

Abstract
The serine protease thrombin plays multiple roles in many important physiological processes, especially coagulation, where it functions as both a pro- and anti-coagulant. The polyanionic glycosaminoglycan heparin modulates thrombin’s activity through binding at exosite II. Sucrose octasulfate (SOS) is often used as a surrogate for heparin, but it is not known whether it is an effective heparin mimic in its interaction with thrombin. We have characterized the interaction of SOS with thrombin in solution and determined a crystal structure of their complex. SOS binds thrombin with a Kd of ~1.4 μM, comparable to that of the much larger polymeric heparin measured under the same conditions. Non-ionic (hydrogen bonding) interactions make a larger contribution to thrombin binding of SOS than to heparin. SOS binding to exosite II inhibits thrombin’s catalytic activity with high potency but with low efficacy. Analytical ultracentrifugation shows that bovine and human thrombins are monomers in solution in the presence of SOS, in contrast to their complexes with heparin, which are dimers. In the x-ray crystal structure, two molecules of SOS are bound non-equivalently to exosites II of a thrombin dimer, in contrast to the 1:2 stoichiometry of the heparin-thrombin complex, which has a different monomer association mode in the dimer. SOS and heparin binding to exosite II of thrombin differ on both chemical and structural levels and, perhaps most significantly, in thrombin inhibition. These differences may offer paths to the design of more potent exosite II binding, allosteric small molecules as modulators of thrombin function.
Thrombin plays a major role in the hemostasis/coagulation system (1 - 4) and is an important mediator of cellular processes, such as activation of platelets (5, 6), monocytes (7), microglial cells (8), and dendritic cells (9), which are implicated in vascular and neural inflammation (8, 10, 11) and tumor metastasis (11). Most of these functions are a consequence of thrombin’s proteolytic activity. In its hemostatic role, thrombin catalyzes the cleavage of fibrinogen to fibrin monomers, which polymerize with the aid of factor XIIIa to form clot. Factor XIIIa and several other proteins of the coagulation cascade, e.g., factors Va and VIIIa, are generated through the proteolytic action of thrombin (11). In direct opposition to this procoagulant role, thrombin also exerts an anticoagulant role by proteolytic processing of protein C in the presence of thrombomodulin, an endothelial cell surface receptor (11, 12). Thrombin’s cellular effects arise through its cleavage of protease-activated receptors (PARs), a family of G-protein coupled receptors present on a large number of cells. Of the four known PARs, thrombin activates isoforms 1, 3 and 4, with major physiological and pathological consequences (5 - 8, 11, 13).
Thrombin’s high specificity interaction with so many substrates is mediated in part by two anion-binding exosites, which are approximately 10 – 20 Å from its active site. Exosite I is formed by several lysines and arginines interspersed with a few hydrophobic residues and is involved in binding to fibrinogen, fibrin, hirudin and other proteins (3, 4, 14). Diametrically opposed to exosite I is another patch of electropositive residues constituting exosite II, also known as the heparin-binding site. Exosite II consists of Arg93, Arg101, Arg126, Arg165, Arg233, Lys235, Lys236 and Lys240 residues (chymotrypsin numbering) and is an expansive area (~25 Å long) that can accommodate a six-unit fragment of heparin. In addition to interacting with heparin, exosite II engages the anionic chondroitin sulfate moiety present in thrombomodulin (15, 16) and also the anionic C-terminal domain of haemadin, a peptide from the leech Haemadipsa sylvestri (14). A group of chemo-enzymatically prepared sulfated low molecular weight lignins has been found to bind in or near exosite II of thrombin (17, 18).
Exosite I – ligand interactions induce conformational changes in the active site of thrombin, thereby influencing the specificity and reactivity of the protease toward its macromolecular substrates (4). The ligands also affect the catalytic efficiency (kcat/KM) of hydrolysis of small peptide substrates, albeit in an unpredictable manner. For example, studies with hirugen, the C-terminal dodecapeptide of hirudin, show that the catalytic efficiency increases or decreases depending on the nature of the chromogenic substrate used to measure activity (19 - 24). Likewise, the effect of exosite II ligands on the kcat/KM of thrombin appears to be ligand dependent. For example, sulfated lignins induce inhibition of proteolytic activity (17), while heparins do not (21, 22), although the latter affect the fluorescence properties of p-aminobenzamidine (PABA) used as an active site probe (23). An exhaustive re-evaluation of allosteric effects on thrombin function revealed significant ligand-specific coupling between each of the two exosites and the active site, but insignificant inter-exosite coupling (24).
Modulation of thrombin’s proteolytic activity by exosite ligands affords an opportunity for the development of specific allosteric effectors. It is surprising that despite the plethora of allosteric interactions reported in the literature, nearly all inhibitors of thrombin have been designed to target its active site (25, 26). A primary reason for this is the difficulty of designing small anionic scaffolds that will specifically target the electropositive exosites of thrombin. The exosites are surface-exposed, shallow depressions on the thrombin surface, rather than well-defined, deep binding sites, which typically are easier to target. The high electropositive character of these exosites is likely to attract practically any collection of anions, thereby degrading the specificity of the interaction, as noted with heparin (23). Finally, the absence of appropriate tools, such as a maneuverable small lead ligand and a molecular modeling protocol, add to the difficulty of designing allosteric effectors.
With the goal of designing specific, small, synthetic allosteric modulators of thrombin function, we studied the interaction of SOS with BT and HT at the molecular level. SOS is a highly sulfated disaccharide known to mimic polymeric heparin (Fig. 1) and to bind in exosite II of thrombin (27). It may thus serve as a possible lead scaffold in structure-based drug design. We have characterized the equilibrium binding of SOS to active thrombin in solution and have studied its effects on thrombin’s catalytic activity. We have parsed the contributions of ionic and hydrogen bond interactions to thrombin-SOS complex stabilization and find significant differences compared to the thrombin-heparin complex. Interestingly, SOS inhibits thrombin to a small extent and is thus the first carbohydrate-based inhibitor of this protease. We also determined the crystal structure of the SOS complex with BT, which shows that two SOS molecules bind in exosite II between the opposed carboxyl termini of the thrombin dimer that constitutes the asymmetric unit of the crystal lattice. This thrombin crystal dimer has a different monomer association mode than that observed in the PPACK-HT-LMWH complex (28). A consequence of this difference is that one SOS molecule in the BT-SOS complex binds at the same locus as heparin in exosite II of HT through interactions almost exclusively with one BT monomer, while a second SOS binds at an adjacent locus, interacting with both exosites II of the juxtaposed BT monomers of the dimer. These studies provide evidence for distinctions in the mode and nature of SOS and heparin binding at exosite II of thrombin and in their effects on catalytic activity. Finally, they affirm the possibility of designing specific, small, synthetic allosteric inhibitors of thrombin targeted to exosite II.
Figure 1.
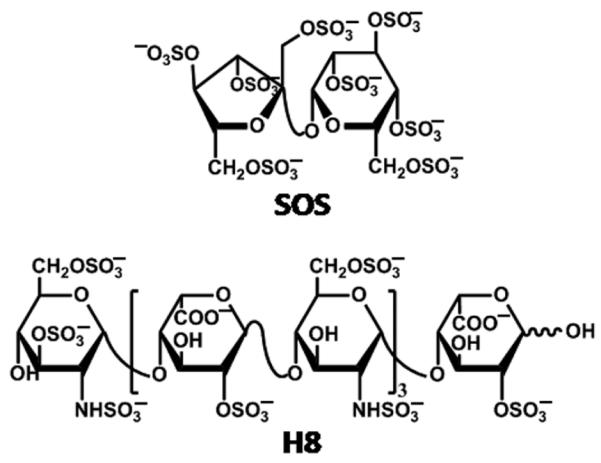
Structures of heparin octasaccharide H8 and sucrose octasulfate.
Experimental Procedures
Materials
Human and bovine α-thrombins were purchased from Haematologic Technologies, Inc. (Essex Junction, VT) as a 10 mg/mL solution in glycerol-water (1:1 v/v) mixture. d-phenylalanyl-l-prolyl-l-arginine chloromethyl ketone and SOS were purchased from Biomol International (now Enzo Life Sciences International, Inc., Plymouth Meeting, PA). Sodium heparin was from Arcos (New Jersey, USA) and Spectrozyme TH was purchased from American Diagnostica (Greenwich, CT). Clinically used Lovenox (enoxaparin, Sanofi-Aventis) was purchased from the VCU Hospital pharmacy. Deionized distilled water (18.2 megohms) from Fisher Scientific (Agawam, MA) was used to prepare all buffers. p-Aminobenzamidine was purchased from Sigma - Aldrich (St. Louis, MO) and other chemicals were from either Sigma – Aldrich or Fisher Scientific (Agawam, MA). All measurements, including the crystal structures, were done at Na+ concentrations far above that for saturation of the Na+ binding site of thrombin (29).
Thermodynamics of Thrombin Binding to SOS
The equilibrium dissociation constant (KD) of the HT – SOS complex at 25 °C was measured in 20 mM sodium phosphate buffer, pH 7.4, containing 0.1 mM EDTA, 0.1% (w/v) PEG 8000 and varying concentrations of NaCl (50 to 250 mM). In this range of sodium concentrations, thrombin will be entirely in the high sodium form. The active site binding probe, PABA, was used as a fluorescent reporter group for ligand binding at exosite II (30, 31). PABA binds non-covalently in the P1 specificity pocket of trypsin-like serine proteases, including thrombin. Although the P1 specificity pocket of thrombin lies on the opposite side of the thrombin molecule from exosite II, at a distance of about 25 Å, PABA bound in that pocket shows fluorescence changes on binding of heparin to exosite II. In the experiments described here, a sub-inhibitory concentration of PABA (10 μM) was used, well below its KD of ~60 - 80 μM (30), so that only a small fraction of thrombin molecules are bound. Fluorescence experiments were performed in PEG 20000 coated cuvettes using a QM4 spectrofluorometer (Photon Technology International, Birmingham, NJ) in ratio mode. The fluorescence of HT – PABA complex (λEM = 370 nm, λEX = 345 nm) as a function of the addition of aliquots of SOS was measured with correction for dilution. SOS does not absorb significantly at either 345 nm or 370 nm and hence no inner filter corrections were necessary. The slit widths on the excitation and emission side were 1 and 2 mm, respectively. The decrease in the fluorescence signal due to SOS binding at exosite 2 was fitted to the quadratic equilibrium binding equation 1 to obtain the KD of interaction:
| eq.1 |
In this equation, ΔF is the change in fluorescence relative to the initial fluorescence FO on formation of the complex following each addition of SOS, and ΔFMAX is the maximal change in fluorescence observed on saturation of thrombin ([T]O). A binding stoichiometry of 1:1 was determined for the SOS – HT complex using stoichiometric titrations with PABA as a probe of interaction (data not shown). This stoichiometry is consistent with the crystal structure of the complex and with the analytical ultracentrifugation results.
The contribution of ionic and non-ionic binding energy to HT – SOS interaction was derived from measurement of KD in buffers of varying ionic strength using equation 2 (23, 26, 32, 37):
| Eq. 2 |
In the absence of any added NaCl, the ionic strength (I) of the buffer is 0.035 at pH 7.4. Higher ionic strengths were achieved by adding NaCl, whose concentration ranged from 50 mM to 250 mM for the measurements made. In equation 2, KD,NI is the dissociation constant at [Na+] = 1 M; Z carries information about the number of charge-charge interactions and Φ is the fraction of monovalent counterions released per negative charge following SOS binding to thrombin.
Direct Inhibition of Thrombin by SOS
The direct inhibition of HT and BT by SOS was determined through a chromogenic substrate (Spectrozyme TH) hydrolysis assay, as previously described for sulfated lignins (17, 18) and also adapted to a 96 well plate format performed on a microplate reader. In the former, a 1 – 10 μL sample of SOS was diluted with 20 mM sodium phosphate or 20 mM Tris buffer, pH 7.4, containing 150 mM NaCl, 0.1 mM CaCl2 or 0.1 mM EDTA, and 0.1 % (v/v) PEG 8000 at room temperature in a PEG 20000-coated polystyrene cuvette. A solution of either 0.5 or 1.0 μM α-thrombin was added followed either immediately or after 10 minutes incubation at room temperature by addition of 15 – 50 μL of 1 – 2 mM Spectrozyme TH. The final volume of the assay was 1000 μl and the final concentrations of α-thrombin and Spectrozyme TH were 5 nM and 50 μM. The initial rate of increase in absorbance at 405 nm was measured for up to 500 seconds. In the 96 well plate format, 187 μl of a solution of SOS in 20 mM phosphate buffer, pH 7.4, 150 mM NaCl, 0.1 M EDTA and 0.1% PEG 8000 was incubated with 5 μl of 500 nM thrombin at 25 °C in wells of a 96 well plate for 10 minutes. Following incubation, 8 μl of 1 mM Spectrozyme TH was added and the initial change in absorbance at 405 nm was measured (final concentration of thrombin and spectrozyme TH were 12.5 nM and 40 mM). The fractional thrombin activity at each concentration of SOS was calculated using the activity measured under otherwise identical conditions in the absence of SOS. Logistic equation 3 was used to fit the dose-dependence of relative thrombin activity to obtain the IC50:
| Eq.3 |
In this equation Y is the ratio of thrombin activity in the presence of SOS to that in its absence, YM and Y0 are the maximum and minimum possible values of the fractional thrombin activity, IC50 is the concentration of SOS that results in 50% inhibition of thrombin, and HS is the Hill Slope. Sigmaplot 8.0 (SPSS, Inc. Chicago, IL) was used to perform non-linear curve fitting in which YM, Y0, IC50 and HS were allowed to float.
SOS Inhibition of Fibrinogen Cleavage By Thrombin
The inhibition of HT cleavage of fibrinogen by SOS was measured in a turbidimetric assay, as previously described for sulfated lignins (33). A 48 μL sample of SOS was diluted with 950 μL of 20 mM sodium phosphate, pH 7.4, containing 40 mg/ml human fibrinogen, 100 mM NaCl, 0.1 mM EDTA, and 0.1 % (v/v) PEG 8000 at room temperature in a PEG 20000-coated polystyrene cuvette. Two μL of 2.65 μM human α–thrombin was added and the increase in turbidity at 600 nm measured immediately for up to 300 seconds.
Competitive Binding Studies with Low Molecular Weight Heparin and SOS
Thrombin inhibition by SOS was performed in the presence of 50 μM LMWH using the 96 well plate format. A 192 μL solution of SOS and thrombin with 50 μM LMWH in 20 mM phosphate buffer, pH 7.4, containing 150 mM NaCl, 0.1 mM EDTA and 0.1 % PEG 8000 was incubated at 25 °C for 10 minutes. Following incubation, 8 μL of 1 mM Spectrozyme TH was added and the initial change in absorbance at 405 nm measured. A 12.5 nM concentration of thrombin was found to give sufficient signal at various concentrations of the chromogenic peptide for reproducible results. The dose-dependence of the fractional residual proteinase activity at each concentration of the competitor was fitted by equation 3 to obtain the apparent concentration of SOS required to reduce thrombin activity to 50% of its initial value (IC50,app). Quantitative comparison of competitive binding was performed using the Dixon-Webb relationship (Eq. 4). In this equation, KLMWH is the dissociation constant of the thrombin–LMWH complex, which has been reported to be ~60 μM (23).
| Eq. 4 |
Thrombin – SOS Crystal Structure Studies
BT was exchanged into 10 mM Tris, pH 8.0 and 50 mM NaCl by repeated dilutions and concentration in an Amicon YM-10 centrifugal concentrator. Aliquots of 10 μl were stored at −80°C and diluted to 7 g/l with the same buffer at time of use. BT–SOS complex was formed by incubation of 20 mM SOS with BT at 7 mg/ml for 1 hour. This incubated solution was then set to crystallize in hanging drops under conditions optimized from those described by Iyaguchi et al. (34). A 4 μl drop of BT-SOS incubation solution was mixed with 4 μl of precipitant reservoir solution consisting of 0.1 M sodium citrate, pH 6.2, 20% (w/v) PEG 3350 and 15% (v/v) 2-propanol and equilibrated against the reservoir solution at room temperature. Isometric crystals of 0.5 mm grew under these conditions and diffracted to 2.2 Å resolution. An identical crystal form was obtained with 1 mM CaCl2 added to the precipitant under otherwise identical conditions. These latter crystals were back-soaked stepwise into the same precipitant solution in which the citrate buffer was replaced by bis-Tris to minimize possible competitive effects of citrate on SOS binding. These crystals diffracted to 2.4 Å resolution. Diffraction intensity data were collected without further cryoprotection from crystals at 100°K on a Rigaku Micromax 007a with Varimax confocal optics and an RAXIS IV++ detector over 180° in Φ in 0.5° steps. Intensity data were integrated, scaled and merged using d*trek and converted to amplitudes with TRUNCATE in the CCP4 program suite. The space group was inferred to be P43212, the same as that of the unliganded BT crystals obtained under these conditions. (The unliganded BT crystals diffracted to only 3.2 Å resolution and had one lattice dimension significantly longer than the SOS-complexed thrombin crystals.) The Matthews coefficient indicated two molecules of BT per asymmetric unit. A molecular replacement solution was found using PHASER 2.1 and the coordinates of the BT light and heavy chains from RCSB entry 1MKX as a search model. The model was initially refined via Cartesian simulated annealing using Phenix.refine and then further refined by alternating cycles of manual fitting in COOT and computational refinement in REFMAC. Statistics for the final refined model structure are shown in Table 2. Coordinates and structure factors for the unliganded BT and SOS-BT crystal structures have been deposited in the Protein Data Bank with accession numbers 3PMB and 3PMA respectively.
Table 2. Crystallographic Data Collection and Refinement.
| 3PMB | 3PMA | |
|---|---|---|
| Data collection | ||
|
| ||
| Resolution | 39.2–2.80 (3.00–2.90) | 29.72–2.20 (2.28–2.20) |
|
| ||
| I/σ(I) (highest resolution shell) | 7.2 (4.4) | 13.5 (6.2) |
|
| ||
| % Completeness (highest resolution shell) | 84.6 (6.6) | 99.8 (100.0) |
|
| ||
| Nobs | 14843 | 38669 |
|
| ||
| Multiplicity (highest resolution shell) | 4.19 (4.62) | 7.88 (9.04) |
| Rmerge (highest resolution shell) | 0.158 (0.314) | 0.083 (0.311) |
| Refinement | ||
|
| ||
| Resolution | 15.0–2.90 (2.97–2.90) | 27.8–2.20 (2.26–2.20) |
|
| ||
| Rwork (highest resolution shell) | 0.242 (0.289) | 0.204 (0.267) |
|
| ||
| Rfree (highest resolution shell) | 0.301 (0.368) | 0.245 (0.327) |
|
| ||
| Nwork | 13968 | 36728 |
| Nfree | 746 | 1938 |
| Natoms | 4514 | 4904 |
| <Bprotein> | 33.4 | 44.8 |
|
| ||
| <Bwater> | 11.9 | 31.4 |
|
| ||
| <Bligand> | 29.6 | 84.2 |
| RMSD from ideal | ||
|
| ||
| Bond length | 0.01 | 0.008 |
|
| ||
| Bond angles | 1.23 | 1.24 |
| Ramachandran Plot | ||
| residues most in most favored regions | 396 | 410 |
|
| ||
| residues in additionally allowed regions | 63 | 59 |
|
| ||
| residues in generously allowed regions | 2 | |
|
| ||
| residues in forbidden regions | 1 | |
Analytical Ultracentrifugation of Thrombin-SOS and Thrombin-Heparin Complexes
BT was diluted to 1.2 mg/ml in 10 mM Tris-HCl buffer pH 8, 50 mM NaCl and human thrombin to 0.5 mg/ml in the same buffer. Samples (420 μl) of thrombin alone and with 20 mM SOS, equimolar LMWH or equimolar unfractionated heparin (UFH) plus a buffer sample were run at 45000 rpm overnight at 20°C in a Beckman Coulter Proteome Lab XL-I analytical ultracentrifuge. Absorbance and interference scans were acquired until the boundary moved to the bottom of the cell. The continuous distribution c(M) analysis was performed using the program SEDFIT (https://sedfitsedphat.nibib.nih.gov). S20,w values were calculated for various oligomer complexes from the crystal structures of the PPACK-HT-LMWH (1XMN) and the BT-SOS complex using Hydropro v. 7.c (35).
Results
Thrombin Binds to SOS with High Affinity
To test whether SOS associates with thrombin, we used a fluorimetric method similar to that used for measuring heparin affinity for thrombin (30). In the original method the interaction of heparin in exosite II of thrombin decreases the fluorescence of the non-covalent, active site bound PABA probe in a saturable manner, which can be fitted by the quadratic binding equation 1 to obtain a KD. The interaction of SOS with exosite II of HT resulted in a maximal decrease in fluorescence at 370 nm of approximately 5% in 20 mM sodium phosphate buffer, pH 7.4, containing 50 mM NaCl, 0.1 mM EDTA and 0.1% PEG8000 (Figure 2A).
Figure 2.

A. Interaction of SOS with human thrombin at pH 7.4 and 25 °C in the presence of 50 (= o), 150 (= □) and 250 mM (= Δ) NaCl. The decrease in fluorescence of the bound external probe, PABA (λEX = 345 nm, λEM = 370 nm) that accompanies binding of SOS was used to calculate the KD,OBS of SOS–thrombin complex. Solid lines represent non-linear fits to the data using quadratic equation 1.
B. Dependence of the KD,OBS on the ionic strength of the medium at pH 7.4. Solid line represents linear regression fits using equation 2.
This maximal decrease was less than that measured for heparin (~13-17%), but was consistent and independent of the concentration of thrombin used in the experiment. Fitting equation 1 to the data gave a KD of 1.4 μM, which is remarkably close to that measured for full-length heparin (1.45 μM) under identical conditions (pH 7.4, I = 0.085, 25 °C) (23). The affinity corresponds to a free energy of binding of 7.9 kcal/mol (Table 1) suggesting a surprisingly high affinity interaction for such a small disaccharide structure. Similar measurements with BT, for which the crystal structure with SOS was determined, gave a comparable KD of 4.0 ± 0.4 μM (data not shown).
Table 1. The interaction of SOS with human α-thrombin at pH 7.4 and 25 °C.
|
KD,OBS (μM)a |
ΔFMAX (%)a |
ΔG° (kcal/mol) |
|
|---|---|---|---|
| 50 mM NaCl | 1.4±0.3b | −4.8±0.1 | 7.9±0.1 |
| 150 mM NaCl | 22±4 | −6.4±0.3 | 6.4±0.2 |
| 250 mM NaCl | 149±25 | −9.2±0.6 | 5.2±0.2 |
Measured spectrofluorometrically using a saturable decrease in fluorescence (λEM = 370 nm) of PABA due to the binding of SOS to thrombin. See ‘Experimental Procedures’ for details.
Errors represent ± 1 S.E.
The High Affinity of SOS for Thrombin Originates from Multiple Ionic Interactions
To determine the nature of the interactions of SOS with HT, the dissociation constant (KD,OBS) was measured as a function of NaCl concentration. According to the protein-polyelectrolyte theory (23,30), log[KD,OBS] is linearly dependent on log[Na+] with an intercept of logKD,NI (see Equation 2), where KD,NI is the equilibrium dissociation constant due to non-ionic forces. This relationship provides the binding energy due to ionic forces (ΔGoIONIC) from the slope and that due to non-ionic forces (ΔGoNON-IONIC) from the intercept of the linear plot.
The affinity of thrombin for SOS at several salt concentrations was measured fluorimetrically, as described above. In all cases, the maximal change in fluorescence was consistently in the range of 5 – 8% (Figure 2A). The logKD,OBS increased linearly with log[Na+] at pH 7.4 for the HT – SOS interaction (Figure 2B). The slope of this line was calculated to be 3.8 ± 0.5 vs. a slope of 4.8 ± 0.2 (23) for full-length heparin, indicating similar ionic interactions of the two polyanions. This corresponds to an ionic binding energy of 4.5 kcal/mol under physiological conditions (pH 7.4, I = 0.15, 25 °C). Full-length heparin was found to make ~6 ionic interactions (5.8 ± 0.2) with HT (26), which corresponds to an ionic binding energy of 5.7 kcal/mol. The comparable ionic binding energies of SOS and heparin to HT indicate that these two ligands make a similar number of ionic bonds in their complexes.
The intercept (log KD,NI) of the linear plot for SOS binding to HT was found to be −1.8 ± 0.4 (Figure 2B). This corresponds to a KD,NON-IONIC of 16.6 ± 6.3 mM, which implies a contribution of 2.4 kcal/mol due to non-ionic forces. This contribution of non-ionic forces to the binding affinity of SOS for HT calculates to approximately 35% of the total binding energy under physiological ionic strength (pH 7.4, I 0.15), a substantial contribution that most probably arises from multiple hydrogen bonds. In comparison, the non-ionic affinity of full-length heparin was measured to be 200 ± 100 mM, suggesting a much lower contribution of non-ionic forces to the heparin – HT complex (31) than the SOS – HT complex. This implies that the SOS – HT complex is characterized by an approximately 12.5-fold higher non-ionic contribution than the heparin – HT complex (23).
SOS Inhibits Thrombin With High Potency, But Weak Efficacy
Although heparin binding to exosite II results in a change in the fluorescence properties of PABA bound in the active site of thrombin, heparin is not a direct (antithrombin-independent) thrombin inhibitor. It has been reported that SOS weakly (≤ 10%) inhibits thrombin hydrolysis of CBS31.39 (CH3SO2-D-Leu-Gly-Arg-p-nitroanilide) (27) and we have further tested this effect using a different substrate. Thrombin activity was measured under pseudo-first order conditions in 20 mM sodium phosphate or 20 mM Tris buffer, pH 7.4, I = 0.15, 25 °C in a spectrophotometric assay using Spectrozyme TH substrate, as previously performed for sulfated lignins (17,18). As the concentration of SOS increased, the proteolytic activity decreased in a sigmoidal manner (on a semi-log plot) (Figure 3A). This decrease could be fitted using the sigmoidal dose-dependence equation 3 to derive an IC50 of 4.5 ± 1.1 μM with a Hill slope of 1.5 ± 0.5. For an allosteric, non-competitive inhibitor, the IC50 correlates well with the inhibition constant (KI), and therefore this result suggests that SOS is a fairly potent inhibitor of thrombin. Considering that most GAG-binding sites are surface exposed and shallow, and are not conducive to high affinity, the low micromolar potency of a small, highly sulfated molecule, SOS, is striking.
Figure 3.

A. Direct inhibition of human α-thrombin by SOS or enoxaparin. Assay conditions were 20 mM sodium phosphate or Tris-HCl buffer, pH 7.4, containing 150 mM NaCl, 0.1% PEG8000 and either 0.1 mM EDTA or 0.1 mM CaCl2 at 25 °C. Thrombin inhibition was measured spectrophotometrically using Spectrozyme TH hydrolysis assay. o = SOS; ■ = enoxaparin. For the SOS data, solid line represents dose-response fit to obtain IC50 and Hill Slope (equation 3), as described in the Experimental Procedures. For the enoxaparin data, solid line is a linear trend line.
B. Fibrinogen Cleavage By Thrombin in the Presence of SOS. The inhibition of HT cleavage of fibrinogen by SOS was followed in time dependent turbidimetric measurement at 600 nm of thrombin catalyzed cleavage of a 40 mg/ml sample of human fibrinogen at different SOS concentrations, as described in Experimental Procedures.
C. Competitive binding of SOS and enoxaparin to human α-thrombin. Thrombin inhibition was performed in the presence of 50 μM enoxaparin in a manner similar to that in its absence and the resulting data were fitted by the dose-response equation to obtain the apparent IC50. 1 = SOS alone; 2 = SOS + 50 μM enoxaparin; 3 = IC50 predicted by Dixon-Webb relationship for ideal competitive binding. Error bars are ±1 S.E. See text for details.
To test whether the ability to inhibit thrombin hydrolysis of small chromogenic substrates translates into the ability to inhibit cleavage of macromolecular substrates, we measured thrombin cleavage of human plasma fibrinogen in the presence of SOS. Fibrinogen cleavage results in the formation of polymeric fibrin, which reduces transmittance at 600 nm. Fibrinogen cleavage profiles measured in the presence of 0.7 – 1.8 mM SOS show a gradual increase in the lag time of cleavage (Figure 3B), suggesting that SOS inhibits the thrombin catalyzed fibrinogen cleavage reaction.
Despite the high potency of thrombin inhibition, the Y0 and YM for inhibition of Spectrozyme TH hydrolysis calculated from Equation 3 indicate a maximal inhibition of only 13.5 ± 0.2% for HT (Figure 3A) and 20 ± 2.0% for BT (not shown). This implies that the efficacy of thrombin inhibition by SOS is weak, but still significant, and contrasts to heparin (full-length or LMWH), which do not inhibit the enzyme even at concentrations as high as 200 μM (18).
SOS Competes with Low Molecular Weight Heparin for Interaction with Human Thrombin
If SOS binding in exosite II of thrombin is the source of thrombin inhibition, then it is expected to compete with full-length or LMWH and show a decrease in the potency of thrombin inhibition. Competition studies were performed by monitoring thrombin’s proteolytic activity as a function of SOS in the presence of a fixed concentration of LMWH. In the presence of 50 μM LMWH, the IC50 increased to 42 ± 3 μM (Figure 3C). Furthermore, the presence of LMWH did not affect the efficacy of SOS inhibition of thrombin, which remained unchanged at ~11%. According to the Dixon-Webb relationship for ideal competitive behavior, the apparent IC50 expected for SOS is 32 μM, very close to that observed. The decrease in SOS inhibition of thrombin in the presence of LMWH confirms that SOS binds at exosite II and that this binding is the source of the inhibition of catalytic activity.
SOS Binds at Two Non-Equivalent Sub-Sites of Exosites II in the Monomer-Monomer Interface of a Bovine Thrombin Dimer
The crystal structure of the BT-SOS complex shows binding of two SOS molecules at the interface of the two BT monomers constituting the crystallographic asymmetric unit. The BT dimer in this crystal lattice is not the same as that for PPACK-HT in its crystal structure with LMWH (28). Figure 4 shows the distinct monomer – monomer association mode of the BT-SOS complex compared to the PPACK-HT-LMWH complex. One monomer of each of these dimer structures was superposed (HT2 and BT2 bottom in pale green) and the relative positions of the other monomers in each of the dimer structures are shown in pink (PPACK-HT-LMWH complex) and wheat (BT-SOS complex). In the PPACK-HT-LMWH structure, the exosites II (blue) of the two HT monomers (pink and light green) in the dimer are almost exactly juxtaposed. The LMWH (red stick) binds in this interface and the local two-fold rotation axis relating the two HT monomers approximately bisects the LMWH (red stick) bound between them. In the BT-SOS complex (wheat and light green), the exosites II (blue) of the opposed monomers are not exactly juxtaposed across the dimer interface, but are skewed so that only parts of the exosites II face each other. This skewing of the two BT monomers leaves much of exosite II on each BT monomer open for unimpeded SOS (green sticks) binding, resulting in two non-equivalent binding sites per BT dimer (sites A and B).
Figure 4.

Differences in monomer contacts between HT-LMWH complex and BT-SOS complex dimers. Monomer AB (HT2) of the AB-GH dimer of the HT-heparin complex was superposed on monomer XD (BT2) of the AB-CD dimer of the BT-SOS complex (light green surface). The relative positions after this superposition of the monomer mates (GH (HT1) in the HT-heparin complex and AB (BT1) in the BT-SOS complex) in each of the dimers are colored pink and wheat respectively. LMWH (red stick) bound to the HT-LMWH dimer and SOS (green stick) bound to sites 1 and 2 of the BT-SOS complex, after superposition of HT2 and BT2 monomers. Both ligands occupy the extended exosite II (residues surface highlighted in blue) shown in exploded view.
The thrombin ligand groups that interact with the sulfate groups of SOS are listed in Table 3 and a detailed comparison of the interactions of BT and HT with SOS and LMWH respectively are shown in Figure 5A and B. The binding of SOS in site A is almost exclusively to charged residues in exosite II of a single BT monomer (Fig. 4, light green monomer; Figure 5A residues labeled in black) and suggests that polyanion binding to this site does not require a thrombin dimer for complex formation. The second SOS ligand in site B makes fewer interactions with BT and these are approximately equally distributed between the two thrombin monomers (Fig. 4, light green and wheat) in the dimer (Figure 5B). The close approach of two sulfate groups, one from each of the two bound SOS, is stabilized by the intervening epsilon amino group of K240. Both sites are inferred to be simultaneously occupied, since the refined occupancies of each of the SOS ligands are 0.75 – 0.80. Three of the residues (K87, K235, R244) that interact with SOS in site B do not interact with LMWH in its complex with PPACK-HT (magenta), though they lie in the zone
Table 3. Thrombin Amino Acid Residues that Interact with Sulfate Groups of Heparin and SOS in their Crystal Complexes.
| K87 | H91 | R93 | R101 | R/K126 | R165 | R233 | K235 | K236 | W237 | K240 | R244 | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Heparin – HT |
|
✓ | ✓ | ✓ | ✓ | ✓ | ✓ |
|
✓ | ✓ | ✓ |
|
| SOS – BT | ✓ | ✓ | ✓ | ✓ | ✓ |
|
✓ | ✓ | ✓ | ✓ | ✓ | |
✓ amino acid interacts with sulfate group in crystal complex
 amino acid does not interact with sulfate group in crystal complex
amino acid does not interact with sulfate group in crystal complex
Figure 5.


Schematic diagram of the interactions (dotted lines) of each of the two SOS ligands with groups on BT in: (A) site 1, and (B) site 2. Distances are shown in Å. The thrombin monomer of each interacting group is denoted in black by (AB) or (CD); double lines from a single group indicate that two possible hydrogen bonds could form simultaneously for the indicated pair; magenta denotes the heparin subunit (H prefix) in the HT-LMWH complex structure to which the indicated residue binds and the thrombin chain in which the residue occurs.
Binding of SOS at site B of a thrombin monomer would likely be weaker than that for binding to site A, because of the smaller number of interactions between SOS and either of the two monomers that contribute to its binding site.
The 4 – 5 ionic interactions between SOS and BT in exosite II (site A) of the crystal structure complex is consistent with multiple ionic interactions calculated from solution binding data. There are several other defined cationic sidechains that could be reoriented to make interactions with sulfates of SOS, but instead adopt alternative conformations. These may be restrained by interactions with solvent molecules, not all of which could be confidently placed at the resolution of our electron density map.
The crystal lattice of unliganded BT, crystallized under the same conditions as those from which the BT-SOS complex crystallized, is closely similar to that of the latter, differing by 7Å along one lattice direction. This implies that the mode of dimerization in the BT-SOS crystals is not due to differences between SOS and LMWH, or to the irreversible inhibition of HT by PPACK in its complex with LMWH. Rather, it is more likely a consequence of the small differences in crystallization conditions, possibly specific to each thrombin, despite their close sequence similarity (86% identical and 92% similar in sequence).
Analytical Ultracentrifugation
Crystals of HT-LMWH and BT-SOS complexes were obtained under non-physiological conditions. To determine the relationship between the oligomeric modes of thrombin-ligand complexes observed in the crystal structures and those that exist under more physiological conditions in solution, we determined the profile of molecular species in these thrombin-ligand mixtures by analytical ultracentrifugation and used the known structures of the crystal complexes to calculate expected S20,w values (Fig. 6).
Figure 6.

Sedimentation velocity profile showing molecular species of thrombin ligand complexes. C(s) is the relative concentration of each species. Black curve is unliganded BT; red is BT + SOS; green is HT + SOS; blue is BT + LMWH.
Sedimentation velocity ultracentrifugation was done on BT and HT alone and incubated with SOS and heparin in 10 mM Tris-HCl buffer pH 8, 50 mM NaCl at high (1.2 mg/ml) concentrations of BT and lower (0.5 mg/ml) concentrations of HT to check for concentration-dependent aggregation. In the presence and absence of the SOS ligand, BT and HT had S20,w values of 3.1 – 3.2. This is in agreement with the S20,w value (3.14) calculated for the thrombin monomers from their crystal structures. The slight shift in mobility of the BT + SOS peak relative to BT alone is consistent with formation of a monomeric BT- SOS complex. In contrast, deconvolution of the broad peak observed for the LMWH complex with BT shows three species. These are consistent with monomer (3.45), dimer (4.45) and tetramer (5.5) complexes of BT with LMWH. The profile of HT + LMWH (not shown) shows only a species with S20,w = 4, which is most consistent with a 2:1 HT:LMWH dimer, similar but not identical to that observed in the crystal structure of this complex, from which an S20,w = 4. 72 is calculated (28). This difference may indicate a difference in frictional ratio between the complex in solution and that in the crystal structure or it may arise from the fact that the crystal complex was composed of PPACK-inhibited HT + LMWH.
Discussion
The high affinity of thrombin for full-length heparin has been attributed to the fact that the polysaccharide is a linear molecule of nearly 10 – 30 disaccharides that represent millions of sequences (Fig. 1). This increases the probability of favorable electrostatic interaction with a high affinity subset of this large population. The probability of interaction is further increased because heparin contains a large number of iduronic acid residues, which confer conformational flexibility (1C4 and 2SO forms) on the polymer.
In contrast to heparin, SOS is considerably smaller and homogeneous with a much more limited distribution of conformations. For these reasons it was expected that SOS would have a low affinity interaction with exosite II of thrombin. It is therefore surprising that SOS, a molecule smaller in size by ~15-fold than full-length heparin, binds to human thrombin with nearly identical affinity and with a greater non-ionic energy contribution. This suggests that SOS has some structural features and charge properties that distinguish it from heparin, and which could confer tight binding to thrombin. Non-ionic binding energy in interactions of sulfate groups is likely to arise from the partial hydrogen bond character of the primarily electrostatic sulfate – arginine (or lysine) ion – pairs. This hydrogen bond character increases the specificity of interaction due to its directionality (36) and could confer a greater specificity on the interaction of SOS with thrombin than is observed for heparin, whose interactions with thrombin are predominantly ionic.
At least 10 unique crystal structures of SOS-protein complexes have been determined (1AFC, 1LR7, 2P39, 2UWN, 2UUS, 2V5E, 3CU1, 3LDJ, 3K6B, 3QRC), but none for thrombin with SOS. The BT-SOS crystal structure described here appears to have a single mode of SOS binding with two SOS molecules bound at the monomer-monomer interface of the thrombin crystal dimer. This 1:1 stoichiometry and the disposition of the two thrombin monomers in the BT-SOS crystal dimer differ from the HT-LMWH complex whose stoichiometry is 1:2 (LMWH:HT). The two exosites II in the HT-LMWH complex sandwich the single LMWH, which lies centered on the non-crystallographic two-fold axis relating the two HT monomers. The SOS bound in site A of the BT-SOS complex interacts with a large fraction of the same anionic groups that LMWH interacts with in exosites II of the HT crystal dimer. However, in the BT-SOS complex these groups, with one exception, are entirely on a single thrombin monomer, in contrast to the HT-LMWH complex, where they are contributed by both thrombin monomers in the dimer. The exception is K240, which lies between proximal sulfate groups of the two adjacent SOS ligands bound in the exosites II of the BT crystal dimer. The analytical ultracentrifugation results are consistent with the crystal structures, showing 1:1 SOS:BT and SOS:HT complexes and a 1:2 LMWH:HT complex.
The reduction in thrombin’s ability to hydrolyze small peptides as a result of its interaction with SOS distinguishes this polyanion ligand from heparin, which does not show this effect. Our measurements show a lower KD for SOS binding to thrombin and somewhat higher levels of thrombin inhibition by SOS than have been reported by others (27). These differences may be due to use of different buffer conditions (ionic strength and presence of Ca+2) and/or different substrates.
There are several possible ways in which SOS binding could alter the activity of thrombin. The higher specificity in the SOS interaction conferred by a greater contribution of non-ionic forces to stabilization of the SOS-thrombin complex may impose a specific local conformation that affects the active site through altered static or dynamic properties of the protein. The available structures of BT and the BT-SOS complex are not of sufficient resolution to reveal subtle static changes in structure that could be responsible for this modest loss of thrombin activity on binding SOS, but any such changes are extremely limited. Alternatively, the higher specificity of SOS interaction with thrombin may alter the dynamics of the thrombin structure and/or the population distribution of active thrombin conformers, which manifests as allostery (37 - 39). Possibly related to such dynamics-driven allostery is the displacement of solvent on binding of heparin and SOS to exosite II. Although SOS has a higher charge density than heparin, heparin binding to thrombin may displace less bound water than SOS due to the presence of hydroxyl groups on heparin and their absence on SOS. Such solvent effects could contribute to the higher affinity of SOS than heparin for thrombin. Finally, the difference in solution oligomeric state of thrombin between the SOS complex (monomer) and the thrombin-LMWH complex (dimer) could also induce dynamic allosteric effects. The closed dimer of thrombin with LMWH may stabilize the two thrombin structures and suppress small structural changes necessary for inhibition of thrombin activity. The inhibition of thrombin catalytic activity by SOS binding in exosite II opens new possibilities for understanding communication between exosite II and the active site and also validates exosite II as a target for the design of more effective thrombin inhibitors.
Acknowledgements
We thank Dr. Carlos Escalante for his generous help with the analytical ultracentrifugation experiments.
This work was supported by grants from the National Institutes of Health (HL099420 and HL090586) and the American Heart Association (EIA 0640053N). Crystallography and computational infrastructure was provided by the VCU Structural Biology Core Facility, supported in part with funding from NIH-NCI Cancer Center Core Support Grant P30 CA016059.
Abbreviations
- HT
human thrombin
- BT
bovine thrombin
- LMWH
low molecular weight heparin
- MOPS
3-morpholinopropane-1-sulfonic acid
- PABA
p-aminobenzamidine
- PAR
protease-activated receptor
- PEG
polyethyleneglycol
- PPACK
d-phenylalanyl-l-prolyl-l-arginine chloromethyl ketone
- SOS
sucrose octasulfate
References
- 1.Di Cera E. Thrombin. Mol. Aspects Med. 2008;29:203–254. doi: 10.1016/j.mam.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Crawley JTB, Zanardelli S, Chion CKNK, Lane DA. The central role of thrombin in hemostasis. J. Thromb. Haemost. 2007;5:95–101. doi: 10.1111/j.1538-7836.2007.02500.x. [DOI] [PubMed] [Google Scholar]
- 3.Davie EW, Kulman JD. An overview of the structure and function of thrombin. Semin. Thromb. Hemostasis. 2006;32:3–15. doi: 10.1055/s-2006-939550. [DOI] [PubMed] [Google Scholar]
- 4.Huntington JA. Molecular recognition mechanisms of thrombin. J. Thromb. Haemost. 2005;3:1861–1872. doi: 10.1111/j.1538-7836.2005.01363.x. [DOI] [PubMed] [Google Scholar]
- 5.Coughlin SR. Protease-activated receptors in hemostasis, thrombosis and vascular biology. J. Thromb. Haemost. 2005;3:1800–1814. doi: 10.1111/j.1538-7836.2005.01377.x. [DOI] [PubMed] [Google Scholar]
- 6.Brass LF. Thrombin and platelet activation. Chest. 2003;124:18S–25S. doi: 10.1378/chest.124.3_suppl.18s. [DOI] [PubMed] [Google Scholar]
- 7.Colognato R, Slupsky JR, Jendrach M, Burysek L, Syrovets T, Simmet T. Differential expression and regulation of protease-activated receptors in human peripheral monocytes and monocyte-derived antigen-presenting cells. Blood. 2003;102:2645–2652. doi: 10.1182/blood-2002-08-2497. [DOI] [PubMed] [Google Scholar]
- 8.Moller T, Weinstein JR, Hanisch U- Activation of microglial cells by thrombin: Past, present, and future. Semin. Thromb. Hemostasis. 2006;32:69–76. doi: 10.1055/s-2006-939556. [DOI] [PubMed] [Google Scholar]
- 9.Li X, Syrovets T, Paskas S, Laumonnier Y, Simmet T. Mature dendritic cells express functional thrombin receptors triggering chemotaxis and CCL18/pulmonary and activation-regulated chemokine induction. J. Immun. 2008;181:1215–1223. doi: 10.4049/jimmunol.181.2.1215. [DOI] [PubMed] [Google Scholar]
- 10.Chen D, Dorling A. Critical roles for thrombin in acute and chronic inflammation. J. Thromb. Haemost. 2009;7:122–126. doi: 10.1111/j.1538-7836.2009.03413.x. [DOI] [PubMed] [Google Scholar]
- 11.Bunnett NW. Protease-activated receptors: How proteases signal to cells to cause inflammation and pain. Semin. Thromb. Hemostasis. 2006;32:39–48. doi: 10.1055/s-2006-939553. [DOI] [PubMed] [Google Scholar]
- 12.Esmon CT. The protein C pathway. Chest. 2003;124:26S–32S. doi: 10.1378/chest.124.3_suppl.26s. [DOI] [PubMed] [Google Scholar]
- 13.Ruf W, Mueller BM. Thrombin generation and the pathogenesis of cancer. Semin. Thromb. Hemostasis. 2006;32:61–68. doi: 10.1055/s-2006-939555. [DOI] [PubMed] [Google Scholar]
- 14.Bode W. The structure of thrombin: A janus-headed proteinase. Semin. Thromb. Hemostasis. 2006;32:16–31. doi: 10.1055/s-2006-939551. [DOI] [PubMed] [Google Scholar]
- 15.Ye J, Liu LW, Esmon CT, Johnson AE. The fifth and sixth growth factor-like domains of thrombomodulin bind to the anion-binding exosite of thrombin and alter its specificity. J. Biol. Chem. 1992;267:11023–11028. [PubMed] [Google Scholar]
- 16.Ye J, Rezaie AR, Esmon CT. Glycosaminoglycan contributions to both protein C activation and thrombin inhibition involve a common arginine-rich site in thrombin that includes residues arginine 93, 97, and 101. J. Biol. Chem. 1994;269:17965–17970. [PubMed] [Google Scholar]
- 17.Monien BH, Henry BL, Raghuraman A, Hindle M, Desai UR. Novel chemo-enzymatic oligomers of cinnamic acids as direct and indirect inhibitors of coagulation proteinases. Bioorg. Med. Chem. 2006;14:7988–7998. doi: 10.1016/j.bmc.2006.07.066. [DOI] [PubMed] [Google Scholar]
- 18.Henry BL, Monien BH, Bock PE, Desai UR. A novel allosteric pathway of thrombin inhibition. J. Biol. Chem. 2007;282:31891–31899. doi: 10.1074/jbc.M704257200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu LW, Vu TK, Esmon CT, Coughlin SR. The region of the thrombin receptor resembling hirudin binds to thrombin and alters enzyme specificity. J. Biol. Chem. 1991;266:16977–16980. [PubMed] [Google Scholar]
- 20.Hortin GL, Trimpe BL. Allosteric changes in thrombin’s activity produced by peptides corresponding to segments of natural inhibitors and substrates. J. Biol. Chem. 1991;266:6866–6871. [PubMed] [Google Scholar]
- 21.Hogg PJ, Jackson CM. Formation of a ternary complex between thrombin, fibrin monomer, and heparin influences the action of thrombin on its substrates. J. Biol. Chem. 1990;265:248–255. [PubMed] [Google Scholar]
- 22.Hogg PJ, Jackson CM, Labanowski JK, Bock PE. Binding of fibrin monomer and heparin to thrombin in a ternary complex alters the environment of the thrombin catalytic site, reduces affinity for hirudin, and inhibits cleavage of fibrinogen. J. Biol. Chem. 1996;271:26088–26095. doi: 10.1074/jbc.271.42.26088. [DOI] [PubMed] [Google Scholar]
- 23.Olson ST, Halvorson HR, Björk I. Quantitative characterization of the thrombin-heparin interaction. Discrimination between specific and nonspecific binding models. J. Biol. Chem. 1991;266:6342–6352. [PubMed] [Google Scholar]
- 24.Verhamme IM, Olson ST, Tollefsen DM, Bock PE. Binding of exosite ligands to human thrombin. J. Biol. Chem. 2002;277:6788–6798. doi: 10.1074/jbc.M110257200. [DOI] [PubMed] [Google Scholar]
- 25.Vacca JP. New advances in the discovery of thrombin and factor Xa inhibitors. Curr. Opin. Chem. Biol. 2000;4:394–400. doi: 10.1016/s1367-5931(00)00112-5. [DOI] [PubMed] [Google Scholar]
- 26.Srivastava S, Goswami LN, Dikshit DK. Progress in the design of low molecular weight thrombin inhibitors. Med. Res. Rev. 2005;25:66–92. doi: 10.1002/med.20016. [DOI] [PubMed] [Google Scholar]
- 27.Sarilla S, Habib SY, Kravtsov DV, Matafonov A, Gailani D, Verhamme IM. Sucrose octasulfate selectively accelerates thrombin inactivation by heparin cofactor II. J. Biol. Chem. 2010;285:8278–8289. doi: 10.1074/jbc.M109.005967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carter WJ, Cama E, Huntington JA. Crystal structure of thrombin bound to heparin. J. Biol. Chem. 2005;280:2745–2749. doi: 10.1074/jbc.M411606200. [DOI] [PubMed] [Google Scholar]
- 29.Kamath P, Huntington JA, Krishnaswamy S. Ligand binding shuttles thrombin along a continuum of zymogen- and proteinase-like states. J. Biol. Chem. 2010;285:28651–28658. doi: 10.1074/jbc.M110.154914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Evans SA, Olson ST, Shore JD. p-Aminobenzamidine as a fluorescent probe for the active site of serine proteases. J. Biol. Chem. 1982;257:3014–3017. [PubMed] [Google Scholar]
- 31.Olson ST. Transient kinetics of heparin-catalyzed protease inactivation by antithrombin III. Linkage of protease-inhibitor-heparin interactions in the reaction with thrombin. J. Biol. Chem. 1988;263:1698–1708. [PubMed] [Google Scholar]
- 32.Cochran S, Li CP, Ferro V. A surface plasmon resonance-based solution affinity assay for heparan sulfate-binding proteins. Glycoconj. J. 2009;26:577–587. doi: 10.1007/s10719-008-9210-0. [DOI] [PubMed] [Google Scholar]
- 33.Henry BL, Abdel Aziz M, Zhou Q, Desai UR. Sulfated, low molecular weight lignins are potent inhibitors of plasmin, in addition to thrombin and factor Xa: Novel opportunity for controlling complex pathologies. Thromb. Haemost. 2010;103:507–515. doi: 10.1160/TH09-07-0454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Iyaguchi D, Yao M, Watanbe N, Tanaka I, Toyota E. Crystallization and preliminary x-ray studies of the unliganded wild-type bovine thrombin. Prot. Pept. Lett. 2007;14:923–924. doi: 10.2174/092986607782110248. [DOI] [PubMed] [Google Scholar]
- 35.García de la Torre J, Huertas ML, Carrasco B. Calculation of hydrodynamic properties of globular proteins from their atomic-level structure. Biophys. J. 2000;78:719–730. doi: 10.1016/S0006-3495(00)76630-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Al-Horani RA, Desai UR. Chemical sulfation of small molecules—Advances and challenges. Tetrahedron. 2010;66:2907–2918. doi: 10.1016/j.tet.2010.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Volkman BF, Lipson D, Wemmer DE, Kern D. Two-state allosteric behavior in a single-domain signaling protein. Science. 2001;291:2429–2433. doi: 10.1126/science.291.5512.2429. [DOI] [PubMed] [Google Scholar]
- 38.Kern D, Zuiderweg ER. The role of dynamics in allosteric regulation. Curr. Opin. Struct. Biol. 2003;13:748–757. doi: 10.1016/j.sbi.2003.10.008. [DOI] [PubMed] [Google Scholar]
- 39.Popovych N, Sun S, Ebright RH, Kalodimos CG. Dynamically driven protein allostery. Nat. Struct. Mol. Biol. 2006;13:831–838. doi: 10.1038/nsmb1132. [DOI] [PMC free article] [PubMed] [Google Scholar]