

Abstract
Bacterial aromatic polyketides that include many antibiotic and antitumor therapeutics are biosynthesized by the type II polyketide synthase (PKS), which consists of 5 – 10 stand-alone enzymatic domains. Hedamycin, an antitumor antibiotic polyketide, is uniquely primed with a hexadienyl group generated by a type I PKS followed by coupling to a downstream type II PKS to biosynthesize a 24-carbon polyketide, whose C9 position is reduced by hedamycin type II ketoreductase (hedKR). HedKR is homologous to the actinorhodin KR (actKR), for which we have conducted extensive structural studies previously. How hedKR can accommodate a longer polyketide substrate than the actKR, and the molecular basis of its regio- and stereospecificities, is not well understood. Here we present a detailed study of hedKR that sheds light on its specificity. Sequence alignment of KRs predicts that hedKR is less active than actKR, with significant differences in substrate/inhibitor recognition. In vitro and in vivo assays of hedKR confirmed this hypothesis. The hedKR crystal structure further provides the molecular basis for the observed differences between hedKR and actKR in the recognition of substrates and inhibitors. Instead of the 94-PGG-96 motif observed in actKR, hedKR has the 92-NGG-94 motif, leading to S-dominant stereospecificity, whose molecular basis can be explained by the crystal structure. Together with mutations, assay results, docking simulations, and the hedKR crystal structure, a model for the observed regio- and stereospecificities is presented herein that elucidates how different type II KRs recognize substrates with different chain lengths, yet precisely reduce only the C9-carbonyl group. The molecular features of hedKR important for regio- and stereospecificities can potentially be applied to biosynthesize new polyketides via protein engineering that rationally controls polyketide ketoreduction.
Polyketides represent a large class of natural products that are diverse both in terms of chemical structure and bioactivity (1). Many of these compounds are pharmaceutically important and can act, for example, as antibiotic, anticancer and antihypercholesterolemic agents (2-4). The ensemble of enzymes contributing to polyketide biosynthesis is known as the polyketide synthase (PKS), which is genetically, structurally and functionally similar to fatty acid synthase (FAS) (5). There are generally three classes of PKS (6) (type I, type II, and type III). In the type I system, enzyme domains are covalently linked together, whereas in type II systems the enzymes are present as discrete proteins. The focus of this study is on enzymes of the type II PKS that are found mainly in bacteria and biosynthesize aromatic polyketides such as the antibiotics actinorhodin and tetracenomycin (Figure 1) (2, 7). In a typical type II PKS system (5) (Figure 2A), a linear poly-β-keto chain is synthesized by the minimal PKS (ketosynthase/chain length factor [KS/CLF] and acyl carrier protein [ACP]) (8). The chain is initiated by an acetate unit derived from decarboxylation of a malonyl-ACP thioester, or by an “initiation module” that supplies an acyl unit ranging from 3 – 6 carbons (9), and iteratively elongated by decarboxylative condensation of malonyl-CoA, to yield the full-length polyketide intermediate. After first-ring cyclization, typically between C7-C12 or C9-C14 (10), an optional ketoreductase (KR) can then catalyze the regiospecific reduction of a single carbonyl group (typically at C9) to a hydroxyl group (11). The KR is an important component of the PKS. Previous studies indicate that KR may direct the C7-C12 first-ring cyclization and introduce stereochemistry in the polyketide intermediate upon reduction (12, 13). The molecular determinants of stereospecificity in various KR domains from type I PKS systems have been characterized through site-directed mutagenesis and biochemical analyses (14-16). However, it is unclear whether the conclusions drawn from type II actKR studies (11, 17) may also be applied to other type II KRs.
Figure 1.

Representative type II polyketides.
Figure 2.

Type II PKS pathway intermediates in the presence or absence of KR. (A) The actinorhodin system, in which the polyketide chain is initiated by an acetyl group and the presence of actKR can lead to 6- or 8-carbon products. (B) The hedamycin system, in which the unique 6-carbon type I polyketide starter unit initiates polyketide chain biosynthesis. In the presence of hedKR, 24-, 22- and 8-carbon products are formed, while 6-, 8-, 10-, 22-, and 24-carbon products are formed in the absence of hedKR. Red numbers are used to denote which carbon of an intermediate can undergo ketoreduction, and are shown for the final products as well.
Hedamycin is a pluramycin-type antitumor antibiotic produced by Streptomyces griseoruber that mediates its biological activity in part through intercalation in DNA (18-20) (Figure 1). One of the unique properties of this aromatic polyketide stems from its biosynthesis. In the actinorhodin (act) system and many other type II PKSs, polyketide chain synthesis is initiated, or primed, by a two-carbon acetate unit and the chain is elongated by the iterative condensation of malonate-derived units (Figure 2A). The polyketide precursor of hedamycin, however, is primed by a unique 2,4-hexadienyl unit that is produced by type I PKSs, HedT and HedU (21), which pass the starter unit to a downstream type II PKS to extend the chain through nine elongation steps and ultimately synthesize a 24-carbon polyketide (Figure 2B). Previous studies on the hedamycin (hed) type II PKS suggest that as part of the type II PKS, hedKR regiospecifically reduces the C9-carbonyl of the polyketide substrate, similar to actKR (22). However, in vivo heterologous expression also implied that hedKR is important for chain length determination, the molecular basis of which is unclear. The goal of this work is to elucidate the biochemical properties of hedKR and determine a possible structural basis for C9-ketoreduction regiospecificity, given the unique hedamycin polyketide substrate that is eight carbons longer than that of actKR. Our previous structural, assay and docking studies of actKR led to the identification of a residue motif (94-PGG-96) that acts as a determinant of stereospecificity (23). However, without further information in the form of crystal structures of additional type II polyketide KRs, it is not possible to assess whether this motif may play a similar role in other type II KRs or to establish a common molecular basis for regiospecific ketoreduction. The HedA KR (hereafter referred to as hedKR) from the hedamycin PKS represents a new target for further structural and biochemical analyses of type II polyketide KRs. Herein we present many interesting differences between actKR and hedKR in regard to substrate/inhibitor specificity, stereospecificity, and ACP-KR interactions. Additionally, the structural and functional studies presented help bridge the current knowledge gap on chain length control in which the type II polyketide KRs may be involved.
MATERIALS AND METHODS
Materials
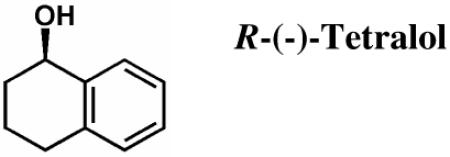
trans-1-Decalone, S-(+)-tetralol, R-(−)-tetralol, cofactor NADP(H), and emodin were purchased from Sigma, and were the highest grade available. Quercetin was purchased from Acros Organics. Custom oligonucleotides were purchased from Operon Biotechnologies. All organic solvents including DMSO were HPLC grade or better and purchased from Fisher.
DNA Manipulation
Plasmid pAD224 consists of the wild type hedA gene cloned into a pET28 expression vector (Novagen). The N92L and N92P single mutations were generated using the QuikChange II Site-Directed Mutagenesis Kit (Stratagene) with pAD224 as the template. E. coli NovaBlue cells were used for plasmid amplification. All mutations were confirmed by sequencing.
HedKR Protein Expression and Purification
Recombinant wild type or mutant hedKR was expressed in E. coli strain BL21(DE3). Following transformation, cells were grown at 37 °C in 1 L Luria-Bertani media supplemented with 50 μg/mL kanamycin to an OD600 of 0.6-0.8. At that point, protein expression was induced by the addition of 0.1 mM IPTG at 18 °C overnight. Cells were harvested by centrifugation (5000 rpm × 30 min), resuspended in 50 mL buffer A (50 mM Tris-Cl pH 7.5, 300 mM NaCl, and 10% glycerol) at 4 °C, and lysed on ice by sonication (5 × 30 sec pulses). Cell debris were removed by centrifugation (14000 rpm × 45 min) followed by binding of lysate supernatant to 3 mL nickel IMAC resin (Bio-Rad) in batch-mode at 4 °C. Bound protein was washed with 10 mL of 10 mM imidazole in buffer A and then with 20 mL of 20 mM imidazole in buffer A. HedKR was eluted at 40, 60, 80, 100, 150, and 250 mM imidazole in buffer A. The last four fractions were pooled together and dialyzed at 4 °C overnight against 4 L buffer A. The protein was concentrated to ~10 mg/mL with Pierce iCON 9000 MWCO protein concentrators.
In Vitro Tetralol Assay for HedKR Stereospecificity
Steady-state kinetic parameters for wild type and mutant hedKR were determined by monitoring the oxidation of S-(+)- or R-(−)-tetralol in the presence of NADP+. The change in absorbance from the conversion of NADP+ to NADPH was monitored at 340 nm (ε340 = 6220 M−1 cm−1) on a DU 800 Spectrophotometer (Beckman Coulter) over 10 minutes. All assays were performed in 400 mM KPi buffer (pH 7.4) and 5% DMSO at 30 °C, and were initiated with the addition of enzyme at a concentration of 1.0-1.7 μM. The Michaelis-Menten constants Km and kcat for each substrate were obtained by varying the substrate concentration in the presence of 375 M NADP+. Data were fitted to the Michaelis-Menten equation using the program KaleidaGraph (Synergy).
ActKS/CLF Expression and Purification
The expression of actKS/CLF from Streptomyces coelicolor CH999/pRJC006 spores has been described previously (24). Briefly, spores were grown in 50 mL Super YEME containing 50 μg/mL kanamycin for 3 days at 30 °C shaking at 250 rpm. The mycelia were then transferred to 500 mL Super YEME containing 50 μg/mL kanamycin and grown as before for 2 days. Protein expression was induced by the addition of 5 μg/mL thiostrepton, and the cell growth continued as before for 1 day. Cells were harvested by centrifugation (5000 rpm × 30 min), resuspended in 40 mL lysis buffer (100 mM KPi pH 7.5, 0.1% Triton X-100, 5 mM TCEP, 1.5 mM benzamidine, 1 tablet EDTA-free protease inhibitor cocktail [Roche], and 10% glycerol), and lysed on ice by sonication (8 × 1 min pulses). Cell debris were removed by centrifugation (14000 rpm × 30 min) followed by binding of lysate supernatant to 3 mL nickel IMAC resin (Bio-Rad) in batch-mode by spinning at 4 °C for 2 hrs. Protein was eluted with increasing concentrations of imidazole in 100 mM KPi pH 7.5, 500 mM NaCl, and 10% glycerol. Fractions containing actKS/CLF were collected at 150 and 500 mM imidazole, pooled, and buffer-exchanged to 100 mM KPi pH 7.2 and 20% glycerol.
Holo-ActACP (C17S) and MAT Expression and Purification
E. coli BAP1 cells (25) expressing pTLF-569 (C17S actI-ORF3 in pET28b, provided by Robert W. Haushalter of the Burkart research group [Department of Chemistry and Biochemistry, University of California, San Diego]) were grown at 37 °C in 1 L Luria-Bertani media supplemented with 50 μg/mL kanamycin to an OD600 of 0.6-0.8. At that point, protein expression was induced by the addition of 0.1 mM IPTG at 18 °C overnight. Cells were harvested by centrifugation (5000 rpm × 30 min), resuspended in 40 mL buffer D (25 mM K2HPO4 pH 7.5, 100 mM NaCl, and 10% glycerol) at 4 °C, and lysed on ice by sonication (5 × 30 sec pulses). The cell debris were removed by centrifugation (14000 rpm × 45 min) followed by binding of lysate supernatant to 5 mL nickel IMAC resin (Bio-Rad) in batch-mode at 4 °C. Protein was eluted with increasing concentrations of imidazole in buffer D. A fraction containing pure holo-actACP (C17S) was eluted at 500 mM imidazole and buffer exchanged to buffer D. S. coelicolor MAT was expressed and purified from E. coli BL21(DE3)/pGFL16 by nickel IMAC as described previously (26).
In Vitro PKS Reconstitution Assay
To determine whether in vitro hedKR can regiospecifically reduce a 16-carbon polyketide substrate at the C9-carbonyl to produce mutactin, 50 μM hedKR was incubated with 10 μM actKS/CLF, 50 μM holo-actACP (C17S), 1 μM MAT, 5 mM malonyl-CoA, and 2 mM NADPH. The total reaction volume was brought to 250 μL by the addition of 100 mM Tris-Cl pH 7.0 and incubated at room temperature overnight in the dark. The mixture was extracted once with 300 μL of 94% ethyl acetate, 5% methanol, and 1% acetic acid. Solvent was evaporated, products resuspended in 100 μL DMSO, and 20 μL subjected to reverse phase HPLC on a Synergi Hydro-RP analytical column (Phenomenex, 4 μ, 150 × 4.6 mm). For comparison, the same assay was conducted using hedKR mutants N92L and N92P, as well as WT actKR purified as described previously (17), in place of hedKR.
Protein Crystallization and Data Collection
Crystals of wild type hedKR were grown in hanging-drops at 15 °C by vapor diffusion. Drops were generated by mixing 1 μL protein with 1 μL well buffer above a well solution of 500 μL. The protein solution consisted of 5 mg/mL hedKR, 2 mM NADPH, and 1 mM DMAC as a potential inhibitor. Crystals were flash-frozen in liquid nitrogen after soaking in 30% glycerol. Data were collected on beamline 8.2.2 at the Advanced Light Source (ALS) to 2.4 Å. Diffraction intensities were indexed, integrated and scaled using HKL-2000 (27).
Phasing by Molecular Replacement and Refinement
The wild type hedKR structure was solved by molecular replacement using Phaser in CCP4i (28). The search model consisted of a homology model of hedKR prepared by SwissModel (29), based on the previously reported wild type actKR structure (PDB accession code 1X7H). Following an initial round of refinement with Refmac5 (30), manual rebuilding was performed and waters were added using Coot (31). Subsequent rounds of refinement in Refmac5 and rebuilding in Coot were conducted until the final R and Rfree were 0.186 and 0.248, respectively. The quality of the final structure was analyzed with Procheck (32). All crystallographic statistics are listed in Table 3.
Table 3.
HedKR Crystallographic Statistics
| HedKR-NADPH | |
| A. Crystallization | 0.1 M imidazole pH 6.2, 50% MPD |
| B. Crystallographic Data | |
| Space group | C121 |
| Cell dimension (Å) | 116.70 57.39 82.11 |
| α = γ =90 °, β = 131.61 ° | |
| Resolution (Å) | 61.39-2.40 |
| Mosaicity | 1.00 |
| No. of observations | 114703 |
| No. of unique reflections | 15891 |
| Completeness % (last shell) | 99.0 (89.0) |
| I/σ(I) (last shell) | 22.9 (6.7) |
| Rmerge % (last shell) | 8.5 (23.5) |
| C. Refinement | |
| Resolution (Å) | 2.40 |
| No. of reflections | 15078 |
| No. of protein atoms | 3696 |
| No. of cofactor atoms | 96 |
| No. of water atoms | 36 |
| Rfree % | 24.8 |
| Rcrys % | 18.6 |
| D. Geometry | |
| RMS bonds (Å) | 0.016 |
| RMS angles (°) | 1.77 |
| RMS B main chain | 0.859 |
| RMS B side chain | 2.335 |
| Ramachandran plot (%) | |
| Most favored | 89.4 |
| Favored | 10.6 |
| Generously allowed | 0 |
Inhibition Kinetics of HedKR
To determine whether quercetin is an inhibitor of hedKR, enzyme activity was measured by varying trans-1-decalone concentration in the presence of 375 μM NADPH and 0, 25, 50, or 75 μM quercetin. Activity was also measured by varying NADPH concentration in the presence of 2 mM trans-1-decalone and 0, 12.5, 25, or 50 μM quercetin. All assays were performed in 400 mM KPi buffer (pH 7.4) containing 2% DMSO at 30 °C and were initiated with the addition of the enzyme at a concentration of 690 nM.
RESULTS AND DISCUSSION
Sequence Alignment Suggests that HedKR and ActKR Have Different Structures and Activities
HedKR is predicted to contain a short-chain dehydrogenase/reductase (SDR) fold (33), similar to other type II polyketide KRs. The hedKR protein sequence was aligned to those of other type II polyketide KRs including actKR, with sequence identity ranging from 61-66% (Figure 3). The characteristic sequence motifs found in SDR enzymes (Table 2 in ref (33)) are conserved in hedKR, such as the cofactor-binding motif TGxxxGxG (hedKR residues 10-17), as well as the active tetrad residues Asn112, Ser142, Tyr155, and Lys159. Notably, the NNAG motif in SDR enzymes becomes 87-NSAG-90 in hedKR. The NNAG motif was proposed to stabilize the central β-sheet (34), and is highly conserved in the other type II polyketide KRs except hedKR. In the bacterial 3β-hydroxysteroid dehydrogenase, an alanine mutation of N87 (equivalent to S88 in hedKR) lowered the reductive and oxidative kcat/Km values by 39 and 83%, respectively (34). Therefore, sequence alignment suggests that hedKR may have lower enzymatic activity than actKR due to the change of the NNAG motif to NSAG (detailed below in the in vitro assay sections).
Figure 3.

Sequence alignment among various type II PKS KRs. Sequences included hedamycin, actinorhodin, frenolicin, granaticin, griseucin, nogalamycin, oxytetracycline, and urdamycin KRs. Key: magenta circles, SDR cofactor-binding motif; blue arrow, arginine patch residue; yellow rectangles, SDR motif involved in the stabilization of the central β-sheet; green-tinted box, “PGG” motif; red stars, catalytic residue.
Table 2.
Kinetic Parameters for the Oxidation of S-(+)-Tetralol and R-(−)-Tetralol by Wild Type and Mutants of Type II Polyketide Ketoreductases (ActKR and HedKR)
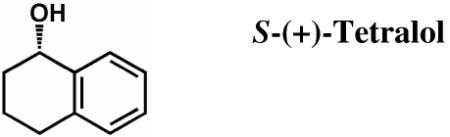 |
 |
|||||
|---|---|---|---|---|---|---|
| kcat (s−1) | Km (mM) | kcat/Km (s−1 mM−1) |
kcat (s−1) | Km (mM) | kcat/Km (s−1 mM−1) |
|
| ActKR | ||||||
| WT | 0.24 ± 0.01 | 6.91 ± 1.2 | 0.035 ± 0.006 | 0.06 ± 0.004 | 6.47 ± 0.90 | 0.010 ± 0.001 |
| P94L | 0.24 ± 0.03 | 6.57 ± 1.8 | 0.036 ± 0.011 | ND | >>40 | ND |
| HedKR | ||||||
| WT | 0.168 ± 0.0109 | 108 ± 7.98 | 0.00156 ± 0.000153 |
ND | ND | ND |
| N92L | 0.169 ± 0.00653 |
4.32 ± 0.521 | 0.0391 ± 0.00496 | ND | ND | ND |
| N92P | 0.0323 ± 0.00166 |
14.1 ± 1.36 | 0.00229 ± 0.000249 |
ND | ND | ND |
ND: No detectable activity
In the active site, > 70% of the residues that define the substrate-binding pocket (based on actKR-emodin crystal structures (17)) are conserved between actKR and hedKR, such as Q147, V149, A152, M192, and V196. However, Phe189 of actKR, which interacts with both emodin and cofactor, becomes Tyr187 of hedKR, indicating subtle changes near the active site. Moreover, the “arginine patch” of actKR, proposed to be the docking point of incoming ACP and PPT-phosphate (23, 35), is not conserved in hedKR. Specifically, Arg65 of actKR becomes Thr63 of hedKR. Therefore, sequence alignment predicts that the interaction between the hedACP and hedKR differs from the corresponding interaction in the act PKS. Corroborating the above hypothesis, the hedACP differs significantly from the actinorhodin ACP (actACP): while the significant majority of type II polyketide ACPs exist as stand-alone proteins, the hedACP is covalently linked to the aromatase/cyclase as the bifunctional protein HedE (21). The difference between the hedKR and actKR arginine patches may therefore be intimately related to the different domain arrangement of hedACP.
In terms of stereospecificity, mutational studies of actKR showed that the “PGG” motif is important for the observed S-dominant stereospecificity, with a 3:1 (S:R) preference in vitro for the oxidation of S- versus R-tetralol (23). In hedKR, “PGG” becomes “NGG,” suggesting that the stereospecificity of hedKR may differ from that of wild type actKR. Altogether, the sequence alignment results show that hedKR has the SDR fold, and its structural features are highly conserved with those of actKR; however, enzymatic activity, interactions with ACP, and stereospecificity of hedKR may differ from those of actKR, reflecting the differences in substrates, domain architecture, and stereochemical requirement of hedamycin versus actinorhodin PKSs.
In Vitro Assays Support that HedKR is Much Less Active than ActKR
To determine in vitro reductase activity, hedKR was assayed with the substrate trans-1-decalone, as had been previously done with actKR (17). The steady-state kinetic parameters for reduction of trans-1-decalone by hedKR or actKR can be found in Table 1. The results show that hedKR can reduce trans-1-decalone in vitro. However, while the Km values are similar, the kcat of hedKR is ~23 times less than that of actKR. Similarly, when we assayed hedKR activity from the reverse direction, through the oxidation of S- and R-tetralol, we found no activity for R-tetralol, and the kcat/Km of hedKR for S-tetralol is 20-fold less than that of actKR (Table 2). The above in vitro assay result is consistent with sequence-based prediction, that the change of the “NNAG” motif of actKR to “NSAG” of hedKR reflects a change in active site geometry, resulting in the decrease in enzymatic activity.
Table 1.
Kinetic Parameters for the Reduction of trans-1-Decalone by Wild Type and Mutants of Type II Polyketide Ketoreductases (ActKR and HedKR)
| kcat (s−1) | Km (mM) | kcat/Km (s−1 mM−1) |
|
|---|---|---|---|
| ActKR | |||
| WT | 2.55 ± 0.121 | 0.790 ± 0.0850 | 3.23 ± 0.318 |
| P94L | 0.720 ± 0.193 | 0.709 ± 0.367 | 1.02 ± 0.592 |
| HedKR | |||
| WT | 0.109 ± 0.00758 | 0.591 ± 0.147 | 0.184 ± 0.0475 |
| N92L | ND | ||
| N92P | ND | ||
ND: No detectable activity
During hedamycin biosynthesis, hedKR reduces a 24-carbon substrate that is primed with a six-carbon starter unit (Figure 2B). To evaluate the substrate specificity and regiospecificity of hedKR, we sought to assess whether hedKR can reduce the 16-carbon octaketide intermediate synthesized by the actinorhodin minimal PKS (act min PKS), and if the ketoreduction still occurs at the C9-carbonyl group. To determine whether the C9-specificity is promoted by hedKR itself, we conducted in vitro PKS reconstitution assays, in which purified actKS/CLF, S. coelicolor MAT, and holo-actACP (C17S) were incubated with or without hedKR or actKR. Here, holo-actACP (C17S) is used instead of the wild type actACP due to the tendency of wild type actACP to dimerize via disulfide bond formation that interferes with experiments. The reconstituted minimal PKS will produce the 16-carbon octaketide chain, but it was unknown whether there would be sufficient effective interactions between hedKR, actACP and actKS/CLF for substrate transfer and ketoreduction to occur. HPLC analysis of the reconstitution products demonstrates that act min PKS + hedKR produces the 16-carbon, C9-reduced mutactin, albeit at a much lower level than in the presence of actKR, presumably due to weaker interactions between hedKR, actACP (C17S), and actKS/CLF (Figure 4). Based on the areas under the peaks corresponding to mutactin, in the same amount of incubation time, hedKR produces ~fivefold less mutactin than actKR does. The reconstitution and HPLC results are consistent with our in vitro assay result of trans-1-decalone, as well as the heterologous expression results by Das and Khosla, in which hedKR is functionally interchangeable with actKR (22). It should be noted that the previous work was based on in vivo protein expression and product characterization; the present study indicates hedKR may not be as active as actKR in vitro, which is consistent with sequence analysis of the SDR “NSAG” motif. Nevertheless, the assays do affirm that hedKR is functional in vitro and is capable of regiospecific C9-ketoreduction of a 16-carbon polyketide chain. The above result supports that the C9-regiospecificity of type II polyketide KR is not closely related to the number of carbons (referred to as “chain length” throughout the text) of the incoming polyketide substrate.
Figure 4.

HPLC analysis of products from in vitro reconstitution assays, demonstrating that hedKR is able to reduce an octaketide intermediate (produced by the act min PKS) at the C9-position to form mutactin (4). HedKR mutants N92L and N92P produce more mutactin, relative to the amount of SEK4 and SEK4b produced, than WT.
Inhibition Kinetic Assays Suggest that Quercetin, but not Emodin, is an Inhibitor of HedKR
We previously conducted extensive inhibition kinetic assays of emodin, which serves as a competitive inhibitor of actKR. Emodin contains three conjugated aromatic rings. We further solved the ternary structures of WT or P94L actKR bound to NADPH and the inhibitor emodin (17). Analyses of these structures revealed that emodin-binding changes the protein conformations of actKR (17). Guided by the insights drawn from the critical structural analysis, we sought to identify an effective inhibitor of hedKR by screening the inhibitory effect of four plant secondary metabolites on the reduction of trans-1-decalone (Figure 5A). Of particular interest were emodin and compounds from green tea extract such as epigallocatechin gallate (EGCG), which has been shown to inhibit the fatty acid KR in bacteria (36). The sequence, structural and functional similarities between fatty acid synthase (FAS) and type II PKS KRs (5) suggests that green tea extract compounds may also act as inhibitors of hedKR. Single-point inhibition kinetic studies showed that the flavonoid quercetin (Figure 5A) displayed the most potent inhibitory effects of the four compounds tested. Further in-depth assays were conducted to identify the mode of inhibition of quercetin (Figure 5B). The compound acts as a competitive inhibitor of trans-1-decalone, with a Ki of approximately 114 μM. Significantly, actKR is inhibited by emodin but not quercetin, while the inhibitory effect of quercetin is much higher than that of emodin toward hedKR. Therefore, although actKR and hedKR were predicted to have similar structures, both sequence alignment and inhibition kinetics predict that the substrate/inhibitor-binding pockets will be different.
Figure 5.

Inhibition of hedKR-catalyzed trans-1-decalone reduction. (A) The four plant secondary metabolites tested as potential inhibitors of hedKR. (B) Inhibition kinetic assays demonstrate that quercetin (22) is a competitive inhibitor of trans-1-decalone reduction.
HedKR Crystallizes under Markedly Different Conditions than ActKR
To establish a structural basis for the observed substrate/inhibitor- and regiospecificities of hedKR, we sought to solve the hedKR crystal structure. Our initial attempts to crystallize hedKR based on conditions that led to effective actKR crystallization were unsuccessful (11, 17, 23). To help stabilize any possible protein flexibility, hedKR was incubated with the cofactor NADPH. After extensive screening, diffraction-quality hedKR crystals were produced in a condition completely different from that of actKR, with different crystal morphology. The differences in growth conditions and crystal morphology between hedKR and actKR suggest that the structural features of the two enzymes, such as protein conformation, may differ as well.
HedKR Overall Architecture is Similar to that of ActKR, but the Substrate Pockets Differ
The 2.4-Å crystal structure of wild type hedKR allows for detailed analysis and comparisons to the previously solved actKR structures. Although hedKR crystallizes in a different space group than actKR, the asymmetric unit similarly consists of two monomers, and the natural hedKR tetramer can be generated across the two-fold symmetry axis (Figure 6A). Each monomer consists of the characteristic Rossmann fold (37) for binding nucleotide cofactors. There is clear electron density for NADPH in each monomer of the crystal structure. An overlay of the two hedKR monomers shows that the catalytic tetrad residues (N112, S142, Y155, and K159) and cofactors adopt the same position and conformation between monomers. A significant difference is that Met192 in monomer B is more extended than in monomer A. This residue lies opposite Asn92 on the other side of the substrate pocket cleft, resulting in an effective closed conformation for monomer B versus the open conformation of monomer A, (Figure 6B).
Figure 6.

Structural analysis of hedKR. (A) The hedKR crystallographic tetramer is formed by rotation of monomers A (teal) and B (purple) about the two-fold symmetry axis shown as a dashed line. (B) Overlay of hedKR monomers A (teal) and B (purple). The zoomed view displays the NADPH cofactor (ball and stick model) and important active site residues (sticks). M192 is more extended in monomer B, possibly interacting with N92 through hydrogen-bonding. (C) Comparison of WT actKR (orange) and WT hedKR (purple) substrate pockets. Portions of the NADPH cofactors are hidden by residues, but outlined in red here for viewing clarity. The hedKR pocket is more constricted and narrower than the actKR pocket, with F215 forming a ridge that juts into the pocket. The F215 sidechain is overlaid on the actKR surface for comparison. (D) Overlay of monomers A of P94L actKR-emodin (gold) and WT hedKR (teal). Key residues are shown as stick models. HedKR Y187 is shown piercing through the emodin molecules, demonstrating how this residue differentiates the hedKR active site from that of actKR. (E) Overlay of WT actKR (orange) and WT hedKR (purple), displaying residues across from the α6-α7 loop region that differ between the two KRs.
The overall hedKR structure is very similar to that of the previously solved actKR structures, namely WT-NADPH, emodin-bound WT-NADPH, P94L-NADPH, and emodin-bound P94L-NADPH (11, 17, 23). The RMS deviations between the hedKR structural dimer and these actKR structures are, respectively, 0.73, 0.75, 0.77, and 0.76 Å. As with the actKR structures, the α6-α7 loop region in the hedKR structure is disordered and displays weak 2Fo-Fc map electron density, although this region in monomer A is better defined than in monomer B. A comparison of the catalytic tetrad residues and cofactors between each hedKR and actKR structure shows that the positions and conformations of these components are similar, suggesting that the observed differences in in vitro enzyme activity between hedKR and actKR are not the result of catalytic residue misalignment, but likely stem from differences in substrate-binding due to different substrate pocket shapes.
Consistent with the above analysis, although the overall structures are similar, we found many structural features in the substrate pockets that differentiate hedKR from actKR (Figure 6C). Across from the α6-α7 loop region in hedKR are three residues with longer sidechains than the corresponding residues in actKR: Glu96, Met150 and Tyr151 (actKR residues Ala98, Val152 and His153) (Figure 6D), which change the substrate pocket shape. Further, hedKR residues Tyr187 and Phe215 point into the substrate pocket, resulting in a more constricted pocket size in actKR. This narrowing of the substrate pocket is especially pronounced when hedKR monomer B is compared to the corresponding WT-NADPH actKR monomer, where the phenyl ring of Phe215 protrudes into the substrate pocket, which can potentially hinder access to the active site from the opening of the enzyme cleft. This difference is readily apparent when the hedKR structure is aligned with P94L-NADPH-emodin actKR, where the actKR Phe189 sidechain points away from the substrate-binding pocket, resulting in a wider pocket for actKR. Therefore, the difference in substrate pocket shape may likely translate to differences between actKR and hedKR in the binding motifs of substrates, inhibitors and transition states. The above analysis corroborates the differences in kcat and kcat/Km between actKR and hedKR for the in vitro reduction of trans-1-decalone and oxidation of R- and S-tetralols (Tables 1-2), as well as differences in inhibitor binding (detailed below).
The conformation of hedKR Met192 versus actKR Met194 differs as well. Compared to both the binary and ternary WT actKR structures, Met192 of hedKR monomer A has a more extended conformation than the corresponding Met194 of actKR. Met192 in hedKR monomer B, however, has a very similar conformation to Met194 in WT-NADPH-emodin actKR monomer B. As previously reported, monomer B of the ternary WT actKR structure has a distinctly closed conformation relative to monomer A (11, 17, 23). The observation that hedKR monomer B also has a closed conformation suggests that in hedKR the methionine opposite the “NGG motif” is important for the open/closed conformation by interacting with Asn92 (Figure 6B). This notion is also supported by a comparison of the hedKR structure with P94L-NADPH actKR: in both structures, monomer A is open and monomer B is closed, while the corresponding methionine residues in both structures have the same side-chain conformation.
Another important difference between hedKR and actKR is the presence of Asn92, which juts into the active site nearly the same distance as Leu94 in the P94L actKR structures does. Based on this observation, it is likely that the stereospecificity of WT hedKR will correspond to that of P94L actKR, as opposed to WT actKR. The structural comparisons between hedKR and actKR indicate that despite a similar overall architecture, the substrate-binding pockets of actKR and hedKR adopt distinct shapes, which offers a strong structural basis to interpret the observed differences in in vitro activity, substrate/inhibitor specificity, and stereospecificity.
Molecular Basis of Inhibitor Specificity between HedKR and ActKR
To understand how different inhibitors can be specifically recognized by actKR versus hedKR, we simulated quercetin binding at the hedKR active site by using the docking program GOLD (38). With no constraint bias, we obtained highly consistent docking solutions that have similar protein conformations, inhibitor positions, and orientations within the substrate pocket (Figure 7A). One of the quercetin hydroxyl groups is oriented within hydrogen-bonding distance of the side chain hydroxyl groups of active site Tyr155 and Ser142, which form the catalytic oxyanion hole. The hydroxyl-bearing carbon of quercetin is also within hydride-transfer distance of C4 of the nicotinamide ring. When quercetin was docked to the P94L-NADPH actKR structure, the docking solutions were not as consistent as within the WT hedKR structure. Reciprocally, when we tried docking the actKR inhibitor emodin into the WT hedKR structure, we also could not obtain a solution whose emodin-binding motif is consistent with those from the reported actKR-NADPH-emodin crystal structures. Moreover, the emodin molecules from the WT and P94L actKR structures did not overlay with any of the top-ten hedKR-emodin docking solutions at all (Figure 7B). In essence, the inhibitors emodin and quercetin serves as “molecular probes”: the wider, tricyclic emodin can inhibit the wider pocket of actKR, but can barely bind the narrower pocket of hedKR, while the reverse is true for hedKR. The above docking results support that the observed differences in inhibitor binding, substrate specificity, and enzymatic activity between hedKR and actKR are reflected by pocket size and shape differences.
Figure 7.

Stereo views of hedKR in silico docking analyses. HedKR monomer A is portrayed in teal in each panel. (A) Quercetin (blue) docking. A quercetin hydroxyl group is within hydrogen-bonding distance of the catalytic S142 and Y155 sidechains, as well as within hydride-transfer distance from C4 of the NADPH nicotinamide moiety. The residues modeled as sticks are targets for site-directed mutagenesis studies to determine long-ranged substrate pocket motifs that affect hedKR stereospecificity. (B) Three examples of emodin docked in hedKR (green, pink and yellow), overlaid with the emodin molecules from the previously solved P94L actKR cocrystal structure (white). The hedKR-emodin docking solutions were not consistent and do not overlap with the emodins in the actKR crystal structures. (C) S-tetralol (green) and R-tetralol (magenta) docked within the hedKR active site. The rigid benzene ring of R-tetralol is sterically hindered by the N92 sidechain that juts into the pocket.
Based on the proximity to the docked quercetin, N92, G186, Y187, V196, and L256 are predicted to define the binding pocket and likely affect the stereospecificity of hedKR (Figure 7C). Therefore, the hedKR crystal structure and docking simulations lead to a structure-based hypothesis on KR stereospecificity that is consistent with the one observed with actKR, that potential steric interference by the active site residues lead to an S-dominant stereospecificity in type II polyketide KRs (11, 17, 23).
Wild Type HedKR Displays High Stereospecificity in Comparison to Wild Type ActKR
To assess the stereospecificity of hedKR, we utilize the ability of a type II KR to catalyze the reverse reaction, namely the oxidation of S- and R-tetralol (39). Previously, we showed that the “PGG” motif in actKR is a key determinant of stereospecificity. While wild type actKR has a threefold preference (kcat/Km) for S- versus R-tetralol, P94L actKR is highly specific for only S-tetralol (Table 2). Because the “PGG” motif of actKR becomes 92-NGG-94 in hedKR, with Asn92 jutting into the active site and creating a similar pocket shape as that of P94L actKR, we predicted that wild type hedKR would have similar stereospecificity to that of P94L actKR, namely S-dominant. Supporting this hypothesis, when assayed separately with S- and R-tetralol, hedKR does displays a dominant stereospecificity for S-tetralol (Table 2). Hence, steric interference is likely to occur in WT hedKR just as in P94L actKR and suggests that the “XGG” motif may be a general determinant of stereospecificity for most type II polyketide KRs.
Analysis of Mutant HedKR Stereospecificity
To determine whether N92 in the “NGG” motif is the most dominant factor that determines the stereospecificity of hedKR, two single mutants were prepared: N92L and N92P hedKR. The rationale for the N92L mutant is to mimic P94L actKR, whereas the N92P mutant was constructed to mimic WT actKR and, in theory, would display the same threefold preference for S- over R-tetralol. The hedKR mutants were subjected to the in vitro assay as before (Tables 1-2). Surprisingly, both mutants displayed very low levels of activity in the presence of R-tetralol. Moreover, based on the catalytic specificity constant, kcat/Km, N92L is about 18 times more active toward S-tetralol than N92P, while both N92L and N92P are much more active than the WT hedKR. It is likely that in N92L a favorable hydrophobic interaction between the Leu side chain and the S-tetralol cyclohexane ring may account for the higher activity; in comparison, the Asn residue in WT hedKR cannot participate in such non-polar interactions, resulting in an elevated Km and hence low kcat/Km. Alternatively, the N92L substrate pocket may adopt a different shape than the WT pocket, perhaps due to non-polar interactions between Leu92 and M192, resulting in increased oxidative activity toward S-tetralol.
The N92L and N92P mutants of hedKR were also assayed through in vitro PKS reconstitution (Figure 4). Based on the HPLC chromatogram peak area corresponding to mutactin, both mutants actually produced more mutactin, relative to the SEK4 and SEK4b peak areas, than WT hedKR. Specifically, N92L produced almost as much mutactin as SEK4 and SEK4b combined. These results correlate with the tetralol kinetic assay results, suggesting that the presence of a hydrophobic residue at position 92 (such as proline or leucine), results in favorable non-polar interactions between residue 92 and M192. In part, these interactions could contribute to the forces that stabilize the closed conformation of a hedKR monomer in order to catalyze polyketide ketoreduction.
The N92P mutant of hedKR was constructed to mimic WT actKR, which displays measurable activity toward R-tetralol. The lack of activity toward R-tetralol by the N92P hedKR mutant may be due to the rate detection limit of the assay, but this result also implies that the “PGG” motif is not a sole determinant of stereospecificity in hedKR. In light of the structural differences between hedKR and actKR, particularly the distinct substrate pocket shapes, it is likely that there are long-ranged effects responsible for guiding the stereospecificity of hedKR. Namely, stereospecificity may be determined by the shape of the enzyme active site pocket as a whole rather than by specific residues. It has been shown that the tropinone reductases, TR-I and TR-II, have the same overall fold, but opposite reaction stereospecificities (40). Nakajima et al. attributed the difference to distinct amino acids lining the substrate pockets of the two enzymes (40). Moreover, it has been suggested that residue side chains across the substrate cavity of 3α-hydroxysteroid dehydrogenase determine the orientations of substrates and inhibitors (41). Therefore, many residues (as opposed to a single motif) that define the overall shape of the hedKR active site pocket and interact with substrates may collectively affect the stereospecificity of a KR. Nevertheless, due to high sequence conservation of pocket residues in type II polyketide KRs, there are likely long-ranged motifs that are important for stereospecificity. Future site-directed mutagenesis studies that focus on such residues can shed light on the molecular basis of long-ranged stereospecificity determinants of hedKR.
Biological Significance: C9 Regiospecificity Versus Substrate Chain Length
The number of carbons in a given type II polyketide, referred to as “chain length,” is closely associated with the specificity of the KS/CLF of the corresponding type II PKS (42). This hypothesis was strongly supported by mutational studies of actCLF residues, in which single mutations completely changed the product chain length, thus validating the dominant influence of KS/CLF (42). However, in the hed type II PKS, both the hedKR and ARO/CYC (HedE) are also important for chain length determination, as concluded from in vivo expression experiments of hedamycin type II PKS in a heterologous host. For example, the expression of hedKS/CLF, the bifunctional HedE (ACP + ARO/CYC), and hedKR resulted in four products (Figure 2B): the full-length, 10 (24 carbons), 11 and 12 (22 carbons), and 13 (8 carbons). In the absence of hedKR, the expression of hedKS/CLF and HedE resulted in five unreduced products with varying chain lengths: 18 (24 carbons), 17 (22 carbons), 16 (10 carbons), 15 (8 carbons), and 14 (6 carbons). Similar results have been reported for the actinorhodin PKS, that in addition to the full-length product (16 carbons), the act PKS also generates truncated products 1-3 (12 carbons) (Figure 2A) (43). In our previous work, we found that the presence of a truncated product is closely associated with the presence of an active actKR (23), and it was hypothesized that the truncated products 2 and 3 result from competition of actKR with actKS/CLF, causing premature reduction of a much shorter chain. In this work, the diverse chain lengths observed in hed PKS offer an excellent opportunity to examine the above hypothesis. The in vivo expression experiments of hed PKS can be explained by two possible reasons: (1) that the pocket size of hedKR is a partial determinant of the polyketide chain lengths, or (2) that similar to act PKS, the chain length is a result of competition between chain elongation (by hedKS/CLF) and ketoreduction (by hedKR). The hedKR crystal structure and enzyme kinetics help distinguish these two possibilities. In this work, we showed that hedKR can reduce both 16- and 24-carbon polyketide substrates. In concert with this result, docking simulations consistently docked the C9-carbonyl group of both 16- or 24-carbon, mono-cyclized polyketides into the hedKR active site. Therefore, while the substrate pockets of hedKR and actKR are different in shape (reflected by their different inhibitor specificities, Figure 6B), the KR active site itself is not specific to the chain length of an incoming substrate. Consequently, the hedKR crystal structure and docking simulations of PPT-tethered polyketides (either 16 or 24 carbons, Figure 8A-B) confirm that the C9-specificity arises from the prerequisite that the first ring is cyclized between C7-C12. Supporting this observation, and consistent with the in vivo expression results by Das and Khosla, docking simulations consistently docked carbon 9 of a C7-C12 cyclized (Figure 8A-B), or carbon 7 of a C5-C10 cyclized polyketide (Figure 8C) to the active site of hedKR. Combining the above together, the hedKR structural and functional studies confirmed that the C9-specificity requires the constraint of a pre-formed ring, and that the observed truncated products reflect a competition between chain elongation (by KS/CLF), first-ring cyclization (by either KS/CLF or KR), and ketoreduction (by KR). Note, also, that the chain length is required to be at least 10 carbons or longer to reach from the surface arginine patch (the ACP-PPT docking site) into the hedKR active site. Therefore, the presence of the 8-carbon, mono-cyclized 13 requires that the ring is pre-formed, thus freeing the chain from the ACP-PPT tether, before reaching the hedKR active site (Figure 7D). Finally, mutational analyses of hedKR confirmed the importance of the “XGG” motif for stereospecificity, but also suggested that more long-ranged motifs may be involved. The above results pave a foundation for future work toward stereospecific engineering of type II PKSs to generate new polyketides.
Figure 8.

Stereo views of various PPT-linked polyketides docked to hedKR, with monomer A portrayed in teal in each panel. (A) C7-C12 cyclized, 16-carbon polyketide (blue); (B) the putative C7-C12 cyclized, 24-carbon hedKR polyketide substrate (pink); and (C) C5-C10 cyclized, 22-carbon polyketide (yellow). These in silico docking analyses demonstrate that hedKR can accommodate the polyketide substrates portrayed in (A)-(C). (D) None of the 8-carbon polyketide (green) docking solutions placed the PPT phosphate within the “arginine patch,” which is the proposed docking site for ACP. These results suggest that the reduced tetraketide compound (13) is formed after release of the thioester-bound polyketide from holo-ACP, spontaneous pyrone ring cyclization, and C7-reduction of the molecule by hedKR.
Acknowledgments
This work is supported by the Pew Foundation and National Institute of General Medicinal Sciences (NIGMS R01GM076330).
Abbreviations
- KR
ketoreductase
- FabG
β-Ketoacyl (Acyl Carrier Protein) Reductase
- act
actinorhodin
- hed
hedamycin
- PKS
polyketide synthase
- NADP
nicotinamide adenine dinucleotide phosphate
- NADPH
reduced nicotinamide adenine dinucleotide phosphate
- SDR
short-chain dehydrogenase/reductase
- ACP
acyl carrier protein
Footnotes
The atomic coordinates have been deposited in the Protein Data Bank (accession code 3SJU).
REFERENCES
- 1.Staunton J, Weissman KJ. Polyketide biosynthesis: a millennium review. Nat Prod Rep. 2001;18:380–416. doi: 10.1039/a909079g. [DOI] [PubMed] [Google Scholar]
- 2.Malpartida F, Hopwood DA. Molecular cloning of the whole biosynthetic pathway of a Streptomyces antibiotic and its expression in a heterologous host. Nature. 1984;309:462–464. doi: 10.1038/309462a0. [DOI] [PubMed] [Google Scholar]
- 3.Otten SL, Stutzman-Engwall KJ, Hutchinson CR. Cloning and expression of daunorubicin biosynthesis genes from Streptomyces peucetius and S. peucetius subsp. caesius. J Bacteriol. 1990;172:3427–3434. doi: 10.1128/jb.172.6.3427-3434.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Manzoni M, Rollini M. Biosynthesis and biotechnological production of statins by filamentous fungi and application of these cholesterol-lowering drugs. Appl Microbiol Biotechnol. 2002;58:555–564. doi: 10.1007/s00253-002-0932-9. [DOI] [PubMed] [Google Scholar]
- 5.Hopwood DA. Genetic Contributions to Understanding Polyketide Synthases. Chem Rev. 1997;97:2465–2498. doi: 10.1021/cr960034i. [DOI] [PubMed] [Google Scholar]
- 6.Shen B. Polyketide biosynthesis beyond the type I, II and III polyketide synthase paradigms. Curr Opin Chem Biol. 2003;7:285–295. doi: 10.1016/s1367-5931(03)00020-6. [DOI] [PubMed] [Google Scholar]
- 7.Motamedi H, Hutchinson CR. Cloning and heterologous expression of a gene cluster for the biosynthesis of tetracenomycin C, the anthracycline antitumor antibiotic of Streptomyces glaucescens. Proc Natl Acad Sci U S A. 1987;84:4445–4449. doi: 10.1073/pnas.84.13.4445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.McDaniel R, Ebert-Khosla S, Fu H, Hopwood DA, Khosla C. Engineered biosynthesis of novel polyketides: influence of a downstream enzyme on the catalytic specificity of a minimal aromatic polyketide synthase. Proc Natl Acad Sci U S A. 1994;91:11542–11546. doi: 10.1073/pnas.91.24.11542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Das A, Khosla C. Biosynthesis of aromatic polyketides in bacteria. Acc Chem Res. 2009;42:631–639. doi: 10.1021/ar8002249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hadfield AT, Limpkin C, Teartasin W, Simpson TJ, Crosby J, Crump MP. The crystal structure of the actIII actinorhodin polyketide reductase: proposed mechanism for ACP and polyketide binding. Structure. 2004;12:1865–1875. doi: 10.1016/j.str.2004.08.002. [DOI] [PubMed] [Google Scholar]
- 11.Korman TP, Hill JA, Vu TN, Tsai SC. Structural analysis of actinorhodin polyketide ketoreductase: cofactor binding and substrate specificity. Biochemistry. 2004;43:14529–14538. doi: 10.1021/bi048133a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.O’Hare HM, Baerga-Ortiz A, Popovic B, Spencer JB, Leadlay PF. High-throughput mutagenesis to evaluate models of stereochemical control in ketoreductase domains from the erythromycin polyketide synthase. Chem Biol. 2006;13:287–296. doi: 10.1016/j.chembiol.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 13.McDaniel R, Ebert-Khosla S, Hopwood DA, Khosla C. Engineered biosynthesis of novel polyketides. Science. 1993;262:1546–1550. doi: 10.1126/science.8248802. [DOI] [PubMed] [Google Scholar]
- 14.Baerga-Ortiz A, Popovic B, Siskos AP, O’Hare HM, Spiteller D, Williams MG, Campillo N, Spencer JB, Leadlay PF. Directed mutagenesis alters the stereochemistry of catalysis by isolated ketoreductase domains from the erythromycin polyketide synthase. Chem Biol. 2006;13:277–285. doi: 10.1016/j.chembiol.2006.01.004. [DOI] [PubMed] [Google Scholar]
- 15.Siskos AP, Baerga-Ortiz A, Bali S, Stein V, Mamdani H, Spiteller D, Popovic B, Spencer JB, Staunton J, Weissman KJ, Leadlay PF. Molecular basis of Celmer’s rules: stereochemistry of catalysis by isolated ketoreductase domains from modular polyketide synthases. Chem Biol. 2005;12:1145–1153. doi: 10.1016/j.chembiol.2005.08.017. [DOI] [PubMed] [Google Scholar]
- 16.Kwan DH, Tosin M, Schlager N, Schulz F, Leadlay PF. Insights into the stereospecificity of ketoreduction in a modular polyketide synthase. Org Biomol Chem. 2011;9:2053–2056. doi: 10.1039/c1ob00022e. [DOI] [PubMed] [Google Scholar]
- 17.Korman TP, Tan YH, Wong J, Luo R, Tsai SC. Inhibition kinetics and emodin cocrystal structure of a type II polyketide ketoreductase. Biochemistry. 2008;47:1837–1847. doi: 10.1021/bi7016427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bradner WT, Heinemann B, Gourevitch A. Hedamycin, a new antitumor antibiotic. II. Biological properties. Antimicrob Agents Chemother (Bethesda) 1966;6:613–618. doi: 10.1128/AAC.6.5.613. [DOI] [PubMed] [Google Scholar]
- 19.Schmitz H, Crook KE, Jr., Bush JA. Hedamycin, a new antitumor antibiotic. I. Production, isolation, and characterization. Antimicrob Agents Chemother (Bethesda) 1966;6:606–612. doi: 10.1128/AAC.6.5.606. [DOI] [PubMed] [Google Scholar]
- 20.Hansen M, Yun S, Hurley L. Hedamycin intercalates the DNA helix and, through carbohydrate-mediated recognition in the minor groove, directs N7-alkylation of guanine in the major groove in a sequence-specific manner. Chem Biol. 1995;2:229–240. doi: 10.1016/1074-5521(95)90273-2. [DOI] [PubMed] [Google Scholar]
- 21.Bililign T, Hyun CG, Williams JS, Czisny AM, Thorson JS. The hedamycin locus implicates a novel aromatic PKS priming mechanism. Chem Biol. 2004;11:959–969. doi: 10.1016/j.chembiol.2004.04.016. [DOI] [PubMed] [Google Scholar]
- 22.Das A, Khosla C. In vivo and in vitro analysis of the hedamycin polyketide synthase. Chem Biol. 2009;16:1197–1207. doi: 10.1016/j.chembiol.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Javidpour P, Korman TP, Shakya G, Tsai SC. Structural and Biochemical Analyses of Regio- and Stereo-Specificities Observed in a Type II Polyketide Ketoreductase. Biochemistry. 2011 doi: 10.1021/bi200335f. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matharu AL, Cox RJ, Crosby J, Byrom KJ, Simpson TJ. MCAT is not required for in vitro polyketide synthesis in a minimal actinorhodin polyketide synthase from Streptomyces coelicolor. Chem Biol. 1998;5:699–711. doi: 10.1016/s1074-5521(98)90663-9. [DOI] [PubMed] [Google Scholar]
- 25.Pfeifer BA, Admiraal SJ, Gramajo H, Cane DE, Khosla C. Biosynthesis of complex polyketides in a metabolically engineered strain of E. coli. Science. 2001;291:1790–1792. doi: 10.1126/science.1058092. [DOI] [PubMed] [Google Scholar]
- 26.Kumar P, Koppisch AT, Cane DE, Khosla C. Enhancing the modularity of the modular polyketide synthases: transacylation in modular polyketide synthases catalyzed by malonyl-CoA:ACP transacylase. J Am Chem Soc. 2003;125:14307–14312. doi: 10.1021/ja037429l. [DOI] [PubMed] [Google Scholar]
- 27.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Method Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 28.The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 29.Bordoli L, Kiefer F, Arnold K, Benkert P, Battey J, Schwede T. Protein structure homology modeling using SWISS-MODEL workspace. Nat Protoc. 2009;4:1–13. doi: 10.1038/nprot.2008.197. [DOI] [PubMed] [Google Scholar]
- 30.Vagin AA, Steiner RA, Lebedev AA, Potterton L, McNicholas S, Long F, Murshudov GN. REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its use. Acta Crystallogr D Biol Crystallogr. 2004;60:2184–2195. doi: 10.1107/S0907444904023510. [DOI] [PubMed] [Google Scholar]
- 31.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 32.Laskowski RA, Rullmannn JA, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: programs for checking the quality of protein structures solved by NMR. J Biomol NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
- 33.Oppermann U, Filling C, Hult M, Shafqat N, Wu X, Lindh M, Shafqat J, Nordling E, Kallberg Y, Persson B, Jornvall H. Short-chain dehydrogenases/reductases (SDR): the 2002 update. Chem Biol Interact. 2003;143-144:247–253. doi: 10.1016/s0009-2797(02)00164-3. [DOI] [PubMed] [Google Scholar]
- 34.Filling C, Berndt KD, Benach J, Knapp S, Prozorovski T, Nordling E, Ladenstein R, Jornvall H, Oppermann U. Critical residues for structure and catalysis in short-chain dehydrogenases/reductases. J Biol Chem. 2002;277:25677–25684. doi: 10.1074/jbc.M202160200. [DOI] [PubMed] [Google Scholar]
- 35.Zhang W, Watanabe K, Wang CC, Tang Y. Heterologous biosynthesis of amidated polyketides with novel cyclization regioselectivity from oxytetracycline polyketide synthase. J Nat Prod. 2006;69:1633–1636. doi: 10.1021/np060290i. [DOI] [PubMed] [Google Scholar]
- 36.Zhang YM, Rock CO. Evaluation of epigallocatechin gallate and related plant polyphenols as inhibitors of the FabG and FabI reductases of bacterial type II fatty-acid synthase. J Biol Chem. 2004;279:30994–31001. doi: 10.1074/jbc.M403697200. [DOI] [PubMed] [Google Scholar]
- 37.Rossmann MG, Argos P. Protein folding. Annu Rev Biochem. 1981;50:497–532. doi: 10.1146/annurev.bi.50.070181.002433. [DOI] [PubMed] [Google Scholar]
- 38.Verdonk ML, Cole JC, Hartshorn MJ, Murray CW, Taylor RD. Improved protein-ligand docking using GOLD. Proteins. 2003;52:609–623. doi: 10.1002/prot.10465. [DOI] [PubMed] [Google Scholar]
- 39.Ostergaard LH, Kellenberger L, Cortes J, Roddis MP, Deacon M, Staunton J, Leadlay PF. Stereochemistry of catalysis by the ketoreductase activity in the first extension module of the erythromycin polyketide synthase. Biochemistry. 2002;41:2719–2726. doi: 10.1021/bi0117605. [DOI] [PubMed] [Google Scholar]
- 40.Nakajima K, Yamashita A, Akama H, Nakatsu T, Kato H, Hashimoto T, Oda J, Yamada Y. Crystal structures of two tropinone reductases: different reaction stereospecificities in the same protein fold. Proc Natl Acad Sci U S A. 1998;95:4876–4881. doi: 10.1073/pnas.95.9.4876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bennett MJ, Albert RH, Jez JM, Ma H, Penning TM, Lewis M. Steroid recognition and regulation of hormone action: crystal structure of testosterone and NADP+ bound to 3 alpha-hydroxysteroid/dihydrodiol dehydrogenase. Structure. 1997;5:799–812. doi: 10.1016/s0969-2126(97)00234-7. [DOI] [PubMed] [Google Scholar]
- 42.Tang Y, Tsai SC, Khosla C. Polyketide chain length control by chain length factor. J Am Chem Soc. 2003;125:12708–12709. doi: 10.1021/ja0378759. [DOI] [PubMed] [Google Scholar]
- 43.Kalaitzis JA, Moore BS. Heterologous biosynthesis of truncated hexaketides derived from the actinorhodin polyketide synthase. J Nat Prod. 2004;67:1419–1422. doi: 10.1021/np0499564. [DOI] [PubMed] [Google Scholar]