI] [PubMed] [Google Scholar]
- Fisher PB, Miranda AF, Babiss LE, Pestka S, Weinstein IB. Opposing effects of interferon produced in bacteria and of tumor promoters on myogenesis in human myoblast cultures. Proc Natl Acad Sci USA. 1983;80:2961–2965. doi: 10.1073/pnas.80.10.2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanegan JB, Pettersson RF, Ambros V, Hewlett MJ, Baltimore D. Covalent linkage of a protein to a defined nucleotide sequence at the 5’-terminus of virion and replicative intermediate RNAs of poliovirus. Proc Natl Acad Sci USA. 1977;74:961–965. doi: 10.1073/pnas.74.3.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredericksen BL, Keller BC, Fornek J, Katze MG, Gale M., Jr Establishment and maintenance of the innate antiviral response to West Nile Virus involves both RIG-I and MDA5 signaling through IPS-1. J Virol. 2008;82(2):609–616. doi: 10.1128/JVI.01305-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman CS, O’Donnell MA, Legarda-Addison D, Ng A, Cardenas WB, Yount JS, et al. The tumour suppressor CYLD is a negative regulator of RIG-I-mediated antiviral response. EMBO Rep. 2008;9(9):930–936. doi: 10.1038/embor.2008.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesen HJ, Stein S, Evinger M, Familletti PC, Moschera J, Meienhofer J, et al. Purification and molecular characterization of human fibroblast interferon. Arch Biochemistry Biophys. 1981;206(2):432–450. doi: 10.1016/0003-9861(81)90111-9. [DOI] [PubMed] [Google Scholar]
- Gack MU, Kirchhofer A, Shin YC, Inn KS, Liang C, Cui S, et al. Roles of RIG-I N-terminal tandem CARD and splice variant in TRIM25-mediated antiviral signal transduction. Proc Natl Acad Sci U S A. 2008;105(43):16743–16748. doi: 10.1073/pnas.0804947105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gee P, Chua PK, Gevorkyan J, Klumpp K, Najera I, Swinney DC, et al. Essential role of the N-terminal domain in the regulation of RIG-I ATPase activity. J Biol Chem. 2008;283(14):9488–9496. doi: 10.1074/jbc.M706777200. [DOI] [PubMed] [Google Scholar]
- Georgel P, Naitza S, Kappler C, Ferrandon D, Zachary D, Swimmer C, et al. Drosophila immune deficiency (IMD) is a death domain protein that activates antibacterial defense and can promote apoptosis. Dev Cell. 2001;1(4):503–514. doi: 10.1016/s1534-5807(01)00059-4. [DOI] [PubMed] [Google Scholar]
- Giacomini P, Aguzzi A, Pestka S, Fisher PB, Ferrone S. Modulation by recombinant DNA leukocyte (α) and fibroblast (β) interferons of the expression and shedding of HLA and tumor associated antigens by human melanoma cells. J Immunol. 1984;133:1649–1655. [PubMed] [Google Scholar]
- Grant S, Bhalla K, Weinstein IB, Pestka S, Mileno MD, Fisher PB. Recombinant human interferon sensitizes resistant myeloid leukemic cells to induction of terminal differentiation. Biochem Biophys Res Commun. 1985;130:379–388. doi: 10.1016/0006-291x(85)90428-0. [DOI] [PubMed] [Google Scholar]
- Greiner JW, Guadagni F, Noguchi P, Pestka S, Colcher D, Fisher PB, et al. Use of recombinant interferon to enhance monoclonal antibody-targeting of carcinoma lesions in vivo. Science. 1987;235:895–898. doi: 10.1126/science.3580039. [DOI] [PubMed] [Google Scholar]
- Greiner JW, Hand PH, Noguchi P, Fisher PB, Pestka S, Schlom J. Enhanced expression of surface tumor-associated antigens on human breast and colon tumor cells after recombinant leukocyte α-interferon treatment. Cancer Res. 1984;44:3208–3214. [PubMed] [Google Scholar]
- Greiner JW, Schlom J, Pestka S, Langer JA, Giacomini P, Kusama M, et al. Modulation of tumor associated antigen expression and shedding by recombinant human leukocyte and fibroblast interferons. Pharmacol & Therapeut. 1985;31:209–236. doi: 10.1016/0163-7258(85)90023-3. [DOI] [PubMed] [Google Scholar]
- Gitlin L, Barchet W, Gilfillan S, Cella M, Beutler B, Flavell RA, et al. Essential role of mda-5 in type I IFN responses to polyriboinosinic:polyribocytidylic acid and encephalomyocarditis picornavirus. Proc Natl Acad Sci U S A. 2006;103(22):8459–8464. doi: 10.1073/pnas.0603082103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerra S, Caceres A, Knobeloch KP, Horak I, Esteban M. Vaccinia virus E3 protein prevents the antiviral action of ISG15. PLoS Pathog. 2008;4(7):e1000096. doi: 10.1371/journal.ppat.1000096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo W, Novick P. The exocyst meets the translocon: a regulatory circuit for secretion and protein synthesis? Trends Cell Biol. 2004;14(2):61–63. doi: 10.1016/j.tcb.2003.12.008. [DOI] [PubMed] [Google Scholar]
- Guo Z, Chen LM, Zeng H, Gomez JA, Plowden J, Fujita T, et al. NS1 protein of influenza A virus inhibits the function of intracytoplasmic pathogen sensor, RIG-I. Am J Respir Cell Mol Biol. 2007;36(3):263–269. doi: 10.1165/rcmb.2006-0283RC. [DOI] [PubMed] [Google Scholar]
- Haasnoot J, de Vries W, Geutjes EJ, Prins M, de Haan P, Berkhout B. The Ebola virus VP35 protein is a suppressor of RNA silencing. PLoS Pathog. 2007;3:794–803. doi: 10.1371/journal.ppat.0030086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habjan M, Andersson I, Klingstrom J, Schumann M, Martin A, Zimmermann P, et al. Processing of genome 5’ termini as a strategy of negative-strand RNA viruses to avoid RIG-I-dependent interferon induction. PLoS ONE. 2008;3(4):e2032. doi: 10.1371/journal.pone.0002032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacker H, Redecke V, Blagoev B, Krathmarova I, Hsu LC, Wang GG, et al. Specificity in Toll-like signalling through distinct effector functions of TRAF3 and TRAF6. Nature. 2006;439:204–207. doi: 10.1038/nature04369. [DOI] [PubMed] [Google Scholar]
- Hakki M, Geballe AP. Double-stranded RNA binding by human cytomegalovirus pTRS1. J Virol. 2005;79(12):7311–7318. doi: 10.1128/JVI.79.12.7311-7318.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann E, Gorlich D, Kostka S, Otto A, Kraft R, Knespel S, et al. A tetrameric complex of membrane proteins in the endoplasmic reticulum. Eur J Biochem. 1993;214(2):375–381. doi: 10.1111/j.1432-1033.1993.tb17933.x. [DOI] [PubMed] [Google Scholar]
- Hornung V, Ellegast J, Kim S, Brzozka K, Jung A, Kato H, et al. 5’-Triphosphate RNA is the ligand for RIG-I. Science. 2006;314(5801):994–997. doi: 10.1126/science.1132505. [DOI] [PubMed] [Google Scholar]
- Hsu H, Huang J, Shu HB, Baichwal V, Goeddel DV. TNF-dependent recruitment of the protein kinase RIP to the TNF receptor-1 signaling complex. Immunity. 1996;4(4):387–396. doi: 10.1016/s1074-7613(00)80252-6. [DOI] [PubMed] [Google Scholar]
- Huang F, Adelman J, Jiang H, Goldstein NI, Fisher PB. Differentiation induction subtraction hybridization (DISH): an approach for cloning genes differentially expressed during growth arrest and terminal differentiation in human melanoma cells. Gene. 1999a;236:125–131. doi: 10.1016/s0378-1119(99)00244-9. [DOI] [PubMed] [Google Scholar]
- Huang F, Adelman J, Jiang H, Goldstein NI, Fisher PB. Identification and temporal expression pattern of genes modulated during irreversible growth arrest and terminal differentiation in human melanoma cells. Oncogene. 1999b;18:3546–3552. doi: 10.1038/sj.onc.1202715. [DOI] [PubMed] [Google Scholar]
- Inohara, Chamaillard, McDonald C, Nunez G. NOD-LRR proteins: role in host-microbial interactions and inflammatory disease. Annu Rev Biochem. 2005;74:355–383. doi: 10.1146/annurev.biochem.74.082803.133347. [DOI] [PubMed] [Google Scholar]
- Isaacs A, Lindenmann J. Virus interference. I. The interferon Proc R Soc Lond B Biol Sci. 1957;147(927):258–267. doi: 10.1098/rspb.1957.0048. [DOI] [PubMed] [Google Scholar]
- Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455(7213):674–678. doi: 10.1038/nature07317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii KJ, Coban C, Kato H, Takahashi K, Torii Y, Takeshita F, Ludwig H, Sutter G, Suzuki K, Hemmi H, Sato S, Yamamoto M, Uematsu S, Kawai T, Takeuchi O, Akira S. A Toll-like recrptor-independent antiviral response induced by double-stranded B-form DNA. Nat Immunol. 2006;7:40–48. doi: 10.1038/ni1282. [DOI] [PubMed] [Google Scholar]
- Jackson WT, Giddings TH, Jr, Taylor MP, Mulinyawe S, Rabinovitch M, Kopito RR, et al. Subversion of cellular autophagosomal machinery by RNA viruses. PLoS Biol. 2005;3(5):e156. doi: 10.1371/journal.pbio.0030156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Fisher PB. Use of a sensitive and efficient subtraction hybridization protocol for the identification of genes differentially regulated during the induction of differentiation in human melanoma cells. Mol Cell Different. 1993;1(3):285–299. [Google Scholar]
- Jiang H, Kang D-c, Alexandre D, Fisher PB. RaSH, A rapid subtraction hybridization approach for identifying and cloning differentially expressed genes. Proc Natl Acad Sci USA. 2000;97:12684–12689. doi: 10.1073/pnas.220431297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin L, Waterman PM, Jonscher KR, Short CM, Reisdorph NA, Cambier JC. MPYS, a novel membrane tetraspanner, is associated with major histocompatibility complex class II and mediates transduction of apoptotic signals. Mol Cell Biol. 2008;28(16):5014–5026. doi: 10.1128/MCB.00640-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joo CH, Shin YC, Gack M, Wu L, Levy D, Jung JU. Inhibition of interferon regulatory factor 7 (IRF7)-mediated interferon signal transduction by the Kaposi’s sarcoma-associated herpesvirus viral IRF homolog vIRF3. J Virol. 2007;81(15):8282–8292. doi: 10.1128/JVI.00235-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jounai N, Takeshita F, Kobiyama K, Sawano A, Miyawaki A, Xin KQ, et al. The Atg5 Atg12 conjugate associates with innate antiviral immune responses. Proc Natl Acad Sci U S A. 2007;104(35):14050–14055. doi: 10.1073/pnas.0704014104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang DC, Gopalkrishnan RV, Lin L, Randolph A, Valerie K, Pestka S, et al. Expression analysis and genomic characterization of human melanoma differentiation associated gene-5, mda-5: a novel type I interferon-responsive apoptosis-inducing gene. Oncogene. 2004;23(9):1789–1800. doi: 10.1038/sj.onc.1207300. [DOI] [PubMed] [Google Scholar]
- Kang DC, Gopalkrishnan RV, Wu Q, Jankowsky E, Pyle AM, Fisher PB. mda-5: An interferon-inducible putative RNA helicase with double-stranded RNA-dependent ATPase activity and melanoma growth-suppressive properties. Proc Natl Acad Sci U S A. 2002;99(2):637–642. doi: 10.1073/pnas.022637199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H, Sato S, Yoneyama M, Yamamoto M, Uematsu S, Matsui K, et al. Cell type-specific involvement of RIG-I in antiviral response. Immunity. 2005;23(1):19–28. doi: 10.1016/j.immuni.2005.04.010. [DOI] [PubMed] [Google Scholar]
- Kato H, Takeuchi O, Mikamo-Satoh E, Hirai R, Kawai T, Matsushita K, et al. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J Exp Med. 2008;205(7):1601–1610. doi: 10.1084/jem.20080091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato H, Takeuchi O, Sato S, Yoneyama M, Yamamoto M, Matsui K, et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441(7089):101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- Kawai T, Takahashi K, Sato S, Coban C, Kumar H, Kato H, et al. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol. 2005;6(10):981–988. doi: 10.1038/ni1243. [DOI] [PubMed] [Google Scholar]
- Kawai T, Akira S. Toll-like receptor and RIG-I-like receptor signaling. Ann N Y Acad Sci. 2008;1143:1–20. doi: 10.1196/annals.1443.020. [DOI] [PubMed] [Google Scholar]
- Kayagaki N, Phung Q, Chan S, Chaudhari R, Quan C, O’Rourke KM, et al. DUBA: a deubiquitinase that regulates type I interferon production. Science. 2007;318(5856):1628–1632. doi: 10.1126/science.1145918. [DOI] [PubMed] [Google Scholar]
- Kelliher MA, Grimm S, Ishida Y, Kuo F, Stanger BZ, Leder P. The death domain kinase RIP mediates the TNF-induced NF-kappaB signal. Immunity. 1998;8(3):297–303. doi: 10.1016/s1074-7613(00)80535-x. [DOI] [PubMed] [Google Scholar]
- Kersse K, Vanden Berghe T, Lamkanfi M, Vandenabeele P. A phylogenetic and functional overview of inflammatory caspases and caspase-1-related CARD-only proteins. Biochem Soc Trans. 2007;35(Pt 6):1508–1511. doi: 10.1042/BST0351508. [DOI] [PubMed] [Google Scholar]
- Kim MJ, Hwang SY, Imaizumi T, Yoo JY. Negative feedback regulation of RIG-I-mediated antiviral signaling by interferon-induced ISG15 conjugation. J Virol. 2008;82(3):1474–1483. doi: 10.1128/JVI.01650-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MJ, Yoo JY. Active caspase-1-mediated secretion of retinoic acid inducible gene-I. J Immunol. 2008;181(10):7324–7331. doi: 10.4049/jimmunol.181.10.7324. [DOI] [PubMed] [Google Scholar]
- Komuro A, Horvath CM. RNA- and virus-independent inhibition of antiviral signaling by RNA helicase LGP2. J Virol. 2006;80(24):12332–12342. doi: 10.1128/JVI.01325-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacsovics M, Martinon F, Micheau O, Bodmer JL, Hofmann K, Tschopp J. Overexpression of Helicard, a CARD-containing helicase cleaved during apoptosis, accelerates DNA degradation. Curr Biol. 2002;12(10):838–843. doi: 10.1016/s0960-9822(02)00842-4. [DOI] [PubMed] [Google Scholar]
- Kumar H, Kawai T, Kato H, Sato S, Takahashi K, Coban C, et al. Essential role of IPS-1 in innate immune responses against RNA viruses. J Exp Med. 2006;203(7):1795–1803. doi: 10.1084/jem.20060792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YF, Nomoto A, Detjen BM, Wimmer E. A protein covalently linked to poliovirus genome RNA. Proc Natl Acad Sci USA. 1977;74:59–63. doi: 10.1073/pnas.74.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lefort S, Soucy-Faulkner A, Grandvaux N, Flamand L. Binding of Kaposi’s sarcoma-associated herpesvirus K-bZIP to interferon-responsive factor 3 elements modulates antiviral gene expression. J Virol. 2007;81(20):10950–10960. doi: 10.1128/JVI.00183-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemaitre B, Kromer-Metzger E, Michaut L, Nicolas E, Meister M, Georgel P, et al. A recessive mutation, immune deficiency (imd), defines two distinct control pathways in the Drosophila host defense. Proc Natl Acad Sci U S A. 1995;92(21):9465–9469. doi: 10.1073/pnas.92.21.9465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leulier F, Vidal S, Saigo K, Ueda R, Lemaitre B. Inducible expression of double-stranded RNA reveals a role for dFADD in the regulation of the antibacterial response in Drosophila adults. Curr Biol. 2002;12(12):996–1000. doi: 10.1016/s0960-9822(02)00873-4. [DOI] [PubMed] [Google Scholar]
- Lin L, Su Z, Lebedeva IV, Gupta P, Boukerche H, Rai T, et al. Activation of Ras/Raf protects cells from melanoma differentiation-associated gene-5-induced apoptosis. Cell Death Differ. 2006;13(11):1982–1993. doi: 10.1038/sj.cdd.4401899. [DOI] [PubMed] [Google Scholar]
- Lin R, Genin P, Mamane Y, Sgarbanti M, Battistini A, Harrington WJ, Jr, et al. HHV-8 encoded vIRF-1 represses the interferon antiviral response by blocking IRF-3 recruitment of the CBP/p300 coactivators. Oncogene. 2001;20(7):800–811. doi: 10.1038/sj.onc.1204163. [DOI] [PubMed] [Google Scholar]
- Lin R, Lacoste J, Nakhaei P, Sun Q, Yang L, Paz S, et al. Dissociation of a MAVS/IPS-1/VISA/Cardif-IKKepsilon molecular complex from the mitochondrial outer membrane by hepatitis C virus NS3-4A proteolytic cleavage. J Virol. 2006;80(12):6072–6083. doi: 10.1128/JVI.02495-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin R, Yang L, Nakhaei P, Sun Q, Sharif-Askari E, Julkunen I, et al. Negative regulation of the retinoic acid-inducible gene I-induced antiviral state by the ubiquitin-editing protein A20. J Biol Chem. 2006;281(4):2095–2103. doi: 10.1074/jbc.M510326200. [DOI] [PubMed] [Google Scholar]
- Lipschutz JH, Lingappa VR, Mostov KE. The exocyst affects protein synthesis by acting on the translocation machinery of the endoplasmic reticulum. J Biol Chem. 2003;278(23):20954–20960. doi: 10.1074/jbc.M213210200. [DOI] [PubMed] [Google Scholar]
- Liu S, Wang H, Jin Y, Podolsky R, Linga Reddy MV, Pedersen J, et al. The IFIH1 polymorphisms are significantly associated with type 1 diabetes and IFIH1 gene expression in peripheral blood mononuclear cells. Hum Mol Genet. 2009;18(2):358–365. doi: 10.1093/hmg/ddn342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu YC, Penninger J, Karin M. Immunity by ubiquitylation: a reversible process of modification. Nat Rev Immunol. 2005;5(12):941–952. doi: 10.1038/nri1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo YM, Fornek J, Crochet N, Bajwa G, Perwitasari O, Martinez-Sobrido L, et al. Distinct RIG-I and MDA5 signaling by RNA viruses in innate immunity. J Virol. 2008;82(1):335–345. doi: 10.1128/JVI.01080-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loo YM, Owen DM, Li K, Erickson AK, Johnson CL, Fish PM, et al. Viral and therapeutic control of IFN-beta promoter stimulator 1 during hepatitis C virus infection. Proc Natl Acad Sci U S A. 2006;103(15):6001–6006. doi: 10.1073/pnas.0601523103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu G, Reinert JT, Pitha-Rowe I, Okumura A, Kellum M, Knobeloch KP, et al. ISG15 enhances the innate antiviral response by inhibition of IRF-3 degradation. Cell Mol Biol (Noisy-le-grand) 2006;52(1):29–41. [PubMed] [Google Scholar]
- Lu LL, Puri M, Horvath CM, Sen GC. Select paramyxoviral V proteins inhibit IRF3 activation by acting as alternative substrates for inhibitor of kappaB kinase epsilon (IKKe)/TBK1. J Biol Chem. 2008;283(21):14269–14276. doi: 10.1074/jbc.M710089200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malathi K, Dong B, Gale M, Jr, Silverman RH. Small self-RNA generated by RNase L amplifies antiviral innate immunity. Nature. 2007;448(7155):816–819. doi: 10.1038/nature06042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malathi K, Paranjape JM, Bulanova E, Shim M, Guenther-Johnson JM, Faber PW, et al. A transcriptional signaling pathway in the IFN system mediated by 2’-5’-oligoadenylate activation of RNase L. Proc Natl Acad Sci U S A. 2005;102(41):14533–14538. doi: 10.1073/pnas.0507551102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matta H, Mazzacurati L, Schamus S, Yang T, Sun Q, Chaudhary PM. Kaposi’s sarcoma-associated herpesvirus (KSHV) oncoprotein K13 bypasses TRAFs and directly interacts with the IkappaB kinase complex to selectively activate NF-kappaB without JNK activation. J Biol Chem. 2007;282(34):24858–24865. doi: 10.1074/jbc.M700118200. [DOI] [PubMed] [Google Scholar]
- Melroe GT, Silva L, Schaffer PA, Knipe DM. Recruitment of activated IRF-3 and CBP/p300 to herpes simplex virus ICP0 nuclear foci: Potential role in blocking IFN-beta induction. Virology. 2007;360(2):305–321. doi: 10.1016/j.virol.2006.10.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menetret JF, Hegde RS, Heinrich SU, Chandramouli P, Ludtke SJ, Rapoport TA, et al. Architecture of the ribosome-channel complex derived from native membranes. J Mol Biol. 2005;348(2):445–457. doi: 10.1016/j.jmb.2005.02.053. [DOI] [PubMed] [Google Scholar]
- Meylan E, Curran J, Hofmann K, Moradpour D, Binder M, Bartenschlager R, et al. Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature. 2005;437(7062):1167–1172. doi: 10.1038/nature04193. [DOI] [PubMed] [Google Scholar]
- Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature. 2007;449(7164):819–826. doi: 10.1038/nature06246. [DOI] [PubMed] [Google Scholar]
- Mibayashi M, Martinez-Sobrido L, Loo YM, Cardenas WB, Gale M, Jr, Garcia-Sastre A. Inhibition of retinoic acid-inducible gene I-mediated induction of beta interferon by the NS1 protein of influenza A virus. J Virol. 2007;81(2):514–524. doi: 10.1128/JVI.01265-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michallet MC, Meylan E, Ermolaeva MA, Vazquez J, Rebsamen M, Curran J, et al. TRADD protein is an essential component of the RIG-like helicase antiviral pathway. Immunity. 2008;28(5):651–661. doi: 10.1016/j.immuni.2008.03.013. [DOI] [PubMed] [Google Scholar]
- Micheau O, Tschopp J. Induction of TNF receptor I-mediated apoptosis via two sequential signaling complexes. Cell. 2003;114(2):181–190. doi: 10.1016/s0092-8674(03)00521-x. [DOI] [PubMed] [Google Scholar]
- Moore CB, Bergstralh DT, Duncan JA, Lei Y, Morrison TE, Zimmermann AG, et al. NLRX1 is a regulator of mitochondrial antiviral immunity. Nature. 2008;451(7178):573–577. doi: 10.1038/nature06501. [DOI] [PubMed] [Google Scholar]
- Moulton TA, Jiang H, Guarini L, Fetell MR, Fisher PB. Induction of growth suppression and modification of gene expression in multidrug resistant human glioblastoma multiforme cells by recombinant human fibroblast and immune interferon. Intl J Cancer. 1992;51:373–378. doi: 10.1002/ijc.2910510307. [DOI] [PubMed] [Google Scholar]
- Mukherjee A, Morosky SA, Shen L, Weber CR, Turner JR, Kim KS, Wang T, Coyne CB. Retinoic Acid-induced Gene-1 (RIG-I) associates with the actin cytoskeleton via caspase activation and recruitment domain-dependent interactions. J Biol Chem. 2009;284(10):6486–6494. doi: 10.1074/jbc.M807547200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naitza S, Rosse C, Kappler C, Georgel P, Belvin M, Gubb D, et al. The Drosophila immune defense against gram-negative infection requires the death protein dFADD. Immunity. 2002;17(5):575–581. doi: 10.1016/s1074-7613(02)00454-5. [DOI] [PubMed] [Google Scholar]
- Narasimhan J, Wang M, Fu Z, Klein JM, Haas AL, Kim JJ. Crystal structure of the interferon-induced ubiquitin-like protein ISG15. J Biol Chem. 2005;280(29):27356–27365. doi: 10.1074/jbc.M502814200. [DOI] [PubMed] [Google Scholar]
- Neznanov N, Chumakov KM, Neznanova L, Almasan A, Banerjee AK, Gudkov AV. Proteolytic cleavage of the p65-RelA subunit of NF-kappaB during poliovirus infection. J Biol Chem. 2005;280(25):24153–24158. doi: 10.1074/jbc.M502303200. [DOI] [PubMed] [Google Scholar]
- Nusslein-Volhard C, Wieschaus E. Mutations affecting segment number and polarity in Drosophila. Nature. 1980;287(5785):795–801. doi: 10.1038/287795a0. [DOI] [PubMed] [Google Scholar]
- Okumura A, Alce T, Lubyova B, Ezelle H, Strebel K, Pitha PM. HIV-1 accessory proteins VPR and Vif modulate antiviral response by targeting IRF-3 for degradation. Virology. 2008;373(1):85–97. doi: 10.1016/j.virol.2007.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opitz B, Rejaibi A, Dauber B, Eckhard J, Vinzing M, Schmeck B, et al. IFNbeta induction by influenza A virus is mediated by RIG-I which is regulated by the viral NS1 protein. Cell Microbiol. 2007;9(4):930–938. doi: 10.1111/j.1462-5822.2006.00841.x. [DOI] [PubMed] [Google Scholar]
- Organesyan G, Saha SK, Guo B, He JQ, Shahanigian A, Zarnegar B, et al. Critical role of TRAF3 in the Toll-like receptor-dependent and -independent antiviral response. Nature. 2006;439:208–211. doi: 10.1038/nature04374. [DOI] [PubMed] [Google Scholar]
- Otsuka M, Kato N, Moriyama M, Taniguchi H, Wang Y, Dharel N, et al. Interaction between the HCV NS3 protein and the host TBK1 protein leads to inhibition of cellular antiviral responses. Hepatology. 2005;41(5):1004–1012. doi: 10.1002/hep.20666. [DOI] [PubMed] [Google Scholar]
- Pan M, Geng S, Xiao S, Ren J, Liu Y, Li X, et al. Apoptosis induced by synthetic retinoic acid CD437 on human melanoma A375 cells involves RIG-I pathway. Arch Dermatol Res. 2008;301(1):15–20. doi: 10.1007/s00403-008-0902-x. [DOI] [PubMed] [Google Scholar]
- Pestka S, Baron S. Definintion and classification of the interferons. Meth Enzymol. 1981;78:3–14. doi: 10.1016/0076-6879(81)78091-1. [DOI] [PubMed] [Google Scholar]
- Peters K, Chattopadhyay S, Sen GC. IRF-3 activation by Sendai virus infection is required for cellular apoptosis and avoidance of persistence. J Virol. 2008;82(7):3500–3508. doi: 10.1128/JVI.02536-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichlmair A, Schulz O, Tan CP, Naslund TI, Liljestrom P, Weber F, et al. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5’-phosphates. Science. 2006;314(5801):997–1001. doi: 10.1126/science.1132998. [DOI] [PubMed] [Google Scholar]
- Plotch SJ, Bouloy M, Ulmanen I, Krug RM. A unique cap(m7GpppXm)-dependent influenza virion endonuclease cleaves capped RNAs to generate the primers that initiate viral RNA transcription. Cell. 1981;23(3):847–858. doi: 10.1016/0092-8674(81)90449-9. [DOI] [PubMed] [Google Scholar]
- Plumet S, Herschke F, Bourhis JM, Valentin H, Longhi S, Gerlier D. Cytosolic 5’-triphosphate ended viral leader transcript of measles virus as activator of the RIG I-mediated interferon response. PLoS ONE. 2007;2(3):e279. doi: 10.1371/journal.pone.0000279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poeck H, Besch R, Maihoefer C, Renn M, Tormo D, Morskaya SS, et al. 5’-Triphosphate-siRNA: turning gene silencing and Rig-I activation against melanoma. Nat Med. 2008;14(11):1256–1263. doi: 10.1038/nm.1887. [DOI] [PubMed] [Google Scholar]
- Rebsamen M, Meylan E, Curran J, Tschopp J. The antiviral adaptor proteins Cardif and Trif are processed and inactivated by caspases. Cell Death Differ. 2008;15(11):1804–1811. doi: 10.1038/cdd.2008.119. [DOI] [PubMed] [Google Scholar]
- Rintahaka J, Wiik D, Kovanen PE, Alenius H, Matikainen S. Cytosolic antiviral RNA recognition pathway activates caspases 1 and 3. J Immunol. 2008;180(3):1749–1757. doi: 10.4049/jimmunol.180.3.1749. [DOI] [PubMed] [Google Scholar]
- Rothenfusser S, Goutagny N, DiPerna G, Gong M, Monks BG, Schoenemeyer A, et al. The RNA helicase Lgp2 inhibits TLR-independent sensing of viral replication by retinoic acid-inducible gene-I. J Immunol. 2005;175(8):5260–5268. doi: 10.4049/jimmunol.175.8.5260. [DOI] [PubMed] [Google Scholar]
- Rubinstein M, Rubinstein S, Familletti PC, Miller R, Waldman AA, Pestka S. Human leukocyte interferon: production, purification to homogeneity, and initial characterization. Proc Natl Acad Sci USA. 1979;76(2):640–644. doi: 10.1073/pnas.76.2.640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saira K, Zhou Y, Jones C. The infected cell protein 0 encoded by bovine herpesvirus 1 (bICP0) induces degradation of interferon response factor 3 and, consequently, inhibits beta interferon promoter activity. J Virol. 2007;81(7):3077–3086. doi: 10.1128/JVI.02064-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito T, Hirai R, Loo YM, Owen D, Johnson CL, Sinha SC, et al. Regulation of innate antiviral defenses through a shared repressor domain in RIG-I and LGP2. Proc Natl Acad Sci U S A. 2007;104(2):582–587. doi: 10.1073/pnas.0606699104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkar D, Desalle R, Fisher PB. Evolution of MDA-5/RIG-I-dependent innate immunity: independent evolution by domain grafting. Proc Natl Acad Sci U S A. 2008;105(44):17040–17045. doi: 10.1073/pnas.0804956105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder M, Baran M, Bowie AG. Viral targeting of DEAD box protein 3 reveals its role in TBK1/IKKepsilon-mediated IRF activation. EMBO J. 2008;27(15):2147–2157. doi: 10.1038/emboj.2008.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen GC, Sarkar SN. The interferon-stimulated genes: targets of direct signaling by interferons, double-stranded RNA, and viruses. Curr Top Microbiol Immunol. 2007;316:233–250. doi: 10.1007/978-3-540-71329-6_12. [DOI] [PubMed] [Google Scholar]
- Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122(5):669–682. doi: 10.1016/j.cell.2005.08.012. [DOI] [PubMed] [Google Scholar]
- Sirén J, Imaizumi T, Sarkar D, Pietilä T, Noah DL, Lin R, Hiscott J, Krug RM, Fisher PB, Julkunen I, Matikainen S. Retinoic acid inducible gene-I and mda-5 are involved in influenza A virus-induced expression of antiviral cytokines. Microbes and Infection. 2006;8(8):2013–2020. doi: 10.1016/j.micinf.2006.02.028. [DOI] [PubMed] [Google Scholar]
- Smith EJ, Marie I, Prakash A, Garcia-Sastre A, Levy DE. IRF3 and IRF7 phosphorylation in virus-infected cells does not require double-stranded RNA-dependent protein kinase R or Ikappa B kinase but is blocked by Vaccinia virus E3L protein. J Biol Chem. 2001;276(12):8951–8957. doi: 10.1074/jbc.M008717200. [DOI] [PubMed] [Google Scholar]
- Smyth DJ, Cooper JD, Bailey R, Field S, Burren O, Smink LJ, et al. A genome-wide association study of nonsynonymous SNPs identifies a type 1 diabetes locus in the interferon-induced helicase (IFIH1) region. Nat Genet. 2006;38(6):617–619. doi: 10.1038/ng1800. [DOI] [PubMed] [Google Scholar]
- Soulat D, Burckstummer T, Westermayer S, Goncalves A, Bauch A, Stefanovic A, et al. The DEAD-box helicase DDX3X is a critical component of the TANK-binding kinase 1-dependent innate immune response. EMBO J. 2008;27(15):2135–2146. doi: 10.1038/emboj.2008.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stetson DB, Medzhitov R. Recognition of cytosolic DNA activates an IRF3-dependent innate immune response. Immunity. 2006;24(1):93–103. doi: 10.1016/j.immuni.2005.12.003. [DOI] [PubMed] [Google Scholar]
- Story RM, Li H, Abelson JN. Crystal structure of a DEAD box protein from the hyperthermophile Methanococcus jannaschii. Proc Natl Acad Sci U S A. 2001;98(4):1465–1470. doi: 10.1073/pnas.98.4.1465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Z-z, Lee S-G, Emdad L, Lebedeva IV, Gupta P, Valerie K, et al. Cloning and characterization of SARI (suppressor of AP-1, regulated by IFN) Proc Natl Acad Sci USA. 2008;105:20906–20911. doi: 10.1073/pnas.0807975106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su ZZ, Sarkar D, Emdad L, Barral PM, Fisher PB. Central role of interferon regulatory factor-1 (IRF-1) in controlling retinoic acid inducible gene-I (RIG-I) expression. J Cell Physiol. 2007;213(2):502–510. doi: 10.1002/jcp.21128. [DOI] [PubMed] [Google Scholar]
- Sumpter R, Jr, Loo YM, Foy E, Li K, Yoneyama M, Fujita T, et al. Regulating intracellular antiviral defense and permissiveness to hepatitis C virus RNA replication through a cellular RNA helicase, RIG-I. J Virol. 2005;79(5):2689–2699. doi: 10.1128/JVI.79.5.2689-2699.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q, Sun L, Liu HH, Chen X, Seth RB, Forman J, et al. The specific and essential role of MAVS in antiviral innate immune responses. Immunity. 2006;24(5):633–642. doi: 10.1016/j.immuni.2006.04.004. [DOI] [PubMed] [Google Scholar]
- Sun YW. RIG-I, a human homolog gene of RNA helicase, is induced by retinoic acid during the differentiation of acute promyelocytic leukemia cell. Shanghi Second Medical University; Shanghai: 1997. [Google Scholar]
- Tait SW, Reid EB, Greaves DR, Wileman TE, Powell PP. Mechanism of inactivation of NF-kappa B by a viral homologue of I kappa b alpha. Signal-induced release of i kappa b alpha results in binding of the viral homologue to NF-kappa B. J Biol Chem. 2000;275(44):34656–34664. doi: 10.1074/jbc.M000320200. [DOI] [PubMed] [Google Scholar]
- Takahasi K, Yoneyama M, Nishihori T, Hirai R, Kumeta H, Narita R, et al. Nonself RNA-sensing mechanism of RIG-I helicase and activation of antiviral immune responses. Mol Cell. 2008;29(4):428–440. doi: 10.1016/j.molcel.2007.11.028. [DOI] [PubMed] [Google Scholar]
- Takeuchi O, Akira S. Recognition of viruses by innate immunity. Immunol Rev. 2007;220:214–224. doi: 10.1111/j.1600-065X.2007.00562.x. [DOI] [PubMed] [Google Scholar]
- Takeuchi O, Akira S. MDA5/RIG-I and virus recognition. Curr Opin Immunol. 2008;20(1):17–22. doi: 10.1016/j.coi.2008.01.002. [DOI] [PubMed] [Google Scholar]
- Talon J, Horvath CM, Polley R, Basler CF, Muster T, Palese P, et al. Activation of interferon regulatory factor 3 is inhibited by the influenza A virus NS1 protein. J Virol. 2000;74(17):7989–7996. doi: 10.1128/jvi.74.17.7989-7996.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd JA, Walker NM, Cooper JD, Smyth DJ, Downes K, Plagnol V, et al. Robust associations of four new chromosome regions from genome-wide analyses of type 1 diabetes. Nat Genet. 2007;39(7):857–864. doi: 10.1038/ng2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valchanova RS, Picard-Maureau M, Budt M, Brune W. Murine cytomegalovirus m142 and m143 are both required to block protein kinase R-mediated shutdown of protein synthesis. J Virol. 2006;80(20):10181–10190. doi: 10.1128/JVI.00908-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkataraman T, Valdes M, Elsby R, Kakuta S, Caceres G, Saijo S, et al. Loss of DExD/H box RNA helicase LGP2 manifests disparate antiviral responses. J Immunol. 2007;178(10):6444–6455. doi: 10.4049/jimmunol.178.10.6444. [DOI] [PubMed] [Google Scholar]
- Wertz IE, O’Rourke KM, Zhou H, Eby M, Aravind L, Seshagiri S, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-kappaB signalling. Nature. 2004;430(7000):694–699. doi: 10.1038/nature02794. [DOI] [PubMed] [Google Scholar]
- Xiang Y, Condit RC, Vijaysri S, Jacobs B, Williams BR, Silverman RH. Blockade of interferon induction and action by the E3L double-stranded RNA binding proteins of vaccinia virus. J Virol. 2002;76(10):5251–5259. doi: 10.1128/JVI.76.10.5251-5259.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, Shu HB. VISA Is an Adapter Protein Required for Virus-Triggered IFN-beta Signaling. Mol Cell. 2005;19(6):727–740. doi: 10.1016/j.molcel.2005.08.014. [DOI] [PubMed] [Google Scholar]
- Yang Y, Liang Y, Qu L, Chen Z, Yi M, Li K, et al. Disruption of innate immunity due to mitochondrial targeting of a picornaviral protease precursor. Proc Natl Acad Sci U S A. 2007;104(17):7253–7258. doi: 10.1073/pnas.0611506104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneyama M, Fujita T. Structural mechanism of RNA recognition by the RIG-I-like receptors. Immunity. 2008;29(2):178–181. doi: 10.1016/j.immuni.2008.07.009. [DOI] [PubMed] [Google Scholar]
- Yoneyama M, Kikuchi M, Matsumoto K, Imaizumi T, Miyagishi M, Taira K, et al. Shared and unique functions of the DExD/H-box helicases RIG-I, MDA5, and LGP2 in antiviral innate immunity. J Immunol. 2005;175(5):2851–2858. doi: 10.4049/jimmunol.175.5.2851. [DOI] [PubMed] [Google Scholar]
- Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, et al. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol. 2004;5(7):730–737. doi: 10.1038/ni1087. [DOI] [PubMed] [Google Scholar]
- Zaragoza C, Saura M, Padalko EY, Lopez-Rivera E, Lizarbe TR, Lamas S, et al. Viral protease cleavage of inhibitor of kappaBalpha triggers host cell apoptosis. Proc Natl Acad Sci U S A. 2006;103(50):19051–19056. doi: 10.1073/pnas.0606019103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Wu X, Lee AJ, Jin W, Chang M, Wright A, et al. Regulation of IkappaB kinase-related kinases and antiviral responses by tumor suppressor CYLD. J Biol Chem. 2008;283(27):18621–18626. doi: 10.1074/jbc.M801451200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang NN, Shen SH, Jiang LJ, Zhang W, Zhang HX, Sun YP, et al. RIG-I plays a critical role in negatively regulating granulocytic proliferation. Proc Natl Acad Sci U S A. 2008;105(30):10553–10558. doi: 10.1073/pnas.0804895105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao C, Denison C, Huibregtse JM, Gygi S, Krug RM. Human ISG15 conjugation targets both IFN-induced and constitutively expressed proteins functioning in diverse cellular pathways. Proc Natl Acad Sci U S A. 2005;102(29):10200–10205. doi: 10.1073/pnas.0504754102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong B, Yang Y, Li S, Wang YY, Li Y, Diao F, et al. The adaptor protein MITA links virus-sensing receptors to IRF3 transcription factor activation. Immunity. 2008;29(4):538–550. doi: 10.1016/j.immuni.2008.09.003. [DOI] [PubMed] [Google Scholar]