

Abstract
Intercellular communication is needed for both the generation of the mesodermal germ layer and its division into distinct subpopulations. To dissect the functions of fibroblast growth factor receptor-1 (FGFR1) during mouse gastrulation as well as to gain insights into its possible roles during later embryonic development, we have introduced specific mutations into the Fgfr1 locus by gene targeting. Our results show functional dominance of one of the receptor isoforms and suggest a function for the autophosphorylation of site Y766 in the negative regulation of FGFR1 activity. Y766F and hypomorphic mutations in Fgfr1 generate opposite phenotypes in terms of homeotic vertebral transformations, suggesting a role for FGFR1 in patterning the embryonic anteriorposterior axis by way of regulation of Hox gene activity.
Keywords: Fibroblast growth factor, somite, Hox gene, homeotic transformation, limb patterning, phospholipase C
Patterning of the mesodermal germ layer is temporally closely linked to its generation during gastrulation. The mesoderm is divided into axial, paraxial, intermediate, lateral, and extraembryonic mesoderm depending on its proximodistal origin in the primitive streak. Transplantation studies with chick embryos have shown that the newly formed paraxial mesoderm, even before its segmentation into somites, also has its anteroposterior (A-P) positional identity determined (Kieny et al. 1972). Consistent with this finding, the mesodermal expression of Hox transcription factor genes governing A-P pattern is activated early in the primitive streak.
Both generation and patterning of the mesoderm appear to involve intercellular communication and work with Xenopus embryos has identified some candidate signaling molecules, including fibroblast growth factors (FGFs). FGFs can induce the expression of mesodermal markers, such as the Brachyury homolog Xbra, in isolated animal cap ectoderm (Slack et al. 1996). Ectopic application or expression of FGFs has also been observed to result in up-regulation of posteriorly expressed members of the Xenopus Hox gene family and a caudal-related gene Xcad3 in anterior mesodermal as well as neuroectodermal tissue, suggesting a role for FGF signaling in A-P patterning (Ruiz i Altaba and Melton 1989; Cho and De Robertis 1990; Cox and Hemmati-Brivanlou 1995; Kengaku and Okamoto 1995; Kolm and Sive 1995; Lamb and Harland 1995; Pownall et al. 1996). Expression of a dominant-negative FGF receptor in Xenopus embryos results in defective posterolateral mesoderm induction and maintenance (Amaya et al. 1991; Kroll and Amaya 1996). Repression or posteriorization of Hox and Xcad3 gene expression has also been observed in embryos expressing the dominant-negative FGF receptor (Pownall et al. 1996; Godsave and Durston 1997). This Hox gene repression can be overcome by ectopic expression of Xcad3, supporting a model where FGFs regulate posterior Hox gene expression through induction of caudal-related transcription factors (Subramanian et al. 1995; Pownall et al. 1996). Loss of caudal-related Cdx1 or reduction of Cdx2 function in mouse leads to homeotic transformations consistent with this model (Subramanian et al. 1995; Chawengsaksophak et al. 1997).
However, to date, there is no clear evidence to support a role for FGF signaling in A-P patterning from the phenotypes caused by mutation of components of the FGF pathways in mice. Consistent with the work with Xenopus, a null mutation in one of the four FGF receptor genes, FGF receptor-1 (Fgfr1), leads to a severe gastrulation defect in mouse embryos (Deng et al. 1994; Yamaguchi et al. 1994). Fgfr1 mutant embryos appear growth retarded and cell migration through the primitive streak is impaired. Even in the most advanced mutants the amount of paraxial mesoderm is greatly reduced and somites are not formed. Analysis of chimeric embryos consisting of a mixture of wild-type and Fgfr1 mutant cells has revealed a cell autonomous function for FGFR1 in the mesodermal and endodermal cell movements through the primitive streak (Ciruna et al. 1997; Deng et al. 1997). However, the requirement for FGFR1 in normal streak morphogenesis obscures any possible additional role in A-P patterning.
To dissect genetically the functions of Fgfr1 during gastrulation and in later developmental processes we have generated a series of hypomorphic and function-specific alleles of the gene. Our results show that alterations in signaling through FGFR1 can result in changes in the vertebral identities independent of defects in the production and segmentation of paraxial mesoderm. Thus, our results provide genetic evidence for the involvement of FGF signaling in regulation of Hox gene activity and A-P pattern. In addition, the hypomorphic alleles reveal a function for FGFR1 in the growth and patterning of the limbs, possibly also involving Hox regulation.
Our series of Fgfr1 mutations also addressed the biological significance of multiple FGFR1 isoforms and signal transduction pathways. We show that one of the two alternatively spliced receptor isoforms (Johnson et al. 1991) is functionally dominant. Mutation of the tyrosine autophosphorylation site Y766, critical for phospholipase Cγ (PLCγ) binding and activation (Mohammadi et al. 1992; Peters et al. 1992), is compatible with survival, but leads to alterations in A-P patterning of the vertebral column in the opposite direction to the hypomorphic alleles. Our results suggest that instead of transducing a net positive signal, a signal starting at phosphorylated Y766 plays a role in the negative regulation of FGFR1 activity in vivo.
Results
Generation of an allelic series of Fgfr1
To create hypomorphic and site-specific alleles of the Fgfr1 gene a series of point mutations as well as neomycin phosphotransferase (neo) expression cassette insertions were introduced into the Fgfr1 locus by gene targeting (Fig. 1A,B; see Materials and Methods). Three types of mutations were produced. First, alleles with a neo cassette in the sense orientation in introns 7 and 15 were generated (alleles n7, IIIcn, IIIbn, n15, and n15YF). These insertions appear to result in a reduction in the amount of full-length Fgfr1 transcript produced (Fig. 1C; see below). Second, the protein coding capacity of the two exons (exons IIIc and IIIb) encoding alternatively spliced isoforms of FGFR1 (Johnson et al. 1991) were desrupted separately by introducing in-frame stop codons into these exons (represented by asterisks in Fig. 1A; alleles IIIcn, IIIcl, IIIbn, and IIIbl). Third, one of the autophosphorylation sites and the PLCγ binding site of FGFR1 (Y766) was inactivated by mutation into phenylalanine (alleles n15YF and Y766Fl). In addition, control alleles carrying loxP sites (recognition site for the site-specific recombinase Cre) in intronic regions were generated (alleles l7 and l15).
Figure 1.
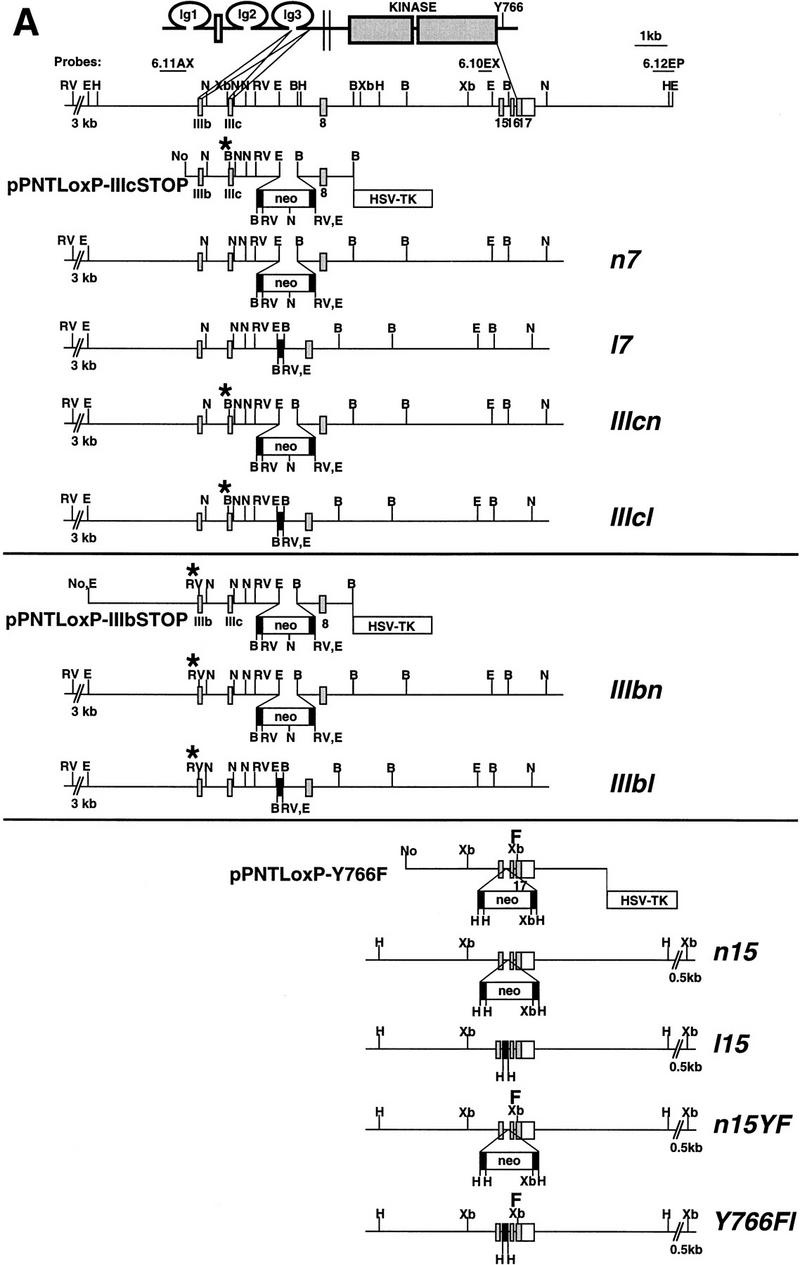


Genomic structure and characterization of the Fgfr1 alleles generated. (A) Schematic structure of the FGFR1 protein is shown at top. The two alternatively spliced receptor isoforms, FGFR1 IIIc and IIIb, differing in their ligand-binding domain, were inactivated separately by introducing stop codons (asterisks) in the beginning of exons 7 and 6, respectively. These stop codons were followed by mutations creating diagnostic restriction enzyme recognition sites BamHI (IIIc) and EcoRV (IIIb). One of the tyrosine autophosphorylation sites of FGFR1, Y766, was mutated into phenylalanine (F) followed by a silent mutation creating a XbaI site. The alleles were produced using three targeting vectors: pPNTLoxP–IIIcSTOP, pPNTLoxP–IIIbSTOP, and pPNTLoxP–Y766F. Depending on the site of homologous recombination, alleles either with (IIIcn, IIIbn, and n15YF) or without (n7 and n15) the above mentioned mutations were generated. The neo cassettes were flanked by loxP sites (black rectangles), allowing their excision in vivo to generate alleles l7, IIIcl, IIIbl, l15, and Y766Fl. (B) BamHI; (E) EcoRI; (H) HindIII; (N) NcoI; (RV) EcoRV; (Xb) XbaI; (Ig) immunoglobulin-like domain; (neo) neomycin phosphotransferase gene driven by PGK-1 promoter and followed by PGK-1 polyadenylation site. (B) Southern blot analysis of the ES cell lines generated. See A for identification of the probes and restriction maps. (C) Northern blot analysis of Fgfr1 mRNA expression in a F2 litter carrying the n7 allele. Total RNA was isolated from E10.5 embryos and analyzed with a Fgfr1 cDNA probe from the kinase domain region and a Gapdh cDNA probe. Quantification of the hybridization signal revealed a three to five fold reduction in the amount of full-length Fgfr1 transcript in the n7 homozygotes compared to their heterozyous or wild-type littermates.
The alleles with the neo cassettes were created in embryonic stem (ES) cells using targeting vectors, where the desired mutations were incorporated in the arms of homology (Fig. 1A; see Materials and Methods). Depending on the site of homologous recombination between the vector arms and the genomic Fgfr1 locus, alleles with or without the specific mutations were generated. In these alleles the neo gene, driven by the phosphoglycerate kinase-1 (PGK-1) promoter and followed by the PGK-1 polyadenylation signal, was inserted in the sense orientation into two different intronic regions of the Fgfr1 gene (introns 7 and 15). These neo cassettes were flanked by loxP sites allowing their removal by crosses with transgenic mice expressing Cre recombinase under the control of a cytomegalovirus enhancer and β-actin promoter (Sauer 1993; Nagy et al. 1998). Therefore, the effects of the site-specific mutations could be analyzed without being complicated by the neo insertion.
IIIc is the major functional isoform of Fgfr1
Embryos homozygous for alleles with a stop codon in exon IIIc (IIIcn and IIIcl) displayed identical phenotypes resembling the embryos homozygous for the putative null alleles (Deng et al. 1994; Yamaguchi et al. 1994). At embryonic day 9.5 (E9.5) IIIcl/IIIcl embryos showed reduced amount of Mox-1-positive paraxial mesoderm, which was not organized into somites (see Fig. 4B; below). On the other hand, mice homozygous for an allele where the exon IIIb was inactivated (IIIbl/IIIbl) were viable and fertile (Table 1). We conclude that IIIc is the dominant isoform and responsible for the majority of FGFR1 functions. However, when the overall activity of Fgfr1 was reduced by a neo cassette insertion into the gene (see following section), an additional mutation in the IIIb exon further enhanced the axial and limb patterning phenotypes observed (cf. the phenotypes of n7 and IIIbn homozygotes, Table 2; Fig. 5, below). In addition, whereas ∼50% of n7/null transheterozygotes survived to term, no newborn IIIbn/null transheterozygotes were recovered (Table 1). Thus, there appears to be some redundancy between the two Fgfr1 isoforms.
Figure 4.
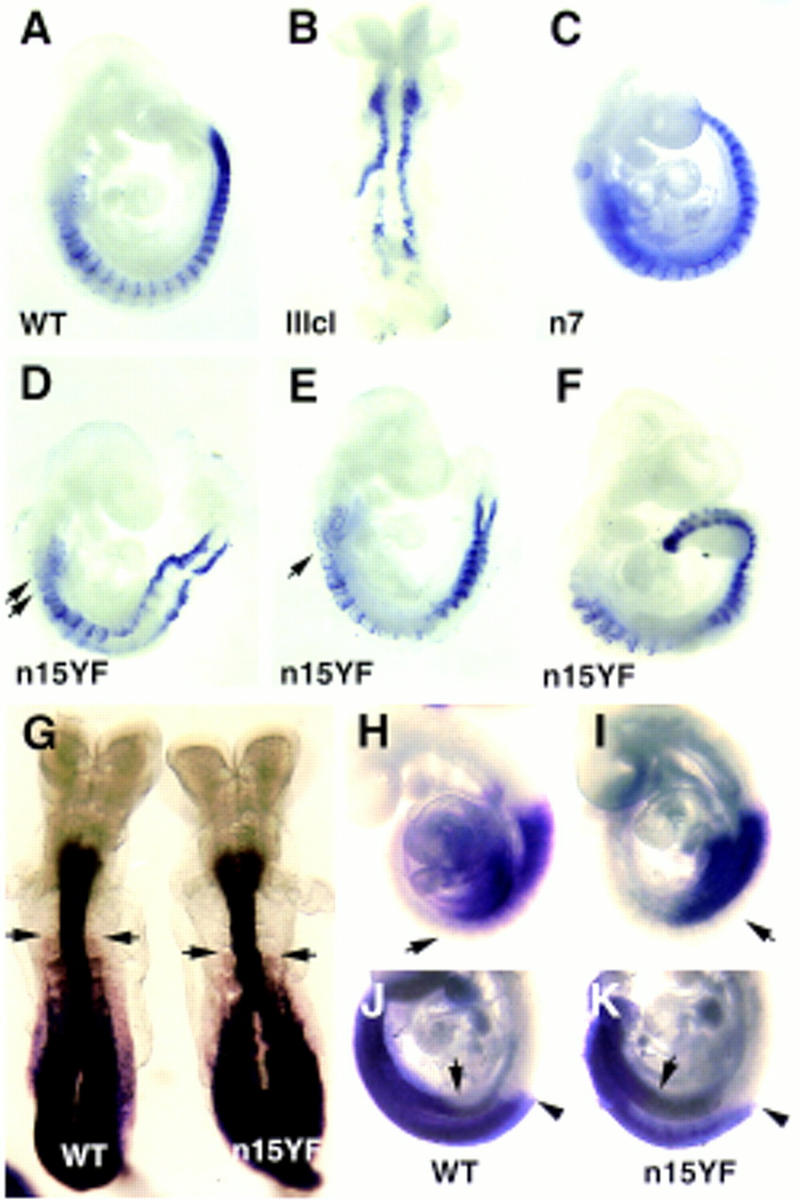
Analysis of Mox-1 and Hox gene expression in the hypomorphic mutants by wholemount mRNA in situ hybridization. Expression of Mox-1 (A–F) in an E9.5 wildtype (A), a IIIcl homozygote (B), a n7 homozygote (C), n15YF homozygotes (D,E), and an E10.5 n15YF homozygote (F). The most anterior somites in n15YF homozygotes were often reduced in size (arrows in D,E). Expression of A-P patterning markers HoxD4 (G), HoxB5 (H,I) and HoxB9 (J,K) in wild type (G,H,J) and n15YF homozygous (G,I,K) embryos. The anterior level of expression of HoxD4 appears shifted posteriorly by one somite level in E8.5 mutant embryo compared to a wild-type littermate (arrows in G). In E9.5 embryos, HoxB5 expression posteriorly was weaker in the n15YF mutants than in the wild-type littermate (arrows in H,I). In a subset of E9.5 embryos hybridized with the HoxB9 probe the anterior level of hybridization appeared posteriorized in the lateral plate mesoderm concomitant to the expansion of the limb fields (arrows in J,K). In all cases the neural expression appeared unchanged (arrowheads in J,K).
Table 1.
Analysis of F2 litters of various Fgfr1 alleles
| Alleles
|
|
+/+
|
+/−
|
−/−
|
Phenotype of −/−a |
|---|---|---|---|---|---|
| n7/n7 | E9.5 | 7 | 22 | 11 | AP(a), L, CR |
| P0 | 94 | 34b | |||
| n7/null | P0 | 45 | 9b | AP(a), L, CR | |
| l7/l7 | weanlings | 15 | 34 | 15 | viable |
| IIIcn/IIIcn | E9.5 | 2 | 5 | 2b,c | GA |
| IIIcl/IIIcl | E9.5 | 16 | 26 | 6b,c | GA |
| IIIbn/IIIbn | E10.5–16.5 | 45 | 85 | 36b,c | AP(a), L, CR |
| P0 | 19 | 30 | 7b | ||
| IIIbn/null | P0 | 43 | 0 | ||
| IIIbl/IIIbl | weanlings | 11 | 39 | 15 | viable |
| n15/n15 | E8.5–11.5 | 17 | 42 | 11b,c | AP(a), L, CR |
| P0 | 67 | 5b | |||
| I15/I15 | P0 | 62 | 20 | viable | |
| n15YF/n15YF | E8.5–11.5 | 84 | 191 | 76b,c | AP(a), L, CR |
| P0 | 67 | 12b | |||
| Y766Fl/Y766Fl | P0 | 116 | 30b | viable, AP(p) | |
| weanlings | 20 | 38 | 16b | ||
| Y766Fl/null | P0 | 49 | 22b | viable, AP(p) | |
[AP(a)] Vertebral transformations predominantly to anterior direction, posterior truncations; (L) distal limb defects; (CR) craniofacial defects, neonatal lethality; (GA) gastrulation defect; no somites; [AP(p)] vertebral transformations to posterior direction.
Distinguishable from the littermates (see text for details).
Also dead/reabsorbed embryos recovered.
Table 2.
Axial skeletal defects observed in newborn mice carrying various Fgfr1 alleles
| Defects
|
+/+ (n = 22)
|
115/115 (n = 16)
|
null/+ (n = 7)
|
n7/n7 (n = 34)
|
IIIbn/IIIbn (n = 6)
|
n7/null (n = 5)
|
n15/n15a (n = 16)
|
Y766FI/+ (n = 27)
|
Y766Fl/ Y766Fl (n = 22)
|
Y766Fl/ null (n = 18)
|
|---|---|---|---|---|---|---|---|---|---|---|
| Anterior transformations | ||||||||||
| C1 → occipital | 4 (12%) | 5 (100%) | 15 (94%) | |||||||
| C2 → C1 | 3 (9%) | 5 (100%) | 13 (81%) | |||||||
| C3 → C2 | 2 (40%) | 5 (31%) | ||||||||
| C7 → C6 | 3 (9%) | 2 (40%) | 8 (50%) | |||||||
| R1 not attached to st. | 11 (32%) | 2 (33%) | 5 (100%) | 16 (100%) | ||||||
| R2 not attached to st. | 2 (40%) | 9 (56%) | ||||||||
| L1 → T13 | 1 (4.5%) | 2 (20%) | 14 (41%) | 4 (67%) | 5 (100%) | N.A. | ||||
| S1 → L6 | 3 (9%) | 3 (50%) | 3 (75%) | N.A. | ||||||
| S2 → S1 | 4 (12%) | 2 (33%) | 3 (75%) | N.A. | ||||||
| Posterior transformations | ||||||||||
| C5 → C6 | 4 (12%) | 1 (17%) | 1 (3.7%) | 2 (9.1%) | 9 (50%) | |||||
| C6 → C7 | 3 (13.6%) | 9 (50%) | ||||||||
| C7 → T1 | 3 (9%) | 5 (19%) | 13 (59%) | 10 (55%) | ||||||
| T1 → T2 | 2 (9.1%) | 4 (22%) | ||||||||
| R7 not attached to st. | 1 (10%) | 20 (59%) | 6 (100%) | 2 (40%) | 6 (38%) | 10 (45%) | 9 (50%) | |||
| T12 → L1 | 1 (4.5%) | 2 (11%) | ||||||||
| T13 → L1 | 2 (7.4%) | 12 (55%) | 8 (44%) | |||||||
| L5 → S1 | 1 (4.5%) | 2 (11%) | ||||||||
| L6 → S1 | 8 (36%) | 1 (6%) | 3 (30%) | 17 (63%) | 16 (73%) | 10 (55%) | ||||
| S1 → S2 | 1 (10%) | 3 (11%) | 13 (59%) | 8 (44%) | ||||||
| S3 → S4 | 5 (23%) | 5 (28%) | ||||||||
| Others | ||||||||||
| C1 malformations | 4 (18%) | |||||||||
| C2 malformations | 11 (32%) | 2 (33%) | 2 (7.4%) | 5 (22%) | 3 (17%) | |||||
| Rib fusions | 16 (47%) | 5 (100%) | 16 (100%) | 9 (21%) | 5 (27%) | |||||
| No. of caudal vertebrae | 31 (s.d. 0.9) | 32 (s.d. 0.89) | 31 (s.d. 0.94) | 25 (s.d. 4.8) | 17 (s.d. 4.6) | 9.8 (s.d. 7.1) | 5 (s.d. 9.4) | 31 (s.d. 0.87) | 31 (s.d. 0.94) | 30 (s.d. 1.4) |
(st.) Sternum; (N.A.) not available; (s.d.) standard deviation.
Combined data from n15/n15 and n15YF/n15YF.
Figure 5.
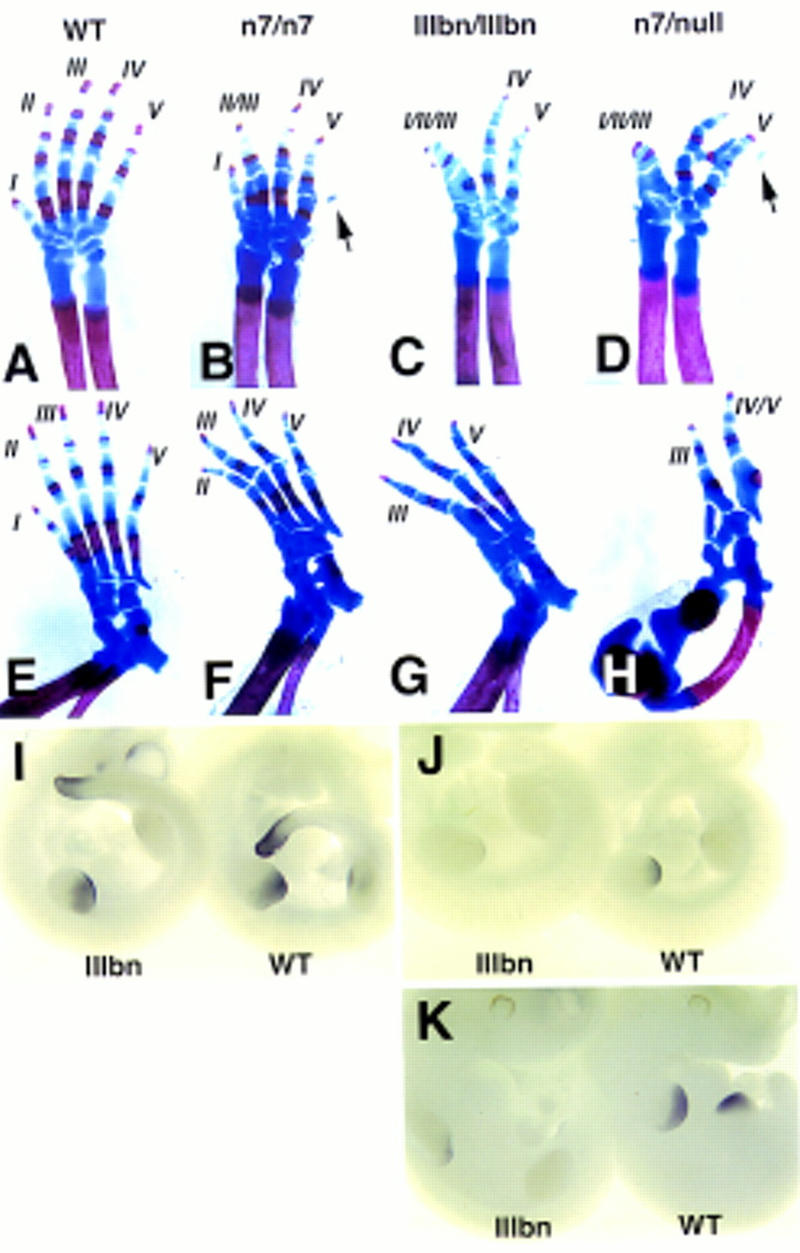
Distal limb defects in the hypomorphic mutants. Skeletal preparations of forelimbs (A–D) and hindlimbs (E–H) of newborn wild-type (A,E), n7 homozygous (B,F), IIIbn homozygous (C,G) and n7/null transheterozygous (D,H) newborn mice. Reduced Fgfr1 function appears to correlate with successive loss of anterior digits. Other defects include syndactyly, delayed ossification of distal phalanges, and occasional appearance of an extra postaxial cartilage condensation in the forelimbs (arrows in B,D). In n7/null transheterozygotes the limb defects are markedly enhanced resulting occasionally in defects in the development of the more proximal limb (H). Analysis of 5′ HoxD gene expression in the limb buds of IIIbn hypomorphic mutants by whole mount mRNA in situ hybridization (I–K). HoxD11 expression at E10.5 (I) in a IIIbn homozygote (IIIbn) and a wild-type littermate (wt), showing no obvious difference. In contrast, HoxD13 expression both at E10.5 (J) and E11 (K) was reduced in the mutants compared to their wild-type littermates.
neo gene insertions into intronic regions create hypomorphic alleles of Fgfr1
The neo cassette insertions into introns 7 and 15 produced alleles (n7, IIIbn, n15YF, and n15), which showed similar phenotypes with quantitative differences. These phenotypes were primarily attributable to the neo cassette insertions themselves, and the associated mutations (IIIb exon and Y766F mutations) had only minor effects on the severity of the phenotypes (Table 1). Mice heterozygous for these alleles appeared normal but when homozygous, all the alleles caused neonatal lethality, defects in craniofacial and limb patterning, and abnormal development of the A-P axis (Table 1). The craniofacial defects, likely contributing to neonatal lethality, result from a marked reduction of the second branchial arch (data not shown). In the case of IIIbn, n15YF, and n15 alleles some of the mutants did not appear to survive to term. To test genetically the nature of these alleles, the n7 heterozygotes were crossed with mice heterozygous for the Fgfr1 null allele (Yamaguchi et al. 1994). All the observed defects were more severe in the n7/null transheterozygotes than in the n7/n7 homozygotes (Table 2 and Fig. 5, below). This suggests that n7, IIIbn, n15YF, and n15 are hypomorphic alleles producing reduced gene activity. Consistent with the genetics, a three- to fivefold reduction in the amount of the full-length Fgfr1 transcript was seen in the n7/n7 embryos compared with the heterozygous or wild-type littermates (Fig. 1C). The exact mechanism for this reduction is unclear but it is possible that the neo cassette (containing the PGK-1 polyadenylation signal) causes premature termination of transcription or that aberrant RNA splicing into the neo cassette results in reduced amounts of Fgfr1 mRNA. Similar reductions in mRNA levels, resulting in hypomorphic alleles, have also been reported after insertions of neo cassettes into the N-myc and Fgf8 loci (Meyers et al. 1998; Nagyet al. 1998) .
Hypomorphic alleles cause posterior truncations and homeotic transformations in the vertebral column
Posterior truncations were observed in newborn mice homozygous for the hypomorphic alleles (Fig. 2A–E and Table 2). The n7 and IIIbn homozygotes had defects in tail development, whereas n15YF and n15 homozygotes displayed more severe posterior deletions, which usually extended into the lumbrosacral level. Posterior to the level of vertebral truncation of the n15 and n15YF homozygotes, hind limbs and occasional remnants of vertebral bodies were observed (Fig. 2E and data not shown).
Figure 2.
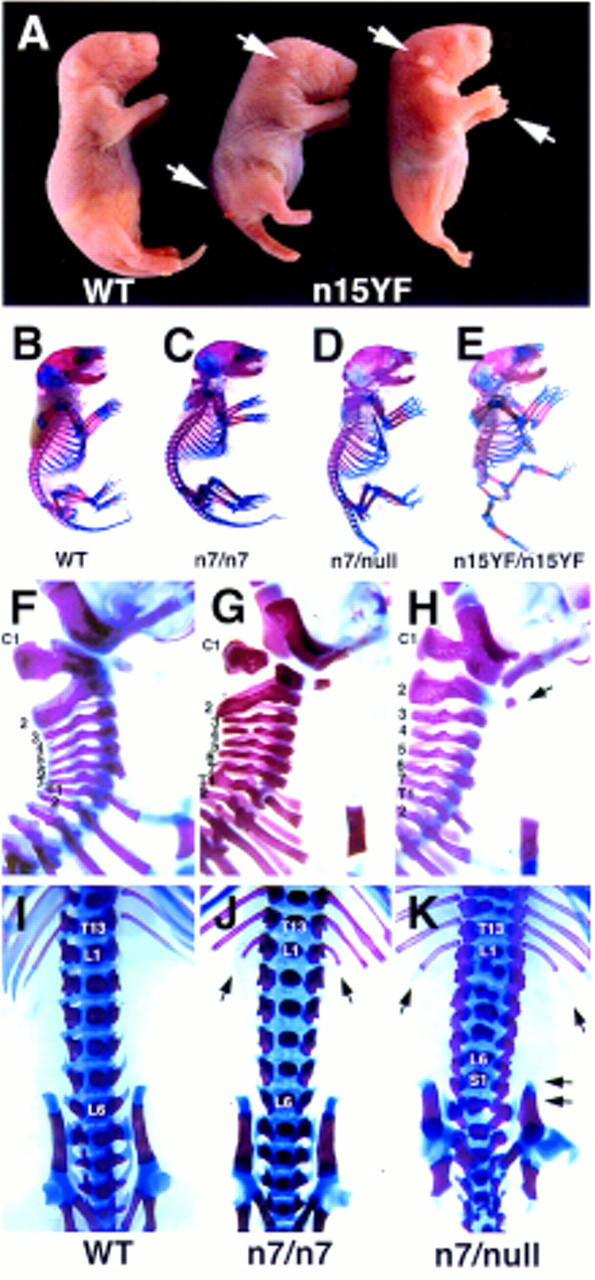
Posterior truncations and homeotic transformations in the Fgfr1 hypomorphs. (A) A newborn wild-type (WT) and two n15YF homozygous (n15YF) pups. Marked reduction in the posterior axis occasionally results in fusion of the hindlimbs (mutant on the right). The mutants also show greatly reduced size of the outer ear, distal limb defects, and severe spina bifida (arrows). Skeletal analysis of a wild type (B), n7 homozygote (C), n7/null transheterozygote (D), and n15YF homozygote (E) demonstrates the series of axial skeletal defects seen in the mutants. Compared to the wild-type cervical vertebrae (F), vertebral malformations are seen with a low frequency in the n7 homozygotes (G). Both the penetrance and expressivity of these defects is increased in the n7/null transheterozygotes (H), which commonly show fusion of the C1 to the occipital bones and partial transformation of C2 into C1 phenotype (arrow). At the lumbrosacral level, compared to the wild type (I), the n7 homozygotes frequently had extra ribs on L1 (J, arrows). Again, these homeotic transformations into the anterior direction were more severe in the n7/null transheterozygotes, which also often showed transformation of S1 into L6 phenotype (K, arrows).
In addition to the posterior truncations, skeletal analysis of the hypomorphic mutants revealed transformations of vertebral identities throughout the remaining vertebral column in both anterior and posterior directions (Fig. 2F–K and Table 2). n7 homozygotes commonly showed an anterior transformation of the lumbar vertebra L1 into a rib-bearing vertebra (Fig. 2J) and a posterior transformation of the thoracic vertebra T7 to the T8 phenotype (not attaching the sternum). In n7/null transheterozygotes as well as n15YF and n15 homozygotes the penetrance and expressivity of the anterior transformations were enhanced (Table 2). At the cervical level, fusion of atlas with occipital bones and partial transformations of axis and other cervical vertebrae were very common (Fig. 2H). A series of anterior transformations was also frequently seen at the lumbar level in the n7/null transheterozygotes (Fig. 2K). In an interesting contrast to the transformations in the anterior direction, the penetrance of the transformations to posterior direction were not increased in n15/n15 or n7/null compared to n7/n7.
Expanded posterior neural folds, widened limb fields, and posterior somite degeneration are seen in the hypomorphic mutants
To characterize the ontogeny of the observed patterning defects, embryos homozygous for the hypomorphic alleles were analyzed at various stages of development (Table 1). At E8.5 expansion of the posterior neural folds was evident in n15/n15 and n15YF/n15YF embryos (Fig. 3A–C). This is consistent with the results of chimeric analysis with Fgfr1 null mutant cells, demonstrating a tendency of the mutant cells to form ectopic posterior neural structures (Ciruna et al. 1997). Defective A-P patterning was also apparent in the lateral plate mesoderm of the n15 and n15YF homozygotes, as evidenced by expansion of the forelimb buds posteriorly at E9.5 and E10.5 (Fig. 3D–F), suggestive of expanded limb fields. Expansion of the hindlimb buds was also observed. In the most extreme cases the widened limb buds divided into as many as three smaller buds. The expanded limb buds resolved later in development, with the posterior extra buds appearing to degenerate leaving only one to develop into a limb (Fig. 3F). This alteration in the lateral mesoderm suggests that positional values are already affected in the mutants before E9.5.
Figure 3.
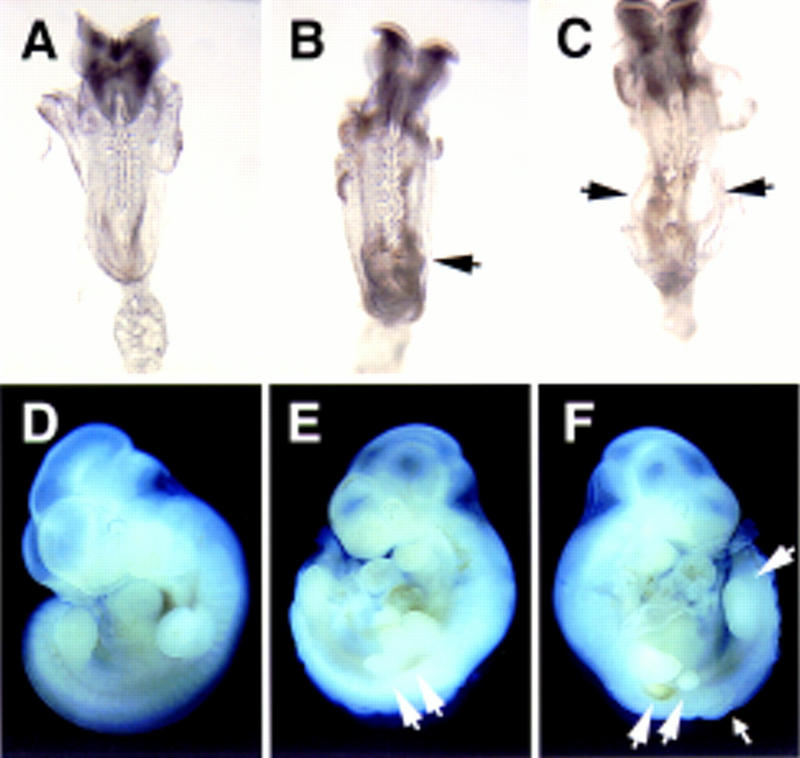
Morphological defects in hypomorphic (n15YF/n15YF) embryos. At E8.5 expansion of the posterior neural folds was seen in n15YF homozygotes (B,C) compared to their wild-type littermates (A). In more severely affected embryos, ectodermal vesicles were often observed (arrows in C). At E10.5, compared to the wild-type embryos (D), the n15YF homozygotes (E,F) showed expansion of both the fore- and hindlimb buds sometimes leading into division of the limb buds into several smaller structures (large arrows). The posteriorly located extra buds appeared to degenerate as seen on the opposite side of the embryo shown in E (F). Expansion of the posterior neural tube was also evident at this stage (small arrow).
Somites were observed posterior to the hindlimb bud level in E10.5 n15 and n15YF homozygotes (Figs. 3E,F and 4F). However, signs of posterior somite degeneration were seen in n15YF and n7 homozygotes at E10.5–E11.5 and E12.5, respectively (data not shown). These observations are consistent with occasional appearance of remnants of vertebral bodies posterior to the lumbrosacral truncations in the skeletal preparations of newborn n15 and n15YF homozygotes, and together suggest that deletion of posterior material contributes to the axial truncations.
Subtle changes in Hox gene expression patterns are seen in the mesoderm of hypomorphic mutants
To analyze the effect of the hypomorphic alleles on somitogenesis we studied the expression of a somitic marker Mox-1 in the hypomorphic mutants by mRNA in situ hybridization (Fig. 4A–F). Posterior somite abnormalities were observed in E9.5 and E10.5 n15YF/n15YF embryos (Fig. 4D–F). The segmentation of the anterior somites appeared normal, but the most anterior somites were smaller than in the wild-type littermates (Fig. 4D–E). This may reflect a partial transformation into the occipital phenotype, but might also result from a more general defect in the generation/proliferation of paraxial mesoderm. The A-P patterning within each somite appeared normal, with Mox-1 expression predominantly in the posterior half-somite.
Homeotic transformations in vertebral identities observed in the hypomorphic mutants are suggestive of alteration in the Hox code. To test this, we studied the expression patterns of Hox genes in the mutants at E8.5 and E9.5 by mRNA in situ hybridization. In the somitic mesoderm of E8.5 embryos, the anterior limit of expression of HoxD4 appeared posteriorized by one somite in n15YF/n15YF mutants (Fig. 4G), consistent with the anterior transformations of atlas and axis observed in the skeletal preparations of the newborn mutants. The anterior expression limit of HoxD4 in the neural tube was unchanged. In contrast to HoxD4, the anterior limit of expression of HoxB5 appeared unchanged in the n15YF/n15YF mutants both in the somitic mesoderm as well as neural tube (Fig. 4H,I). Interestingly, however, the expression of HoxB5 in the somites did not extend as far posteriorly as in the wild-type littermates. This may reflect some of the transformations in the posterior direction, which were also seen in the hypomorphic mutants (Table 2). In fact, transformations to both anterior and posterior directions are sometimes also seen in single Hox gene mutants (Jeannotte et al. 1993; Small and Potter 1993; Horan et al. 1995).
In two of five E9.5 n15YF/n15YF embryos the widening of the limb buds was accompanied by a posterior shift in the expression of HoxB9 in lateral mesoderm (Fig. 4J,K). This is consistent with A-P mispatterning of the lateral plate mesoderm, but the incomplete correlation argues against a causal link between HoxB9 expression change and the widening of the limb fields. It is possible, however, that altered expression of several Hox genes in the lateral plate (one of which might be HoxB9) results in the limb field mispositioning (Cohn et al. 1997). Alternatively, the limb bud widening may reflect similar changes in the intermediate mesoderm, which has been suggested to be responsible for limb bud induction.
As members of the Cdx gene family have been implicated in the regulation of the Hox gene expression, we analyzed Cdx1 and Cdx4 mRNA expression in the n15YF/n15YF embryos at E8.5. No significant change in their expression levels was detected (data not shown).
Hypomorphic alleles cause defects in the patterning of distal limbs
In addition to the primary body axis, reduced FGFR1 signaling also affected development and patterning of the distal limb structures (Figs. 2A and 5). Skeletal preparations of newborn hypomorphic mutants showed syn- and oligodactyly, delayed ossification of distal phalanges, and postaxial cartilage condensations (Fig. 5A–H). Also the development of the carpal and tarsal bones was abnormal. With various alleles and allele combinations, a series of phenotypes was observed. The development of the anterior digits was affected in the n7/n7 (n = 34) mice and the defects were successively more severe in IIIbn/IIIbn (n = 6) and n7/null (n = 5) mice. The development of the proximal limbs was normal in the mutants except for occasional shortening of the tibia seen in the n7/null mice (Fig. 5H).
Similar limb phenotypes are also seen in some of the 5′ Hox gene mutants (Dolle et al. 1993; Davis and Capecchi 1996; Fromental-Ramain et al. 1996; Mortlock et al. 1996). Furthermore, FGF signal from the apical ectodermal ridge together with Sonic Hedgehog from the zone of polarizing activity has been implicated in the regulation of Hox gene expression in the limb mesenchyme (Niswander 1996; Johnson and Tabin 1997). Therefore, we analyzed the expression of HoxD11 and HoxD13 in IIIbn/IIIbn mutants. Expression of HoxD11 appeared unaffected, but expression of HoxD13 was reduced both during the “early phase” at the posterior margin of the limb bud at E10.5 and during the “late phase” at E11 in the entire autopod (Fig. 5 I–K). This result is consistent with the restricted skeletal defects in the distal limbs of the IIIbn/IIIbn mice.
Y766F mutation creates a semidominant Fgfr1 allele causing homeotic transformations in a posterior direction
To test the biological significance of the tyrosine autophosphorylation site Y766 and PLCγ signal transduction pathway, we generated an allele where Y766 was mutated to phenylalanine (Y766Fl, see Fig. 1A). Mice homozygous for the Y766Fl allele were viable and fertile and did not show obvious craniofacial, limb, or tail defects. Interestingly, however, skeletal analysis of newborn Y766Fl homozygotes revealed frequent transformations throughout the vertebral column (Table 2; Fig. 6). In contrast to the hypomorphic mutants, these transformations occurred exclusively in the posterior direction. For example, Y766Fl homozygotes often showed a posterior transformation of the last cervical vertebra into a rib-bearing vertebra (Fig. 6A–C) and the last thoracic vertebra into a lumbar phenotype (Fig. 6D–F). These transformations appeared independent of each other. None of the transformations occurred in all of the mutants, but 21 of 22 analyzed mutants showed one or more of the vertebral transformations. Interestingly, these posterior transformations were also seen in the Y766Fl heterozygotes with a lower penetrance (Table 2). The phenotype of Y766Fl/null mice was very similar to Y766Fl homozygotes, further arguing against a possibility that Y766F mutation creates a hypomorphic allele. These observations together with the opposite phenotypes of the Y766Fl and hypomorphic alleles suggest that the Y766F mutation creates a gain-of-function allele. This conclusion is also supported by the observation that the prenatal lethality caused by neo insertion into intron 15 appears to be less penetrant if the allele carries the Y766F mutation (Table 1, cf. alleles n15 and n15YF, P < 0.05).
Figure 6.
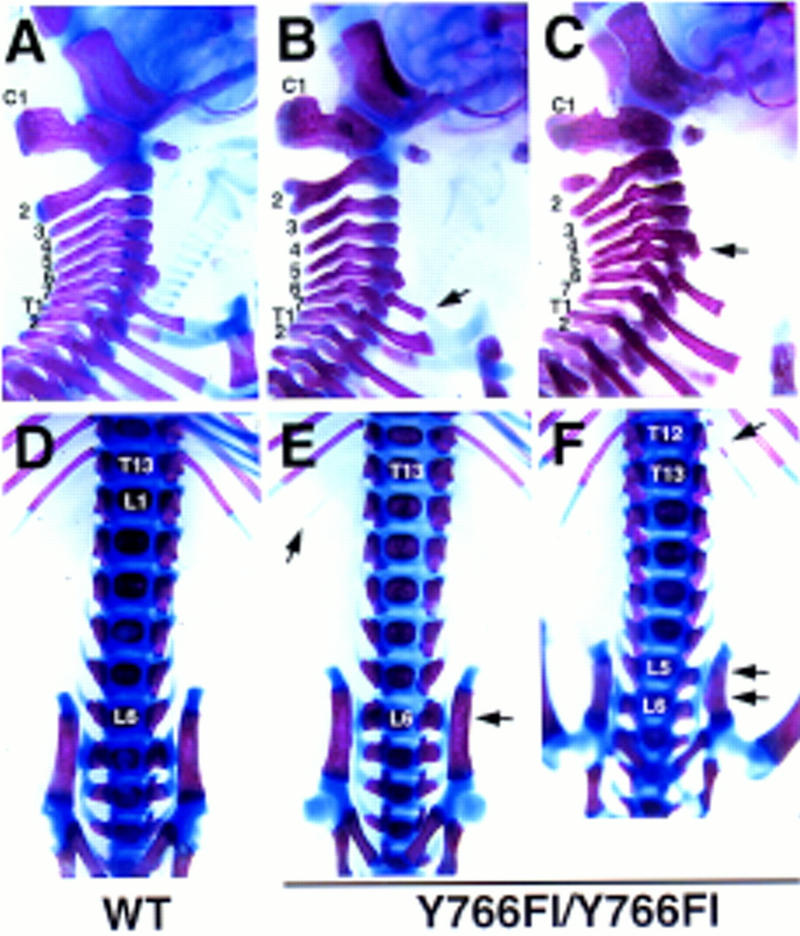
Homeotic transformations in posterior direction in the Y766F mutants. Cervical (A–C) and lumbrosacral (D–F) vertebrae of newborn wild-type (A,D) and Y766Fl homozygous mice (B,C,E,F) are shown. In the Y766F mutants the cervical vertebra C7 often has ribs fusing to ribs on the T1 (B) or to the sternum (C). Other transformations include shift of anterior trabeculi from the C6 to the C5 (arrow in C). At the thoracolumbar level, the T13 often loses its ribs taking a L1 phenotype (E,F, arrow in E points to a remaining distal part of a rib). L6 is frequently fused to the iliac bones (arrow in E), although this phenotype is sometimes seen also in wild-type mice. In Y766F mutants these transformations can occur also over two segments (arrows in F).
Discussion
The correct establishment of vertebrate Hox gene expression, leading to the development of distinct structures along the A-P axis, is an interesting but still poorly understood phenomenon. This complex process appears to be regulated at multiple levels (van der Hoeven et al. 1996). Opening of the chromatin structure of Hox complexes might provide a mechanism for competence of gene expression and perhaps is regulated by the polycomb and trithorax family members (Schumacher and Magnuson 1997). Whether the competence leads to actual Hox gene activation appears to be regulated by intercellular signals acting through enhancer sequences within the complexes. Retinoic acid and peptide growth factors, such as FGFs and activin family members, are extensively studied candidates for such signals. The involvement of retinoic acid in the regulation of Hox gene expression in mice has been supported by transgenic and gene targeting studies of the retinoic acid receptors and retinoic acid response elements present in the Hox complexes (Kastner et al. 1995; Dupe et al. 1997).
Analysis of the involvement of FGFs in the A-P patterning is complicated by the diverse functions of FGFs and their receptors. We have been able to partially circumvent this problem by generating a series of hypomorphic and gain-of-function alleles of Fgfr1. The hypomorphic mutants died neonatally and showed posterior truncations, homeotic transformations in the vertebral column predominantly to the anterior direction, as well as expansion of the limb fields and later distal limb defects. Transformations exclusively to the posterior direction were seen in the gain-of-function Y766F mutants in the absence of other abnormalities. In this respect, the phenotype of Y766F mutants resembles mice carrying loss-of-function mutations in polycomb family members, which are thought to act as negative regulators of Hox complexes (Schumacher and Magnuson 1997). Our results are consistent with the data from Xenopus (Ruiz i Altaba and Melton 1989; Cho and De Robertis 1990; Kolm and Sive 1995; Pownall et al. 1996), suggesting a role for FGFs in the positive regulation of Hox gene expression and thus assignment of positional values (Fig. 7). The wide spectrum of transformations seen in the Fgfr1 mutants suggests a global role for FGFR1 in the Hox gene regulation. In agreement with this model subtle changes in the early expression patterns of Hox genes, such as HoxD4, HoxB5, and HoxB9, were detected when FGFR1 signal was reduced. These early changes in Hox gene expression and morphological defects suggestive of A-P mispatterning (i.e., limb field expansion) as early as at E8.5 and E9.5, suggest that the function of FGFR1 is in the initial assignment of positional values rather than later in the readout of this information. However, in addition to an early function during gastrulation, FGFR1 may also play a separate role later during vertebral development.
Figure 7.
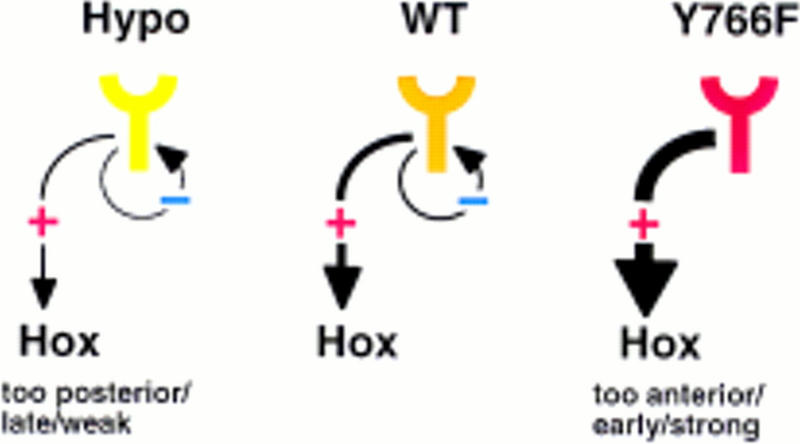
A model for FGFR1 function during A-P patterning and the effects of hypomorphic and Y766F mutations on it. The signals through FGFR1 appear to regulate the expression and activity of Hox gene products. In the hypomorphic situation homeotic transformations are seen predominantly to anterior direction, which are consistent with decreased Hox activity. In the absence of a negative regulatory signal, involving the Y766 autophosphorylation site, homeotic transformations to posterior direction are seen, consistent with increased Hox activity.
There are at least three possibilities, which are not mutually exclusive, how FGFR1 could be involved in the early regulation of Hox gene expression. First, signaling through FGFR1 could regulate directly the expression or activity of transcription factors, such as the caudal-related factors Cdx1, Cdx2, and Cdx4 implicated in the regulation of Hox gene expression (Gamer and Wright 1993; Subramanian et al. 1995; Chawengsaksophak et al. 1997), or proteins regulating the chromatin structure at the Hox complexes, such as the members of the polycomb and trithorax families (Schumacher and Magnuson 1997). This possibility is supported by the studies by Pownall et al. (1996), who showed that in Xenopus embryos the effect of dominant-negative FGF receptor could be rescued partially by overexpression of a caudal-related factor Xcad3. No significant changes in Cdx1 or Cdx4 mRNA expression levels were seen in the Fgfr1 hypomorphs. However, because of the dynamic expression of these genes, it is very difficult to tell with certainty whether their pattern of expression has been altered in the mutants. The second possibility is that changes in cell migratory properties in the primitive streak, shown to occur in Fgfr1 null mutants (Deng et al. 1994; Yamaguchi et al. 1994; Ciruna et al. 1997), lead into heterochrony and abnormal exposure to other patterning signals resulting in changes in cell fates. Third, it has been proposed that cellular proliferation and perhaps the speed of the cell cycle is an important regulator of the Hox gene activation, maybe through regulation of opening of the chromatin structure (Duboule 1994). By regulating the rate of proliferation of paraxial mesoderm cells and their stem cells in the primitive streak, FGFR1 may have an effect on the Hox complexes. In the weak hypomorphs (n7) and Y766F mutants the production and segmentation of paraxial mesoderm appears normal, suggesting that Fgfr1 signaling is directly involved in A-P patterning. However, we cannot exclude minor effects on the timing of mesodermal proliferation and migration. Whether the effect involves a change in cell behavior or is more direct, the fact that opposite changes in A-P patterning are seen with hypomorphic and gain-of-function alleles argues for an intimate link between the FGFR1-regulated process and establishment or maintenance of Hox gene activity.
A series of posterior truncations somewhat similar to the Fgfr1 mutants is seen with Wnt-3a/vestigial tail alleles (Greco et al. 1996). However, no homeotic transformations were observed in the Wnt-3a mutants, suggesting that WNT-3a and FGF signals have distinct functions during axis elongation. In contrast to the Wnt-3a mutants, in which the level of axial truncations correlates with a failure in somitogenesis, Fgfr1 hypomorphs showed signs of posterior somite degeneration. Thus, it is possible that, similarly to retinoic acid-treated embryos (Kessel 1992), mispatterning of the posterior somites contributes to the observed axial truncations.
It is of interest that disruption of the murine type IIB activin receptor (ActRIIB), one of the cell surface receptors for activin/BMP family signaling molecules, was shown recently to cause posterior shifts in Hox gene expression domains resulting in anterior homeotic transformations in the vertebral column (Oh and Li 1997). In addition to FGFs, work with Xenopus has implicated these TGFβ superfamily members in the mesoderm induction and patterning, and collaboration of FGF and activin-like signals in these processes has been demonstrated (LaBonne and Whitman 1994; Cornell et al. 1995). Work with ActRIIB, as well as our present work, provide genetic evidence for the involvement of the same signals in formation of the mesoderm and its patterning in the mouse. In contrast to the hypomorphic Fgfr1 mutants, the transformations in the ActRIIB mutants are confined to the lower thoracic and lumbar levels and posterior truncations are not seen in the ActRIIB mutants. This appears consistent with the finding that activin and FGF are able to activate different members of the Hox gene family in the Xenopus animal cap explants (Cho and De Robertis 1990). Therefore, it is likely that the TGFβ superfamily and FGF family members deliver distinct inputs to the mesodermal cells to guide the correct establishment of the Hox code.
In addition to generating and patterning the primary (A-P) axis, there is abundant evidence, mostly from gain-of-function type experiments with chick embryos, suggesting functions for FGFs at various stages of development of the secondary axes of limbs, including limb induction, outgrowth, and patterning (Niswander 1996; Johnson and Tabin 1997). Genes of the Hox complexes are also essential for the growth and patterning of the limbs. The distal limb phenotypes seen in our hypomorphic mutants resemble some of the mutant phenotypes of the 5′ Hox genes, such as loss of anterior digits seen in hypodactyly (Hd) and HoxA13 nullmutant mice (Fromental-Ramain et al. 1996; Mortlock et al. 1996) as well as delayed ossification of phalanges and post axial digit rudiments seen in HoxD13 mutants (Dolle et al. 1993; Davis and Capecchi 1996). Furthermore, reduction of HoxD13 expression is seen in the hypomorph limb buds, making it tempting to speculate that FGFR1 function during limb development may be similar to its role during A-P patterning. However, growth and patterning are linked intimately in the developing limb, making interpretation more difficult. In the Fgfr1 hypomorphs it appears as if the potentially FGF4-induced proliferation and anterior expansion (Vargesson et al. 1997) of HoxD13-expressing cells in the autopods is disturbed. Whether the observed reduction in HoxD13 expression is a cause or merely an effect of the deficient proliferation remains unclear. It appears that two FGF receptors, FGFR1 and FGFR2, have distinct functions during limb development. Whereas FGFR2 is required for the formation of the apical ectodermal ridge and early limb outgrowth (Xu et al. 1998), our results demonstrate a function for FGFR1 in the growth and patterning of the more distal limb mesenchyme.
Our studies also have implications for the understanding of the molecular biology of FGFR1 function. The two alternatively spliced FGFR1 isoforms, IIIb and IIIc, have been demonstrated to have distinct ligand-binding characteristics and somewhat different expression patterns (Werner et al. 1992; Ornitz et al. 1996). However, our results show that the IIIc isoform carries out the majority of the biological functions of the Fgfr1 gene, whereas IIIb plays a minor and to some extent redundant role. The situation may be different with other FGF receptors, which also have similar isoforms. For example, in the case of Fgfr2 the two isoforms appear to have mutually exclusive expression patterns and important functions have been assigned to the IIIb isoform (Peters et al. 1994). Fgfr1, Fgfr2, and Fgfr3 appear to share a common ancestral gene encoding two alternatively spliced isoforms. Apparently, during the gene duplication events and subsequent evolution, the IIIb isoform of Fgfr1 has not acquired a separate function but remains subsidiary to the other isoform and other receptors. This may have happened even more clearly to the Fgfr4 gene, which does not have the exon encoding the IIIb isoform, and may have lost it during evolution (Vainikka et al. 1992).
Abundant evidence demonstrates the requirement of the Ras mitogen-activated protein kinase pathway for signal transduction by FGF receptors to induce mesoderm differentiation as well as other cellular effects (MacNicol et al. 1993; LaBonne et al. 1995; Umbhauer et al. 1995). Another signal transduction pathway of FGFR1 involves receptor autophosphorylation at a carboxy-terminal tyrosine residue, Y766, leading into binding and activation of the PLCγ, which in turn results in activation of the protein kinase C (PKC) and the calcium signal. In vitro functional assays using a Y766F mutant FGFR1 have failed to show a requirement for this pathway for any of the FGFR1 induced responses (Mohammadi et al. 1992; Peters et al. 1992; Clyman et al. 1994; Muslin et al. 1994; Spivak-Kroizman et al. 1994). Biochemical characterization of the signal transduction properties of the Y766F mutant receptor has suggested both positive (Huang et al. 1995) and negative roles (Sorokin et al. 1994; Landgren et al. 1995) for Y766 pathway in the modulation of FGFR1-induced signal. Our results provide a clear in vivo demonstration that the PLCγ pathway of FGFR1 is not required for normal pre- or postnatal development or viability. Interestingly however, the Y766F mutation produces a semidominant allele causing an A-P patterning defect, which is mostly opposite to the phenotype of the hypomorphic mutants. Therefore, it is likely that the Y766F mutation results in overactive ligand-dependent FGFR1 function, at least during the A-P patterning process.
Consistent with our findings, the phosphorylation of Y766 of FGFR1 has been implicated in the internalization and degradation of the receptor (Sorokin et al. 1994). In addition, a FGFR1 variant lacking a putative PKC phosphorylation site has been shown to be resistant to phorbol ester-induced down-regulation of the receptor activity in Xenopus oocytes (Gillespie et al. 1995). PKC has also been suggested to be involved in phosphorylation and negative regulation of Src-family kinases, which are other downstream targets of FGFR1 (Landgren et al. 1995). It is thus possible that in the Y766F mutants a negative signal (maybe involving PLCγ and PKC), which regulates the activity of the FGFR1 itself or its downstream signal transduction cascade, is disrupted leading into extended and amplified signaling through FGFR1 (Fig. 7). Consistent with this model the phenotype caused by Y766F mutation is suppressed more by expression of wild-type receptors than by reduction of the mutant receptor levels (the phenotype of Y766Fl/+ is less severe than Y766Fl/null). This might involve heterodimer formation between mutant and wild-type receptors or transduction of the negative signal to downstream targets by the wild-type FGFR1. Interestingly, a Y766-related autophosphorylation site capable of binding PLCγ regulates negatively the biological activity of the Drosophila torso receptor tyrosine kinase (Cleghon et al. 1996). A tyrosine phosphorylation site regulating negatively the transforming activity of the NEU receptor tyrosine kinase has also been identified (Dankort et al. 1997). Therefore, these receptor tyrosine kinases may use a common feedback mechanism to regulate their ligand-induced activities.
Growth and patterning during vertebrate embryonic development are, for the most part, inseparable phenomena. Therefore, it seems only logical that the same intercellular signals regulate both processes. Our partial loss-of-function and gain-of-function alleles have provided evidence that Fgfr1, which is required for generation of paraxial mesoderm, also has an additional function in the assignment of its A-P positional values. The limb phenotypes of our hypomorphic Fgfr1 mutants further suggest that a similar link between growth and patterning may also exist during later Fgfr1-regulated developmental processes.
Materials and methods
Targeting vectors
For introduction of the desired mutations into the Fgfr1 locus targeting vectors were generated, in which the mutations were engineered into one of the arms of homology and the selectable marker neo was located in an intronic region (pPNTLoxP–IIIcSTOP, pPNTLoxP–IIIbSTOP, and pPNTLoxP–Y766F; see Fig. 1A). The mutations were introduced into genomic clones of Fgfr1 (Yamaguchi et al. 1994) by Altered Sites in vitro mutagenesis system (Promega). To inactivate isoform IIIb, codon 314 (5 nucleotides from the beginning of exon 6) was changed to a stop codon and an EcoRV site was introduced 5 nucleotides after it. To inactivate isoform IIIc, codon 314 (4 nucleotides from the beginning of exon 7) was changed to a stop codon and a BamHI site was introduced 4 nucleotides after it. Tyrosine 766 was changed to phenylalanine followed after 4 nucleotides by a neutral mutation, which created a XbaI site.
The targeting vectors were generated as follows. pPNTLoxP–IIIbSTOP and pPNTLoxP–IIIcSTOP: a 1.7-kb BamHI fragment was subcloned as the 3′ arm into pPNTloxP vector (having the neo gene flanked by loxP sites; Shalaby et al. 1995). In the case of pPNTLoxP–IIIbSTOP, a 6-kb EcoRI fragment containing a mutation in exon 6 (see above) was inserted as the 5′ arm. In the case of pPNTLoxP–IIIcSTOP, a 3-kb XhoI–EcoRI fragment containing a mutation in exon 7 (see above) was inserted as a 5′ arm. pPNTLoxP–Y766F: a 3-kb BamHI–BglII fragment containing the Y766F mutation (see above) was subcloned into pPNTloxP as the 3′ arm. A 3-kb BamHI fragment was inserted as the 5′ arm.
Gene targeting in ES cells and generation of the allelic series
The targeting vectors were linearized with NotI and electroporated into R1 ES cells. G418r Gancr clones were screened by Southern blot hybridization for targeting events and introduction of the mutations. With the pPNTLoxP–IIIcSTOP vector five targeted clones were obtained (of 97 colonies screened), two of which contained the mutation in the exon 7 identified by the presence of the BamHI site (alleles n7 and IIIcn; see Fig. 1A,B). With the pPNTLoxP–IIIbSTOP vector nine targeted clones were obtained (of 144 colonies screened), four of which contained the mutation in the exon 6 identified by the presence of the EcoRV site (allele IIIbn). With the pPNTLoxP–Y766F vector three targeted clones were obtained (of 62 colonies screened), two of which contained the Y766F mutation diagnosed by the adjacent XbaI site (alleles n15 and n15YF). The positive clones were analyzed for correct homologous recombination of both 5′ and 3′ arms (Fig. 1B; data not shown).
ES cell lines containing the alleles n7 (1), IIIbn (2), IIIcn (2), n15 (1), and n15YF (2) were used to generate chimeric mice (Wood et al. 1993), which transmitted the alleles through the germ line. Phenotypic analysis of the mutants was performed in a mixed 129sv/CD-1 background. To remove the neo cassettes from the alleles, chimeras transmitting the alleles n7, IIIbn, IIIcn, n15, and n15YF were bred with CD-1 mice carrying a transgene expressing Cre recombinase under the control of cytomegalovirus enhancer and β-actin promoter (Sauer 1993; Nagy et al. 1998) to generate alleles l7, IIIbl, IIIcl, l15, and Y766Fl, respectively.
Fgfr1 mRNA analysis
The level of Fgfr1 mRNA in a n7 F2litter was analyzed by Northern blotting and hybridization with a Fgfr1 cDNA probe, which contained the 3′ region of the gene. Total RNA from E10.5 embryos was isolated using the Trizol reagent (GIBCO BRL). The hybridization signals were quantified using a phosphorImager (Molecular Dynamics) with Imagequant software.
Skeletal analysis
Skeletal preparations of newborn mice were made by standard techniques (Dolle et al. 1993).
RNA in situ hybridization
Whole mount RNA in situ hybridization with Mox-1, HoxD4, HoxB5, HoxB9, HoxD11, HoxD13, Cdx1, and Cdx4 riboprobes was carried out as described. The protocol by Henrique et al. (1995) was used for E8.5 and E9.5 embryos. The protocol by Conlon and Rossant (1992) was used for E10.5 and E11 embryos.
Acknowledgments
We thank Sue McMaster, Celine Champigny, and Ken Harpal for expert technical assistance. We also thank Chris Wright for the Mox-1 and Cdx4 probes, Rob Krumlauf for the HoxB5 and HoxB9 probes, Mark Featherstone for the HoxD4 probe, Peter Gruss for the Cdx1 probe, and Denis Duboule for HoxD11 and HoxD13 probes. This work was supported by a Terry Fox Program Project grant from the National Cancer Institute of Canada (NCIC). J.R. is a Terry Fox Research Scientist of NCIC and International Research Scholar of the Howard Hughes Medical Institute. J.P. was supported by fellowships from the Academy of Finland, European Molecular Biology Organization, and the Medical Research Council of Canada.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL rossant@mshri.on.ca; FAX (416) 586-8588.
References
- Amaya E, Musci TJ, Kirschner MW. Expression of a dominant negative mutant of the FGF receptor disrupts mesoderm formation in Xenopus embryos. Cell. 1991;66:257–270. doi: 10.1016/0092-8674(91)90616-7. [DOI] [PubMed] [Google Scholar]
- Chawengsaksophak K, James R, Hammond VE, Kontgen F, Beck F. Homeosis and intestinal tumours in Cdx2 mutant mice. Nature. 1997;385:84–87. doi: 10.1038/386084a0. [DOI] [PubMed] [Google Scholar]
- Cho KW, De Robertis EM. Differential activation of Xenopus homeobox genes by mesoderm-inducing growth factors and retinoic acid. Genes & Dev. 1990;4:1910–1916. doi: 10.1101/gad.4.11.1910. [DOI] [PubMed] [Google Scholar]
- Ciruna BG, Schwartz L, Harpal K, Yamaguchi TP, Rossant J. Chimeric analysis of fibroblast growth factor receptor-1 (Fgfr1) function: A role for FGFR1 in morphogenetic movement through the primitive streak. Development. 1997;124:2829–2841. doi: 10.1242/dev.124.14.2829. [DOI] [PubMed] [Google Scholar]
- Cleghon V, Gayko U, Copeland TD, Perkins LA, Perrimon N, Morrison DK. Drosophila terminal structure development is regulated by the compensatory activities of positive and negative phosphotyrosine signaling sites on the Torso RTK. Genes & Dev. 1996;10:566–577. doi: 10.1101/gad.10.5.566. [DOI] [PubMed] [Google Scholar]
- Clyman RI, Peters KG, Chen YQ, Escobedo J, Williams LT, Ives HE, Wilson E. Phospholipase C gamma activation, phosphotidylinositol hydrolysis, and calcium mobilization are not required for FGF receptor-mediated chemotaxis. Cell Adhes Commun. 1994;1:333–342. doi: 10.3109/15419069409097264. [DOI] [PubMed] [Google Scholar]
- Cohn MJ, Patel K, Krumlauf R, Wilkinson DG, Clarke JD, Tickle C. Hox9 genes and vertebrate limb specification. Nature. 1997;387:97–101. doi: 10.1038/387097a0. [DOI] [PubMed] [Google Scholar]
- Conlon RA, Rossant J. Exogenous retinoic acid rapidly induces anterior ectopic expression of murine Hox-2 genes in vivo. Development. 1992;116:357–368. doi: 10.1242/dev.116.2.357. [DOI] [PubMed] [Google Scholar]
- Cornell RA, Musci TJ, Kimelman D. FGF is a prospective competence factor for early activin-type signals in Xenopus mesoderm induction. Development. 1995;121:2429–2437. doi: 10.1242/dev.121.8.2429. [DOI] [PubMed] [Google Scholar]
- Cox WG, Hemmati-Brivanlou A. Caudalization of neural fate by tissue recombination and bFGF. Development. 1995;121:4349–4358. doi: 10.1242/dev.121.12.4349. [DOI] [PubMed] [Google Scholar]
- Dankort DL, Wang Z, Blackmore V, Moran MF, Muller WJ. Distinct tyrosine autophosphorylation sites negatively and positively modulate neu-mediated transformation. Mol Cell Biol. 1997;17:5410–5425. doi: 10.1128/mcb.17.9.5410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis AP, Capecchi MR. A mutational analysis of the 5′ HoxD genes: Dissection of genetic interactions during limb development in the mouse. Development. 1996;122:1175–1185. doi: 10.1242/dev.122.4.1175. [DOI] [PubMed] [Google Scholar]
- Deng CX, Wynshaw-Boris A, Shen MM, Daugherty C, Ornitz DM, Leder P. Murine FGFR-1 is required for early postimplantation growth and axial organization. Genes & Dev. 1994;8:3045–3057. doi: 10.1101/gad.8.24.3045. [DOI] [PubMed] [Google Scholar]
- Deng C, Bedford M, Li C, Xu X, Yang X, Dunmore J, Leder P. Fibroblast growth factor receptor-1 (FGFR-1) is essential for normal neural tube and limb development. Dev Biol. 1997;185:42–54. doi: 10.1006/dbio.1997.8553. [DOI] [PubMed] [Google Scholar]
- Dolle P, Dierich A, LeMeur M, Schimmang T, Schuhbaur B, Chambon P, Duboule D. Disruption of the Hoxd-13 gene induces localized heterochrony leading to mice with neotenic limbs. Cell. 1993;75:431–441. doi: 10.1016/0092-8674(93)90378-4. [DOI] [PubMed] [Google Scholar]
- Duboule D. Temporal colinearity and the phylotypic progression: A basis for the stability of a vertebrate Bauplan and the evolution of morphologies through heterochrony. Development. 1994;(Suppl):135–142. [PubMed] [Google Scholar]
- Dupe V, Davenne M, Brocard J, Dolle P, Mark M, Dierich A, Chambon P, Rijli FM. In vivo functional analysis of the Hoxa-1 3′ retinoic acid response element (3′RARE) Development. 1997;124:399–410. doi: 10.1242/dev.124.2.399. [DOI] [PubMed] [Google Scholar]
- Fromental-Ramain C, Warot X, Messadecq N, LeMeur M, Dolle P, Chambon P. Hoxa-13 and Hoxd-13 play a crucial role in the patterning of the limb autopod. Development. 1996;122:2997–3011. doi: 10.1242/dev.122.10.2997. [DOI] [PubMed] [Google Scholar]
- Gamer LW, Wright CV. Murine Cdx-4 bears striking similarities to the Drosophila caudal gene in its homeodomain sequence and early expression pattern. Mech Dev. 1993;43:71–81. doi: 10.1016/0925-4773(93)90024-r. [DOI] [PubMed] [Google Scholar]
- Gillespie LL, Chen G, Paterno GD. Cloning of a fibroblast growth factor receptor 1 splice variant from Xenopus embryos that lacks a protein kinase C site important for the regulation of receptor activity. J Biol Chem. 1995;270:22758–22763. doi: 10.1074/jbc.270.39.22758. [DOI] [PubMed] [Google Scholar]
- Godsave SF, Durston AJ. Neural induction and patterning in embryos deficient in FGF signaling. Int J Dev Biol. 1997;41:57–65. [PubMed] [Google Scholar]
- Greco TL, Takada S, Newhouse MM, McMahon JA, McMahon AP, Camper SA. Analysis of the vestigial tail mutation demonstrates that Wnt-3a gene dosage regulates mouse axial development. Genes & Dev. 1996;10:313–324. doi: 10.1101/gad.10.3.313. [DOI] [PubMed] [Google Scholar]
- Henrique D, Adam J, Myat A, Chitnis A, Lewis J, Ish-Horowicz D. Expression of a Delta homologue in prospective neurons in the chick. Nature. 1995;375:787–790. doi: 10.1038/375787a0. [DOI] [PubMed] [Google Scholar]
- Horan GS, Ramirez-Solis R, Featherstone MS, Wolgemuth DJ, Bradley A, Behringer RR. Compound mutants for the paralogous hoxa-4, hoxb-4, and hoxd-4 genes show more complete homeotic transformations and a dose-dependent increase in the number of vertebrae transformed. Genes & Dev. 1995;9:1667–1677. doi: 10.1101/gad.9.13.1667. [DOI] [PubMed] [Google Scholar]
- Huang J, Mohammadi M, Rodrigues GA, Schlessinger J. Reduced activation of RAF-1 and MAP kinase by a fibroblast growth factor receptor mutant deficient in stimulation of phosphatidylinositol hydrolysis. J Biol Chem. 1995;270:5065–5072. doi: 10.1074/jbc.270.10.5065. [DOI] [PubMed] [Google Scholar]
- Jeannotte L, Lemieux M, Charron J, Poirier F, Robertson EJ. Specification of axial identity in the mouse: Role of the Hoxa-5 (Hox1.3) gene. Genes & Dev. 1993;7:2085–2096. doi: 10.1101/gad.7.11.2085. [DOI] [PubMed] [Google Scholar]
- Johnson DE, Lu J, Chen H, Werner S, Williams LT. The human fibroblast growth factor receptor genes: A common structural arrangement underlies the mechanisms for generating receptor forms that differ in their third immunoglobulin domain. Mol Cell Biol. 1991;11:4627–4634. doi: 10.1128/mcb.11.9.4627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RL, Tabin CJ. Molecular models for vertebrate limb development. Cell. 1997;90:979–990. doi: 10.1016/s0092-8674(00)80364-5. [DOI] [PubMed] [Google Scholar]
- Kastner P, Mark M, Chambon P. Nonsteroid nuclear receptors: What are genetic studies telling us about their role in real life? Cell. 1995;83:859–869. doi: 10.1016/0092-8674(95)90202-3. [DOI] [PubMed] [Google Scholar]
- Kengaku M, Okamoto H. bFGF as a possible morphogen for the anteroposterior axis of the central nervous system in Xenopus. Development. 1995;121:3121–3130. doi: 10.1242/dev.121.9.3121. [DOI] [PubMed] [Google Scholar]
- Kessel M. Respecification of vertebral identities by retinoic acid. Development. 1992;115:487–501. doi: 10.1242/dev.115.2.487. [DOI] [PubMed] [Google Scholar]
- Kieny M, Mauger A, Sengel P. Early regionalization of the somitic mesoderm as studied by the development of the axial skeleton of the chick embryo. Dev Biol. 1972;28:142–161. doi: 10.1016/0012-1606(72)90133-9. [DOI] [PubMed] [Google Scholar]
- Kolm PJ, Sive HL. Regulation of the Xenopus labial homeodomain genes, HoxA1 and HoxD1: Activation by retinoids and peptide growth factors. Dev Biol. 1995;167:34–49. doi: 10.1006/dbio.1995.1005. [DOI] [PubMed] [Google Scholar]
- Kroll KL, Amaya E. Transgenic Xenopus embryos from sperm nuclear transplantations reveal FGF signaling requirements during gastrulation. Development. 1996;122:3173–3183. doi: 10.1242/dev.122.10.3173. [DOI] [PubMed] [Google Scholar]
- LaBonne C, Whitman M. Mesoderm induction by activin requires FGF-mediated intracellular signals. Development. 1994;120:463–472. doi: 10.1242/dev.120.2.463. [DOI] [PubMed] [Google Scholar]
- LaBonne C, Burke B, Whitman M. Role of MAP kinase in mesoderm induction and axial patterning during Xenopus development. Development. 1995;121:1475–1486. doi: 10.1242/dev.121.5.1475. [DOI] [PubMed] [Google Scholar]
- Lamb TM, Harland RM. Fibroblast growth factor is a direct neural inducer, which combined with noggin generates anterior-posterior neural pattern. Development. 1995;121:3627–3636. doi: 10.1242/dev.121.11.3627. [DOI] [PubMed] [Google Scholar]
- Landgren E, Blume-Jensen P, Courtneidge SA, Claesson-Welsh L. Fibroblast growth factor receptor-1 regulation of Src family kinases. Oncogene. 1995;10:2027–2035. [PubMed] [Google Scholar]
- MacNicol AM, Muslin AJ, Williams LT. Raf-1 kinase is essential for early Xenopus development and mediates the induction of mesoderm by FGF. Cell. 1993;73:571–583. doi: 10.1016/0092-8674(93)90143-e. [DOI] [PubMed] [Google Scholar]
- Meyers EN, Lewandoski M, Martin GR. An Fgf8 mutant allelic series generated by Cre- and Flp-mediated recombination. Nature Genet. 1998;18:136–141. doi: 10.1038/ng0298-136. [DOI] [PubMed] [Google Scholar]
- Mohammadi M, Dionne CA, Li W, Li N, Spivak T, Honegger AM, Jaye M, Schlessinger J. Point mutation in FGF receptor eliminates phosphatidylinositol hydrolysis without affecting mitogenesis. Nature. 1992;358:681–684. doi: 10.1038/358681a0. [DOI] [PubMed] [Google Scholar]
- Mortlock DP, Post LC, Innis JW. The molecular basis of hypodactyly (Hd): A deletion in Hoxa13 leads to arrest of digital arch formation. Nature Genet. 1996;13:284–289. doi: 10.1038/ng0796-284. [DOI] [PubMed] [Google Scholar]
- Muslin AJ, Peters KG, Williams LT. Direct activation of phospholipase C-gamma by fibroblast growth factor receptor is not required for mesoderm induction in Xenopus animal caps. Mol Cell Biol. 1994;14:3006–3012. doi: 10.1128/mcb.14.5.3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy A, Moens CB, Ivanyi E, Pawling J, Gertsenstein M, Hadjantonakis A-K, Pirity M, Rossant J. Dissecting the role of N-myc in development using a single targeting vector to generate a series of alleles. Curr Biol. 1998;8:661–664. doi: 10.1016/s0960-9822(98)70254-4. [DOI] [PubMed] [Google Scholar]
- Niswander L. Growth factor interactions in limb development. Ann N Y Acad Sci. 1996;785:23–26. doi: 10.1111/j.1749-6632.1996.tb56239.x. [DOI] [PubMed] [Google Scholar]
- Oh SP, Li E. The signaling pathway mediated by the type IIB activin receptor controls axial patterning and lateral asymmetry in the mouse. Genes & Dev. 1997;11:1812–1826. doi: 10.1101/gad.11.14.1812. [DOI] [PubMed] [Google Scholar]
- Ornitz DM, Xu J, Colvin JS, McEwen DG, MacArthur CA, Coulier F, Gao G, Goldfarb M. Receptor specificity of the fibroblast growth factor family. J Biol Chem. 1996;271:15292–15297. doi: 10.1074/jbc.271.25.15292. [DOI] [PubMed] [Google Scholar]
- Peters KG, Marie J, Wilson E, Ives HE, Escobedo J, Del Rosario M, Mirda D, Williams LT. Point mutation of an FGF receptor abolishes phosphatidylinositol turnover and Ca2+ flux but not mitogenesis. Nature. 1992;358:678–681. doi: 10.1038/358678a0. [DOI] [PubMed] [Google Scholar]
- Peters K, Werner S, Liao X, Wert S, Whitsett J, Williams L. Targeted expression of a dominant negative FGF receptor blocks branching morphogenesis and epithelial differentiation of the mouse lung. EMBO J. 1994;13:3296–3301. doi: 10.1002/j.1460-2075.1994.tb06631.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pownall ME, Tucker AS, Slack JM, Isaacs HV. eFGF, Xcad3 and Hox genes form a molecular pathway that establishes the anteroposterior axis in Xenopus. Development. 1996;122:3881–3892. doi: 10.1242/dev.122.12.3881. [DOI] [PubMed] [Google Scholar]
- Ruiz i Altaba A, Melton DA. Interaction between peptide growth factors and homeobox genes in the establishment of antero-posterior polarity in frog embryos. Nature. 1989;341:33–38. doi: 10.1038/341033a0. [DOI] [PubMed] [Google Scholar]
- Sauer B. Manipulation of transgenes by site-specific recombination: Use of Cre recombinase. Methods Enzymol. 1993;225:890–900. doi: 10.1016/0076-6879(93)25056-8. [DOI] [PubMed] [Google Scholar]
- Schumacher A, Magnuson T. Murine Polycomb- and trithorax-group genes regulate homeotic pathways and beyond. Trends Genet. 1997;13:167–170. [PubMed] [Google Scholar]
- Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, Breitman ML, Schuh AC. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature. 1995;376:62–66. doi: 10.1038/376062a0. [DOI] [PubMed] [Google Scholar]
- Slack JM, Isaacs HV, Song J, Durbin L, Pownall ME. The role of fibroblast growth factors in early Xenopus development. Biochem Soc Symp. 1996;62:1–12. [PubMed] [Google Scholar]
- Small KM, Potter SS. Homeotic transformations and limb defects in Hox A11 mutant mice. Genes & Dev. 1993;7:2318–2328. doi: 10.1101/gad.7.12a.2318. [DOI] [PubMed] [Google Scholar]
- Sorokin A, Mohammadi M, Huang J, Schlessinger J. Internalization of fibroblast growth factor receptor is inhibited by a point mutation at tyrosine 766. J Biol Chem. 1994;269:17056–17061. [PubMed] [Google Scholar]
- Spivak-Kroizman T, Mohammadi M, Hu P, Jaye M, Schlessinger J, Lax I. Point mutation in the fibroblast growth factor receptor eliminates phosphatidylinositol hydrolysis without affecting neuronal differentiation of PC12 cells. J Biol Chem. 1994;269:14419–14423. [PubMed] [Google Scholar]
- Subramanian V, Meyer BI, Gruss P. Disruption of the murine homeobox gene Cdx1 affects axial skeletal identities by altering the mesodermal expression domains of Hox genes. Cell. 1995;83:641–653. doi: 10.1016/0092-8674(95)90104-3. [DOI] [PubMed] [Google Scholar]
- Umbhauer M, Marshall CJ, Mason CS, Old RW, Smith JC. Mesoderm induction in Xenopus caused by activation of MAP kinase. Nature. 1995;376:58–62. doi: 10.1038/376058a0. [DOI] [PubMed] [Google Scholar]
- Vainikka S, Partanen J, Bellosta P, Coulier F, Birnbaum D, Basilico C, Jaye M, Alitalo K. Fibroblast growth factor receptor-4 shows novel features in genomic structure, ligand binding and signal transduction. EMBO J. 1992;11:4273–4280. doi: 10.1002/j.1460-2075.1992.tb05526.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Hoeven F, Zakany J, Duboule D. Gene transpositions in the HoxD complex reveal a hierarchy of regulatory controls. Cell. 1996;85:1025–1035. doi: 10.1016/s0092-8674(00)81303-3. [DOI] [PubMed] [Google Scholar]
- Vargesson N, Clarke JD, Vincent K, Coles C, Wolpert L, Tickle C. Cell fate in the chick limb bud and relationship to gene expression. Development. 1997;124:1909–1918. doi: 10.1242/dev.124.10.1909. [DOI] [PubMed] [Google Scholar]
- Werner S, Duan DS, de Vries C, Peters KG, Johnson DE, Williams LT. Differential splicing in the extracellular region of fibroblast growth factor receptor 1 generates receptor variants with different ligand- binding specificities. Mol Cell Biol. 1992;12:82–88. doi: 10.1128/mcb.12.1.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood SA, Allen ND, Rossant J, Auerbach A, Nagy A. Non-injection methods for the production of embryonic stem cell-embryo chimaeras. Nature. 1993;365:87–89. doi: 10.1038/365087a0. [DOI] [PubMed] [Google Scholar]
- Xu X, Weinstein M, Li C, Naski M, Cohen R, Ornitz D, Leder P, Deng C. Fibroblast growth factor receptor 2 (FGFR2)-mediated reciprocal regulation loop between FGF8 and FGF10 is essential for limb induction. Development. 1998;125:753–765. doi: 10.1242/dev.125.4.753. [DOI] [PubMed] [Google Scholar]
- Yamaguchi TP, Harpal K, Henkemeyer M, Rossant J. fgfr-1 is required for embryonic growth and mesodermal patterning during mouse gastrulation. Genes & Dev. 1994;8:3032–3044. doi: 10.1101/gad.8.24.3032. [DOI] [PubMed] [Google Scholar]