

Abstract
Sam68 (Src-associated protein in mitosis 68 kDa) is a multifunctional protein, known to govern cellular signal transduction, transcription, RNA metabolism, proliferation, apoptosis and HIV-1 replication. Although intrinsic mechanisms that modulate Sam68 function are beginning to emerge, the regulatory events contributing to its expression remain elusive. We previously reported that heat shock protein-22 (Hsp22) antagonizes Sam68 function in rev-response element (RRE)-mediated gene expression. We now demonstrate that Sam68 levels correlate inversely with Hsp22 in a variety of cells, including U87, Jurkat, 293T and U-937. In U87 glioblastoma cells, which contained high levels of Hsp22 than other cell lines tested, Hsp22 knockdown dramatically increased both Sam68 mRNA and protein, altered cellular morphology and enhanced cell proliferation. This heightened proliferation was associated with a sharp decrease in G0/G1 and a corresponding increase in S and G2/M phases in exponentially growing cultures. The increased S phase population in turn correlated with enhanced expression of cell cycle regulatory proteins such as cyclin E, cyclin A, ribonucleotide reductase (RNR) and proliferating cell nuclear antigen (PCNA), which are required for the transition of cells from G1 to S phase. Collectively, our results demonstrate for the first time that Hsp22 regulates Sam68 expression and the ratio of Sam68 to Hsp22 may determine the proliferative potential of glioblastoma cells.
Keywords: Hsp22 knockdown, Sam68, Cell morphology, Proliferation, Cell cycle analysis
Introduction
Sam68 (Src-associated in mitosis 68 kDa), a target for tyrosine kinase c-Src during mitosis, belongs to the signal transduction and activation of RNA (STAR) family of RNA-binding proteins, which are critical for normal physiology (Richard, 2010). Sam68 is involved in a wide range of cellular processes including signal transduction, transcription, RNA metabolism, cell cycle progression and apoptosis (Bielli et al., 2011). In addition, we reported that Sam68 can functionally substitute and/or synergize with HIV-1 Rev in RRE-mediated gene expression and virus production (Reddy et al., 1999; Suhasini and Reddy, 2009), confirming its pleiotropic nature.
Sam68 controls cell cycle progression and proliferation by regulating alternate splicing pathways and is modulated by post-translational modifications (Elliott and Rajan, 2010). Pre-mRNA splicing targets of Sam68 include: i) transmembrane glycoprotein CD44; ii) cyclin D1, a G1 phase cyclin; and iii) an anti-apoptotic protein, Bcl-x (Matter et al., 2002; Paronetto et al., 2007, 2010). It was reportedly upregulated in breast cancer cells (Song et al., 2010), and in human prostate carcinoma, which contributed to proliferation and survival of cancer cells (Busa et al., 2007). Sam68 has been shown to co-activate androgen receptor (AR) transcriptional activity independent of its RNA binding, while ligand-activated AR repressed Sam68-dependent splicing of CD44 (Clark et al., 2008), suggesting cross-talk between Sam68 and AR in regulating the genes involved in proliferation and survival of prostate cancer cells. Although it is evident from these observations that changes in Sam68 levels can both stimulate and alter development and progression of a variety of diseases, it remains to be established how Sam68 expression is regulated.
In an effort to understand the mechanism by which Sam68 regulates HIV-1 Rev function, we discovered that Hsp22 interacts with Sam68 and inhibits Sam68/RRE-mediated gene expression (Badri et al., 2006). Hsp22 displays chaperone-like activity, and has been implicated in cell proliferation, apoptosis and carcinogenesis, exerting either pro- or anti-apoptotic effects depending on the cell (Shemetov et al., 2008). We tested Jurkat, U937, 293T, HeLa, U87 and Cos-1 cell lines and observed high levels of Hsp22 mRNA in U87 glioblastoma cells (Badri et al., 2006). Diminished viral production in U87 cells has been attributed to low levels of Sam68 (Li et al., 2002), which is required for HIV-1 Rev function (Modem et al., 2005). Since Hsp22 inhibits Sam68 function and U87 cells express high levels of Hsp22 (Badri et al., 2006), we reasoned that Hsp22 knockdown could restore Sam68 function and stimulate viral production. To test this possibility, we generated stable sub-lines of U87 glioblastoma cells with Hsp22 knockdown and found increased expression of both Sam68 mRNA and protein along with dramatically increased cell proliferation, which in turn was associated with an increase in S phase and a corresponding decrease in G0/G1 phase cells in exponentially growing cultures. This finding was corroborated by the increased expression of the regulatory proteins necessary for transition of cells from G1 to S phase and a marked decrease in G1-specific cyclin D1. To the best of our knowledge, this is the first direct evidence implicating Hsp22 in Sam68 regulation, which may determine the proliferative status of not only U87 glioblastoma cells but other lines as well.
Materials and Methods
Plasmids
Stable Hsp22 knockdown U87 cells were generated using the SuppressorNeo-IMG-800 RNAi expression vector (Imgenex, San Diego, CA). Hsp22 primers (forward: 5′-TCGAGGTGTGTGTGAATGTGCACAGAGTACTGTGTGCACATTCACACACACTTTT T-3′ and reverse: 5′-CTAGAAAAAGTGTGTGTGAATGTGCACACAGTACTCTGTGCAC ATTCACACACACC-3′) were annealed and cloned into the Sal I and Xba I enzyme sites of pIMG-800 to generate pHsp22-RNAi.
Cells, transfections, and western analysis
All cell lines were obtained from ATCC. U87MG glioblastoma and 293T cell lines were maintained in DMEM, whereas U937 and Jurkat cells were maintained in RPMI supplemented with 10% FBS. U87 cells were transfected with 1 to 3 μg pHsp22-RNAi vector or control vector pIMG-800, using Fugene HD (Roche) reagent. To create stable Hsp22 knockdown cell lines, cells were selected by G418 (Gibco). Knockdown was assessed in G418-resistant colonies by Western blot using anti-Hsp22 antibodies. Similarly, cell extracts were analyzed by western blot analysis with antibodies against the target protein(s) using an enhanced chemi-luminescence reagent (Michigan Diagnostics).
Sam68 and Hsp22 Real-Time RT-PCR
Total RNA isolated using a PARIS RNA isolation kit (Ambion) was reverse-transcribed with random hexamer primers using a Transcriptor First Strand cDNA synthesis kit (Roche). qRT-PCR was performed using TaqMan Gene Expression Assay kit for Sam68 (Assay ID: Hs00173141_m1), Hsp22 (Hs00205056 _m1), and 18S (Hs03928985 _g1) from Applied Biosystems with an Applied Biosystems 7500 Real-Time PCR system. Each reaction was studied in triplicate and consisted of 1/10th volume of RT product. Thermal cycling conditions were 95°C for 10 min, followed by 40 cycles at 95°C for 15 sec, and 60°C for 1 min. Relative expression of Sam68 and Hsp22 was calculated by normalizing to 18S expression using the 2(−ΔΔCt) method.
Cell morphology and proliferation
Cells were grown on chamber slides and fixed with 4% paraformaldehyde, then examined under a Zeiss Axioplan 2 microscope. To assess cell proliferation, 1×105 cells were seeded in 12-well culture plates containing DMEM supplemented with 10% FBS. Over time (24-, 48- and 72-hr intervals), cells were trypsinized and mixed gently with trypan blue solution (Sigma). Cells were counted in triplicate using a hemocytometer. Dead cells were identified by their trypan blue staining. Cell cycle progression of propidium iodide-stained cells was performed using FACS Calibur (BD Biosciences) (Busa et al., 2007) and data were analyzed by CELL Quest software (BD Biosciences).
Results
Hsp22 regulates Sam68 expression
Since Sam68 plays a critical role in HIV-1 production (Modem et al., 2005) and Hsp22 suppresses Sam68 function (Badri et al., 2006), we examined the levels of these proteins in cells known to support (Jurkat, 293T and U-937) and restrict (U87) HIV-1 production. Consistent with its significant role in HIV-1 production, we observed high levels of Sam68 protein in Jurkat, 293T and U-937 cells. In contrast, Hsp22 was barely detectable in these cells, when compared to U87 cells (Fig. 1A), suggesting that Hsp22 might suppress Sam68 expression and function, and inhibit HIV-1 production. To further investigate the effect of Hsp22 knockdown on Sam68 regulation, we employed RNAi strategy to generate stable Hsp22 knockdown sub-lines of U87 cells (HKU-1, HKU-2, and HKU-3) and found that all three had undetectable levels of Hsp22 protein compared to vector control (VC) sub-line. Interestingly, a dramatic increase of Sam68 protein was detected in all three HKU cells as compared to VC sub-line (Fig. 1B). To investigate whether the increase in Sam68 protein was due to transcriptional or translational regulation, we quantified Sam68 mRNA and found that it was 3- to 6-fold higher, while Hsp22 mRNA was 3- to 8-fold lower (Fig. 1C) in HKU cells. This suggests that knocking down Hsp22 mRNA reduced expression of the protein, therefore implying that Hsp22 knockdown increased both Sam68 mRNA and protein. It is evident from these observations that in addition to influencing Sam68 function through direct interaction (Badri et al., 2006), Hsp22 may also regulate Sam68 expression in U87 cells.
Figure 1. Hsp22 knockdown upregulates Sam68 expression.
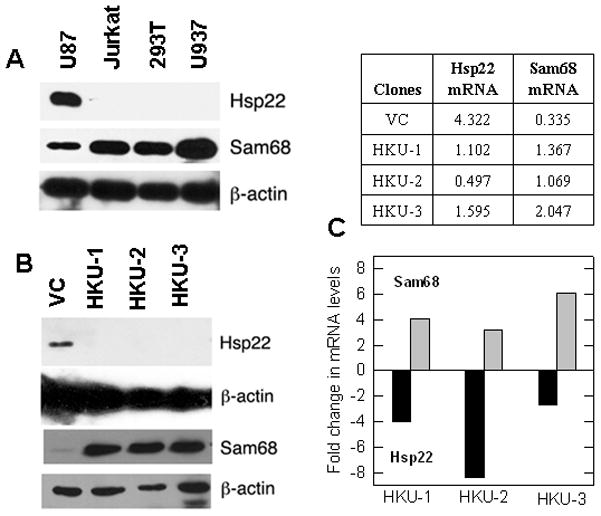
(A) U87 cells express high levels of Hsp22. Cell extracts (10 μg) from various cells were analyzed for Hsp22, Sam68 and actin expression by Western analysis. (B) Stable Hsp22 knockdown U87 cells express high levels of Sam68. Expression of Hsp22 and Sam68 in the lysates (10 μg and 5 μg) of HKU cells was assessed by Western analysis. VC: vector control; HKU-1, HKU-2 and HKU-3: Hsp22 knockdown U87 cells. (C) qRT-PCR of Sam68 and Hsp22 mRNA: Quantitative real time PCR on total RNA was performed. Table shows arbitrary units of individual mRNAs. All values are the average of three qRT-PCR reactions, which varied by less than 5%. Bar graph shows fold change in mRNA levels in HKU clones compared to vector controls.
Hsp22 knockdown alters U87 cell morphology and stimulates proliferation
As Hsp22 knockdown became apparent, the phenotypic appearance of HKU cells was distinct from the characteristic neuronal morphology of the VC cells, i.e., stellate with long extensions from the cell body. All HKU sub-lines exhibited epithelial morphology and lacked noticeable extensions. Moreover, unlike control cells, which were large and adhered firmly to the culture dish, the HKU cells were small and loosely attached to the dish, forming clusters resembling cobblestones (Fig. 2A). We also observed that HKU cells became confluent much sooner than the VC cells, suggesting a much higher rate of proliferation. They multiplied 3- to 4-fold faster than the vector control U87 cells (Fig. 1B), resulting in a change in the doubling time from ~68 hrs in VC cells to ~20 hrs in HKU cells (Fig. 2B), which correlates with a corresponding increase in Sam68 levels (Fig. 1B).
Figure 2. Knockdown of Hsp22 alters the morphology and proliferation rate of U87 cells.
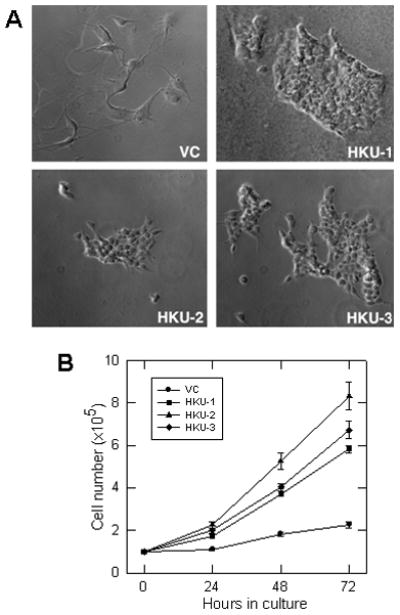
(A) Morphological changes in Hsp22 knockdown clones. Cells were grown on chamber slides, fixed with paraformaldehyde and examined under microscope. (B) Knockdown of Hsp22 expression stimulated U87 cell proliferation. Cells were counted as a function of time, indicated in the figure. Each error bar represents the average + SD of three independent experiments.
Increased proliferation of Hsp22 knockdown cells correlates with fewer cells in G0/G1 phase
The dramatic increase in HKU cell proliferation prompted us to examine their distribution in various phases of the cell cycle. Flow cytometry revealed that the number of cells in G0/G1 phase decreased from 62% in VC cells to 35% in HKU cells, while S phase cells increased from 24% VC to 45% in HKU, and G2/M phase cells increased from 14% VC to 20% in HKU (Fig. 3). It is possible that a significant decrease in G0/G1 phase is due to a rapid transition from G1 to S and G2/M phases, which contributed to the increased rate of HKU proliferation.
Figure 3. Cell cycle progression of Hsp22 knockdown U87 cells analyzed by FACS.
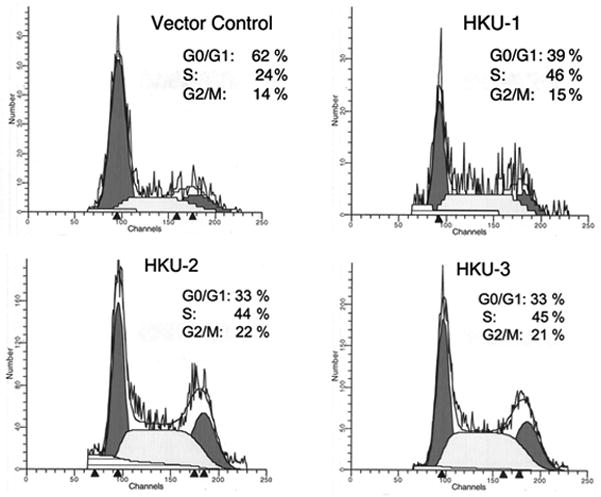
Exponentially growing vector control, HKU-1, HKU-2 and HKU-3 cells were stained with propidium iodide and cell cycle progression analyzed as described in the Materials and Methods.
Cell cycle regulatory proteins responsible for transition from G1 to S phase are increased in Hsp22 knockdown cells
To determine whether increased HKU proliferation was due to rapid transition from G1 to S phase, we analyzed the expression of several regulatory proteins required for this process, including cyclin E, cyclin A, RNR, PCNA and cyclin-dependent kinase inhibitor p21Cip1. While they all increased appreciably in HKU cells, cyclin D1, a regulatory protein normally expressed in G1 phase cells, was significantly down modulated (Fig. 4), consistent with rapid progression through the cell cycle. We also observed increased expression of the anti-apoptotic protein Hax-1 (Suzuki et al., 1997), which was barely detectable in the vector controls (Fig. 4); however, with longer exposure, a weak Hax-1 signal was detected (data not shown). Additionally, we detected high levels of calcium-dependent cell adhesion molecules, such as E-cadherin (epithelial), N-cadherin (neural) and β-catenin in HKU cells compared to VC cells (Fig. 4).
Figure 4. Analysis of proteins associated with cell cycle progression.
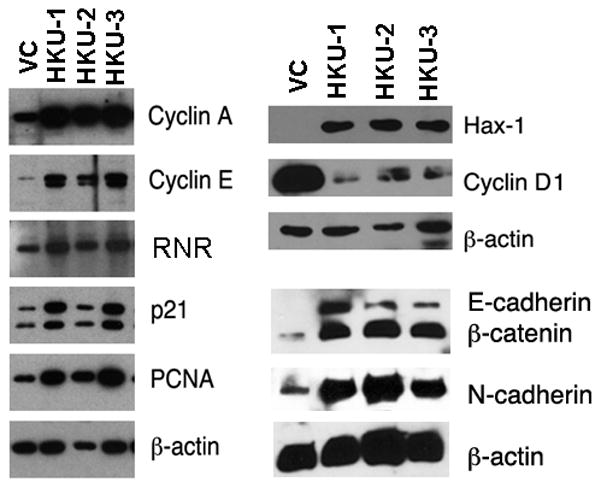
Cell extracts of stable clones (20 μg of total protein for cyclin A, cyclin E, RNR and PCNA; 5 μg for Hax-1, cyclin D1, E-cadherin, N-cadherin and β-catenin) were analyzed by Western blot using respective antibodies. β-actin was used as loading control. Fig. 1B Sam68 blot was reused for Hax-1 and cyclin D1 expressions.
Discussion
The intracellular levels of Sam68 have been reported to be crucial for cancer progression and HIV-1 production, and it remains critical to both cell proliferation and viability; yet its regulation remains elusive. Since low expression of Sam68 reportedly impeded HIV-1 production in U87 cells (Li et al., 2002), and we observed that Hsp22 inhibited the Sam68 activity (Badri et al., 2006) required for HIV-1 production (Modem et al., 2005), we used U87 cells to determine the effect of Hsp22 knockdown on Sam68 expression. Our data demonstrates for the first time that Hsp22 knockdown dramatically increased Sam68 expression in U87 cells, while simultaneously inducing morphological changes and greatly increasing cell proliferation. We observed that U87 cells express high levels of Hsp22 and low levels of Sam68 (Fig. 1A), while downregulation of Hsp22 stimulated both Sam68 mRNA (Fig. 1C) and protein (Fig. 1B), suggesting that Hsp22 regulates Sam68 expression. Although there are no reports implicating Hsp22 in transcriptional regulation, as a chaperone, it may regulate nuclear localization and function of the transcription factors responsible for Sam68 expression. In addition, Hsp22/HspB8 reportedly blocked mRNA translation by phosphorylating the alpha subunit of the translation initiation factor eIF2 (Carra et al., 2009), so that Hsp22 knockdown may suppress eIF2-α phosphorylation and restore Sam68 synthesis.
While it is unclear why Hsp22 knockdown U87 cells are morphologically distinct (Fig 2A), Sam68 could help regulate epithelial-to-mesenchymal transition (EMT) (Valacca et al., 2010). In addition, aggressive glioblastomas expressing high levels of E-cadherin have also been shown to resemble the epithelium (Lewis-Tuffin et al., 2010). Consistent with these reports, we detected high levels of E-cadherin in HKU cells when compared to VC cells (Fig. 4), suggesting that upregulation of E-cadherin results in the epithelial morphology of HKU cells. However, it is unclear whether Sam68 regulates cadherin synthesis. Alternatively, Hsp22 knockdown may result in enhanced E-cadherin expression, which may contribute to mesenchymal-to-epithelial transition; or it could trigger a signaling pathway such as STAT3 that can alter morphology. Transient activation of STAT3 is shown to be required for astrocytes differentiation (Rajan and McKay, 1998).
Our results demonstrate that Hsp22 knockdown stimulated proliferation of U87 cells (Fig. 2B); yet Hsp22/H11 is reportedly required for proliferation of fibroblasts and cardiac cells (Sui et al., 2009; Wadhwa et al., 2010). It is likely that the anti- and pro-proliferative effects of Hsp22 are cell type-specific and may also depend on the level of Sam68 expression. Given the growing evidence supporting Sam68 as a marker of proliferation in various cancers (Elliott and Rajan, 2010), any pro-proliferative actions conferred to HKU cells could be attributed to increased Sam68 levels. Sam68 knockdown reportedly suppressed proliferation of breast and prostate cancer cells (Song et al., 2010; Busa et al., 2007), and yet Sam68 overexpression was cited as suppressing fibroblast proliferation (Taylor et al., 2004). These seemingly contradictory findings suggest that changes in Sam68 and Hsp22 levels could have either a positive or negative effect on proliferation and survival depending on the cell type.
A significantly high number of HKU cells were found in the S and G2/M phases of the cell cycle (Fig. 3), suggesting that Sam68 overexpression stimulates progression from G1 to S phase. Consistent with this possibility, Sam68 knockdown was reported as delaying progression of prostate cancer cells from G1 to S phase (Busa et al., 2007; Song et al., 2010). Accelerated progression of HKU cells from G1 to S phase was also corroborated by our observation that several regulatory proteins required for transition from G1 to S phase were elevated in HKU cells; however, cyclin D1, which is considered essential for linking extracellular mitogenic signals to core cell cycle machinery, decreased dramatically in HKU cells (Fig. 4). Although cyclin D1 is important during the G1 phase, other cyclins expressed later in the cell cycle are able to fill in for cyclin D1 function and trigger entry from G1 to S phase (Reddy, 1999). Consistent with this possibility is the observation that increased cyclin E expression obviated the role of cyclin D1 during normal brain development in cyclin D1 knockout mice (Chen et al., 2005). Thus, cyclin E overexpression in HKU cells may have compensated for the decrease in cyclin D1 function and stimulated progression from G1 to S phase.
It is unclear whether reduced Hsp22 and/or elevated Sam68 levels caused downregulation of cyclin D1. HspB8/Hsp22 reportedly upregulated cyclin D1 and contributed to the progression of estrogen receptor-positive breast cancer (Trent et al., 2007); however, it is not clear whether the Hsp22-induced increase in cyclin D1 was mediated by changes in Sam68 levels. In prostate cancer cells, Sam68 reportedly downregulated cyclin D1 expression (Paronetto, et al., 2010), and its knockdown resulted in accumulation of cyclin D1 mRNA and protein (Busa et al., 2007). Similarly, Sam68 overexpression suppressed cyclin D1 expression in NIH-3T3 (Taylor et al., 2004). Together these observations suggest that overexpression of Sam68 is responsible for the dramatically decreased cyclin D1 expression in Hsp22 knockdown glioblastoma cells.
The observed upregulation of the cyclin-dependent kinase-2 inhibitor p21Cip1 in HKU cells (Fig. 4) may be advantageous to survival of rapidly proliferating tumor cells, as increased expression of p21 has been associated with increased proliferation and progression of invasive ductal carcinoma (Wong et al., 2001). Upregulation of Hax-1 (Fig. 4), an anti-apoptotic protein in HKU cells, suggests that Hax-1 protects cells from apoptosis. We propose that increased p21 and Hax-1 together with decreased cyclin D1 may increase survival of HKU cells, which multiplied 3- to 4 times faster than the parental U87 cells. This is reflected in our observation that there was no noticeable death of exponentially growing HKU cells as determined by trypan blue exclusion (data not shown). Additionally, increased levels of N-cadherin and β-catenin in HKU cells suggest their involvement in cell proliferation. In line with our observations, knockdown of N-cadherin and β-catenin was shown to delay S phase entry and reduce Schwann cell proliferation (Gess et al., 2008). These results further validate that Hsp22 exhibits anti-proliferative property in U87 cells.
In summary, our results provide direct evidence that Hsp22 is involved in the regulation of Sam68 expression, as Hsp22 knockdown stimulated Sam68 expression and proliferation of glioblastoma cells. Since Sam68 has been implicated in growth and survival of cancer cells (Busa et al., 2007) and HIV-1 production (Modem et al., 2005), it is of importance to focus future studies on elucidating the molecular mechanism by which Hsp22 regulates Sam68 expression in cancer and HIV permissive cells.
Acknowledgments
We wish to thank Dr. Avindra Nath (Johns Hopkins University) for his constant support. This work was supported by National Institutes of Health Grants AI46240 and NS060650 (to TRR).
Literature Cited
- Badri KR, Modem S, Gerard HC, Khan I, Bagchi M, Hudson AP, Reddy TR. Regulation of Sam68 activity by small heat shock protein 22. Journal of cellular biochemistry. 2006;99(5):1353–1362. doi: 10.1002/jcb.21004. [DOI] [PubMed] [Google Scholar]
- Bielli P, Busà R, Paronetto MP, Sette C. The RNA binding protein Sam68 is a multifunctional player in human cancer. Endocr Relat Cancer. 2011 May 12; doi: 10.1530/ERC-11-0041. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Busa R, Paronetto MP, Farini D, Pierantozzi E, Botti F, Angelini DF, Attisani F, Vespasiani G, Sette C. The RNA-binding protein Sam68 contributes to proliferation and survival of human prostate cancer cells. Oncogene. 2007;26(30):4372–4382. doi: 10.1038/sj.onc.1210224. [DOI] [PubMed] [Google Scholar]
- Carra S, Brunsting JF, Lambert H, Landry J, Kampinga HH. HspB8 participates in protein quality control by a non-chaperone-like mechanism that requires eIF2{alpha} phosphorylation. The Journal of biological chemistry. 2009;284(9):5523–5532. doi: 10.1074/jbc.M807440200. [DOI] [PubMed] [Google Scholar]
- Chen Z, Duan RS, Zhu Y, Folkesson R, Albanese C, Winblad B, Zhu J. Increased cyclin E expression may obviate the role of cyclin D1 during brain development in cyclin D1 knockout mice. Journal of neurochemistry. 2005;92(5):1281–1284. doi: 10.1111/j.1471-4159.2004.02934.x. [DOI] [PubMed] [Google Scholar]
- Clark EL, Coulson A, Dalgliesh C, Rajan P, Nicol SM, Fleming S, Heer R, Gaughan L, Leung HY, Elliott DJ, Fuller-Pace FV, Robson CN. The RNA helicase p68 is a novel androgen receptor coactivator involved in splicing and is overexpressed in prostate cancer. Cancer research. 2008;68(19):7938–7946. doi: 10.1158/0008-5472.CAN-08-0932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott DJ, Rajan P. The role of the RNA-binding protein Sam68 in mammary tumourigenesis. The Journal of pathology. 2010;222(3):223–226. doi: 10.1002/path.2753. [DOI] [PubMed] [Google Scholar]
- Gess B, Halfter H, Kleffner I, Monje P, Athauda G, Wood PM, Young P, Wanner IB. Inhibition of N-cadherin and beta-catenin function reduces axon-induced Schwann cell proliferation. J Neurosci Res. 2008;86(4):797–812. doi: 10.1002/jnr.21528. [DOI] [PubMed] [Google Scholar]
- Lewis-Tuffin LJ, Rodriguez F, Giannini C, Scheithauer B, Necela BM, Sarkaria JN, Anastasiadis PZ. Misregulated E-cadherin expression associated with an aggressive brain tumor phenotype. PloS one. 2010;5(10):e13665. doi: 10.1371/journal.pone.0013665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Liu Y, Park IW, He JJ. Expression of exogenous Sam68, the 68-kilodalton SRC-associated protein in mitosis, is able to alleviate impaired Rev function in astrocytes. Journal of virology. 2002;76(9):4526–4535. doi: 10.1128/JVI.76.9.4526-4535.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matter N, Herrlich P, Konig H. Signal-dependent regulation of splicing via phosphorylation of Sam68. Nature. 2002;420(6916):691–695. doi: 10.1038/nature01153. [DOI] [PubMed] [Google Scholar]
- Modem S, Badri KR, Holland TC, Reddy TR. Sam68 is absolutely required for Rev function and HIV-1 production. Nucleic acids research. 2005;33(3):873–879. doi: 10.1093/nar/gki231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paronetto MP, Achsel T, Massiello A, Chalfant CE, Sette C. The RNA-binding protein Sam68 modulates the alternative splicing of Bcl-x. The Journal of cell biology. 2007;176(7):929–939. doi: 10.1083/jcb.200701005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paronetto MP, Cappellari M, Busa R, Pedrotti S, Vitali R, Comstock C, Hyslop T, Knudsen KE, Sette C. Alternative splicing of the cyclin D1 proto-oncogene is regulated by the RNA-binding protein Sam68. Cancer research. 2010;70(1):229–239. doi: 10.1158/0008-5472.CAN-09-2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajan P, McKay RD. Multiple routes to astrocytic differentiation in the CNS. J Neurosci. 1998;18(10):3620–3629. doi: 10.1523/JNEUROSCI.18-10-03620.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy GP. DNA replication and S phase. In: Stein GS, Giordano A, Denhart DT, editors. The molecular basis of cell cycle and growth control. Wiley-Liss, Inc; 1999. pp. 80–154. [Google Scholar]
- Reddy TR, Xu W, Mau JK, Goodwin CD, Suhasini M, Tang H, Frimpong K, Rose DW, Wong-Staal F. Inhibition of HIV replication by dominant negative mutants of Sam68, a functional homolog of HIV-1 Rev. Nature medicine. 1999;5(6):635–642. doi: 10.1038/9479. [DOI] [PubMed] [Google Scholar]
- Richard S. Reaching for the stars: Linking RNA binding proteins to diseases. Advances in experimental medicine and biology. 2010;693:142–157. [PubMed] [Google Scholar]
- Shemetov AA, Seit-Nebi AS, Gusev NB. Structure, properties, and functions of the human small heat-shock protein HSP22 (HspB8, H11, E2IG1): a critical review. Journal of neuroscience research. 2008;86(2):264–269. doi: 10.1002/jnr.21441. [DOI] [PubMed] [Google Scholar]
- Song L, Wang L, Li Y, Xiong H, Wu J, Li J, Li M. Sam68 up-regulation correlates with, and its down-regulation inhibits, proliferation and tumourigenicity of breast cancer cells. The Journal of pathology. 2010;222(3):227–237. doi: 10.1002/path.2751. [DOI] [PubMed] [Google Scholar]
- Suhasini M, Reddy TR. Cellular proteins and HIV-1 Rev function. Current HIV research. 2009;7(1):91–100. doi: 10.2174/157016209787048474. [DOI] [PubMed] [Google Scholar]
- Sui X, Li D, Qiu H, Gaussin V, Depre C. Activation of the bone morphogenetic protein receptor by H11kinase/Hsp22 promotes cardiac cell growth and survival. Circulation research. 2009;104(7):887–895. doi: 10.1161/CIRCRESAHA.108.192328. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Demoliere C, Kitamura D, Takeshita H, Deuschle U, Watanabe T. HAX-1, a novel intracellular protein, localized on mitochondria, directly associates with HS1, a substrate of Src family tyrosine kinases. J Immunol. 1997;158(6):2736–2744. [PubMed] [Google Scholar]
- Taylor SJ, Resnick RJ, Shalloway D. Sam68 exerts separable effects on cell cycle progression and apoptosis. BMC cell biology. 2004;5:5. doi: 10.1186/1471-2121-5-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trent S, Yang C, Li C, Lynch M, Schmidt EV. Heat shock protein B8, a cyclin-dependent kinase-independent cyclin D1 target gene, contributes to its effects on radiation sensitivity. Cancer research. 2007;67(22):10774–10781. doi: 10.1158/0008-5472.CAN-07-1475. [DOI] [PubMed] [Google Scholar]
- Valacca C, Bonomi S, Buratti E, Pedrotti S, Baralle FE, Sette C, Ghigna C, Biamonti G. Sam68 regulates EMT through alternative splicing-activated nonsense-mediated mRNA decay of the SF2/ASF proto-oncogene. The Journal of cell biology. 2010;191(1):87–99. doi: 10.1083/jcb.201001073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadhwa R, Ryu J, Gao R, Choi IK, Morrow G, Kaur K, Kim I, Kaul SC, Yun CO, Tanguay RM. Proproliferative functions of Drosophila small mitochondrial heat shock protein 22 in human cells. The Journal of biological chemistry. 2010;285(6):3833–3839. doi: 10.1074/jbc.M109.080424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong SC, Chan JK, Lee KC, Hsiao WL. Differential expression of p16/p21/p27 and cyclin D1/D3, and their relationships to cell proliferation, apoptosis, and tumour progression in invasive ductal carcinoma of the breast. The Journal of pathology. 2001;194(1):35–42. doi: 10.1002/path.838. [DOI] [PubMed] [Google Scholar]