

Non-technical summary
The response to stress is orchestrated by parvocellular neuroendocrine cells in the paraventricular nucleus of the hypothalamus. These cells integrate synaptic information from a number of brain regions and release hormone, yet little is known about the basic rules of synaptic communication at glutamate synapses that provide the dominant excitatory input to these cells. Here we describe experiments aimed at providing insights into how these synapses behave when recruited at different activation frequencies and what impact this has on cell firing.
Abstract
Abstract
Glutamatergic synaptic inputs onto parvocellular neurosecretory cells (PNCs) in the paraventricular nucleus of the hypothalamus (PVN) regulate the hypothalamic-pituitary-adrenal (HPA) axis responses to stress and undergo stress-dependent changes in their capacity to transmit information. In spite of their pivotal role in regulating PNCs, relatively little is known about the fundamental rules that govern transmission at these synapses. Furthermore, since salient information in the nervous system is often transmitted in bursts, it is also important to understand the short-term dynamics of glutamate transmission under basal conditions. To characterize these properties, we obtained whole-cell patch clamp recordings from PNCs in brain slices from postnatal day 21–35 male Sprague–Dawley rats and examined EPSCs. EPSCs were elicited by electrically stimulating glutamatergic afferents along the periventricular aspect. In response to a paired-pulse stimulation protocol, EPSCs generally displayed a robust short-term depression that recovered within 5 s. Similarly, trains of synaptic stimuli (5–50 Hz) resulted in a frequency-dependent depression until a near steady state was achieved. Application of inhibitors of AMPA receptor (AMPAR) desensitization or the low-affinity, competitive AMPAR antagonist failed to affect the depression due to paired-pulse and trains of synaptic stimulation indicating that this use-dependent short-term synaptic depression has a presynaptic locus of expression. We used cumulative amplitude profiles during trains of stimulation and variance–mean analysis to estimate synaptic parameters. Finally, we report that these properties contribute to hamper the efficiency with which high frequency synaptic inputs generate spikes in PNCs, indicating that these synapses operate as effective low-pass filters in basal conditions.
Introduction
The paraventricular nucleus of the hypothalamus (PVN) is an important site for the integration of hypothalamo-pituitary-adrenal (HPA) axis stress responses. The neuroendocrine response to stressors is both initiated and terminated by afferents from limbic, brainstem and hypothalamic regions that synapse onto parvocellular neurosecretory cells (PNCs) in the PVN (Ulrich-Lai & Herman, 2009). In response to stress, the activation of PNCs results in the release of corticotrophin-releasing hormone and subsequent elevations in circulating glucocorticoids. The activity of PNCs is tightly controlled by GABAergic synaptic inputs (Decavel & Van den Pol, 1990; Roland & Sawchenko, 1993); release from this substantial inhibitory tone is necessary for the initiation of the stress response (Cole & Sawchenko, 2002; Hewitt et al. 2009). It is becoming increasingly clear, however, that glutamatergic synaptic transmission also plays an important role in mounting a stress response. PNCs receive robust glutamatergic input (van den Pol et al. 1990) and several studies have demonstrated that central injection of glutamate activates the HPA axis (Makara & Stark, 1975; Darlington et al. 1989; Jezováet al. 1995), whereas application of glutamate receptor antagonists inhibits stress-induced corticosterone release (Ziegler & Herman, 2000).
Furthermore, glutamate synapses may also be particularly important in retaining information encoded by specific stress challenges. Specifically, following exposure to a stressor, glutamate synapses onto PNCs undergo a remarkable change in their ability to express short-term synaptic plasticity in response to trains of high frequency stimulation (Kuzmiski et al. 2010). This activity-dependent, short-term synaptic potentiation is mediated by an increase in the synaptic release of glutamate that culminates in the synchronous release of multiple, glutamate-filled vesicles. In addition to this post-tetanic potentiation, the majority of glutamatergic synapses display a marked depression of the second evoked current during paired-pulse stimulation (Wamsteeker et al. 2010; Kuzmiski et al. 2010). Short-term plasticity of synaptic strength can be regulated by a number of mechanisms including postsynaptic receptor desensitization, saturation, depletion of transmitter-filled vesicles or alterations in release probability (Zucker & Regehr, 2002). Considering the importance of excitatory transmission in mounting an appropriate stress response, surprisingly little is known about the functional properties of glutamate synapses onto PNCs, the mechanisms that contribute to short-term synaptic dynamics under basal conditions and how these combine to impact firing of the postsynaptic neuron.
To address this gap in our understanding, we obtained whole-cell recordings from PNCs in the PVN and examined the properties of excitatory synaptic transmission. We show that glutamate synapses display a frequency-dependent short-term depression, which is dependent on vesicle depletion or a decrease in release probability. This creates a low-pass filter and ensures these synapses induce spiking with greater fidelity at lower rates of synaptic activity.
Methods
Slice preparation
All experiments were performed according to protocols approved by the University of Calgary Animal Care and Use Committee in accordance with the guidelines established by the Canadian Council on Animal Care. Male Sprague–Dawley rats (postnatal day 21–35) were anaesthetized with sodium pentobarbital (30 mg (kg body weight)-1i.p.) and then decapitated. The brain was quickly removed and placed in ice-cold slicing solution containing (in mm): 87 NaCl, 2.5 KCl, 25 NaHCO3, 0.5 CaCl2, 7 MgCl2, 1.25 NaH2PO4, 25 glucose and 75 sucrose, saturated with 95% O2 and 5% CO2. Coronal slices (300 μm) were cut with a vibrating slicer (Leica, Nussloch, Germany) from a block of tissue containing the hypothalamus. Slices containing the PVN were hemisected along the midline (3rd ventricle) and allowed to recover for at least 1 h at 32.5°C in artificial cerebrospinal fluid (ACSF) containing (in mm): 126 NaCl, 2.5 KCl, 26 NaHCO3, 2.5 CaCl2, 1.5 MgCl2, 1.25 NaH2PO4, 10 glucose, saturated with 95% O2 and 5% CO2.
Electrophysiology
Hypothalamic slices were transferred to a recording chamber where they were submerged and continuously perfused with ACSF at 32.5°C at a flow rate of 1–2 ml min-1. Whole-cell patch clamp recordings were performed on PNCs visually identified using infrared differential interference contrast optics (BX50WI, Olympus Optical, Tokyo, Japan). PNCs were identified based on morphology and well-defined electrophysiological characteristics (Luther et al. 2002). Briefly, putative neurosecretory parvocellular neurons were identified by the absence of a pronounced dampening of the membrane-charging curve that causes a delay to action potential generation characteristic of magnocellular neuroendocrine cells. PNCs were further differentiated by the absence of a low threshold spike indicative of non-neurosecretory cells. Patch clamp recording pipettes (3–5 MΩ) were pulled from borosilicate glass and filled with a solution containing (in mm): 108 potassium gluconate, 2 MgCl2, 8 sodium gluconate, 8 KCl, 1 K2-EGTA, 4 K2-ATP and 0.3 Na3-GTP buffered with 10 Hepes.
EPSCs were evoked using a small-diameter (1 μm) ACSF-filled glass electrode placed in the neuropil surrounding the cell, to the periventricular aspect (Fig. 1A). AMPAR-mediated EPSCs were isolated by holding the postsynaptic neuron at –60 mV to block voltage-dependent postsynaptic NMDA receptors (NMDARs) and including the GABAA receptor antagonist picrotoxin (100 μm) in the ACSF. For determining current–voltage relationships of AMPAR currents, 3-((R)-2-carboxypiperazin-4-yl)-propyl-1-phosphonic acid (CPP; 10 μm) was added to the bath and 0.1 mm spermine was included in the patch pipette solution. All other experiments investigating AMPAR-dependent synaptic currents did not include NMDAR antagonists. EPSCs were evoked at 0.2 Hz and paired-pulse ratios (PPRs) were evoked by two stimuli of equal intensity given at 20–5000 ms intervals. Access resistance (<20 MΩ) was continuously monitored and recordings were accepted for analysis if changes were <15%.
Figure 1. AMPA receptor-mediated synaptic transmission in parvocellular neurosecretory cells.
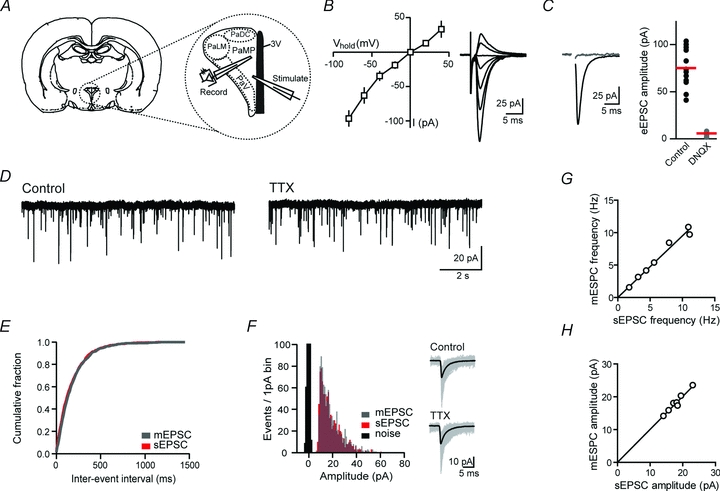
A, schematic diagram of coronal brain slice illustrating the location of stimulating and recording electrodes in expanded view of PVN. Adapted from Paxinos & Watson (2005) and reproduced from Wamsteeker et al. (2010) with permission. B, current–voltage relationship (left) and AMPA receptor-mediated synaptic currents (right). C, sample traces (left) and summary plot of effect of DNQX (10 μm) on synaptic currents. D, sample quantal events in control and after application of TTX (1 μm). E, cumulative fraction plot of sEPSC and mESPC inter-event intervals from the neuron in (D). F, amplitude distribution histogram of sEPSCs and mEPSCs from the neuron in D; inset shows individual (grey) and average (black) quantal events in control and after application of TTX. G and H, summary plots of sEPSC versus mEPSC frequency (G, slope = 0.95 ± 0.03) and amplitude (H, slope = 1.01 ± 0.01).
Data analysis
Synaptic currents were amplified using the Multiclamp 700B amplifier (Molecular Devices, Union City, CA, USA), low-pass filtered at 1 kHz and digitized at 10–20 kHz with the Digidata 1322 (Molecular Devices). Evoked EPSC amplitude was calculated from the baseline current prior to stimulation to the peak synaptic current (Clampfit 9.2, Molecular Devices). PPR was expressed as the ratio of the amplitude of the second synaptic response to the first synaptic response (P2/P1) during paired stimulation. For experiments where stimulation trains were delivered, a minimum of 10 sweeps at each frequency were collected and averaged. Each sweep was separated by a 10 s interval.
To obtain an estimate for the size of the readily releasable pool (RRP) of synchronous release (RRPsyn) and the probability that any given synaptic vesicle in the RRP will be released (Pr,ves), the cumulative amplitudes of the EPSCs evoked by a 1 s, 50 Hz or 100 Hz train of presynaptic stimulation were calculated (Schneggenburger et al. 1999). Individual amplitudes were calculated by measuring the maximal current difference in the period between two consecutive stimulations. The current amplitudes reached a steady state by the 15th stimulation suggesting that the pool was effectively depleted within 250 ms. The RRPsyn was calculated by back-extrapolating the linear fit of the steady-state phase of the cumulative amplitude profile to time 0. Pr,ves was calculated by dividing the amplitude of the first evoked (e)EPSC in the train by the size of the RRP and the total number of synaptic vesicles ready for release (Nq) was calculated as the ratio between RRPsyn and the mean quantal amplitude.
For estimating synaptic parameters during short-term plasticity, variance–mean (V–M) relationships were constructed using a non-stationary analysis (Meyer et al. 2001; Scheuss et al. 2002). Trains of stimuli (50 Hz, ≥50 repetitions, 4 mm Ca2+o) were applied to induce depression at intervals of 10 s (sufficient recovery time between trains). The stability of the data was assessed by fitting a straight line to the amplitudes of the first stimulus in the train and data were accepted if there was <20% change in the regression line. The amplitude of a response in a train is given by Npq, where p is the release probability, N is number of functional release sites and q is the quantal size. The relationship between V and M can be described by the equation: V = qM−M2/N. Plots of the V–M data were fitted with this equation and estimates of q* and N* were obtained from the initial slope and width of the parabolic fit, respectively. These estimates were subsequently corrected for the variability of spontaneous (s)EPSC amplitude distributions to give corrected quantal sizes and number of release sites according to:
and
Here, CV denotes the average coefficient of variation (SD/mean) measured from sEPSC distributions and W is the fraction of quantal variance that is caused by intrasite variability (Frerking & Wilson, 1996).
Quantal synaptic transmission was detected using a variable threshold (MiniAnalysis, Synaptosoft, Decatur, GA, USA). Kolmogorov–Smirnov tests were used for comparing two cumulative distributions. sEPSC amplitude histograms were plotted with 1 pA bins. All data are presented as mean ± standard error of the mean (SEM) and statistical analyses were performed with non-parametric tests. A value of P < 0.05 was considered statistically significant.
Chemicals
All drugs were bath applied. Appropriate stock solutions were made and diluted with ACSF just before application. Drugs used were 3-((R)-2-carboxypiperazin-4-yl)-propyl-1-phosphonic acid (CPP), ifenprodil, cyclothiazide, aniracetam, γ-d-glutamylglycine (γ-DGG) (Tocris, Park Ellisville, MO, USA), tetrodotoxin (TTX) (Alomone Labs, Israel), 6,7-dinitroquinoxaline-2,3-dione (DNQX), picrotoxin and BAPTA (Sigma, St Louis, MO, USA).
Results
Properties of glutamatergic receptor-mediated excitatory synaptic transmission
We first obtained whole-cell patch clamp recordings of evoked (e)EPSCs. To activate glutamatergic synapses, a stimulation electrode was positioned in the neuropil surrounding the cell to the periventricular aspect (Fig. 1A). We pharmacologically isolated AMPA receptor-mediated synaptic currents by bath application of CPP (10 μm) to block NMDA receptors and picrotoxin (100 μm) to block GABAA receptors. AMPA receptor evoked (e)EPSCs had a reversal potential of –1.7 ± 1.2 mV (n = 9) and displayed a linear current–voltage relationship indicating the presence of GluR2-containing AMPA receptors (Fig. 1B). Application of the AMPA/kainate receptor antagonist DNQX (10 μm) completely blocked the eEPSCs (Vhold–60 mV; amplitude control 78.9 ± 6.0 pA, DNQX 5.7 ± 0.6 pA, n = 12, P < 0.001, Fig. 1C).
Quantal glutamate transmission onto PNCs
Next, to probe the properties of glutamatergic transmission onto PNCs, we recorded quantal synaptic transmission. Both sEPSCs and miniature EPSCs (mEPSCs; recorded in the presence of 1 μm TTX) were examined. The average frequency and amplitude of sEPSCs was 6.5 ± 1.4 Hz and 18.1 ± 1.1 pA, respectively (n = 7, Fig. 1D). After bath application of TTX, there was no change in either the frequency or amplitude of quantal events (mEPSC frequency 6.1 ± 1.3 Hz, P = 0.22 vs. sEPSCs, amplitude 18.2 ± 1.2 pA, P = 0.69 vs. sEPSCs) (Fig. 1E–H). In addition, there was no change in either the decay or 10–90 rise times of quantal events following application of TTX (decay time sEPSC 2.9 ± 0.2 ms, mEPSC 2.9 ± 0.3 ms, n = 7, P = 0.94; 10–90 rise time sEPSC 0.9 ± 0.1 ms, mEPSC 0.9 ± 0.1 ms, P = 0.29). These data show that quantal glutamate release onto PNCs, in a coronal slice, is TTX insensitive.
Short-term synaptic dynamics of glutamate synapses
At glutamate synapses onto PNCs, paired stimulation consistently produces paired-pulse depression (Wamsteeker et al. 2010; Kuzmiski et al. 2010). To characterize the dynamic changes in synaptic efficacy at glutamate synapses onto PNCs, we studied the time course of synaptic depression and recovery. We used a paired-pulse stimulation protocol consisting of two stimuli of equal intensity applied at varying interstimulus intervals in 2.5 mm/1.5 mm Ca2+o/Mg2+o. At short interstimulus intervals, the PPR was less than 1 and as the interval increased in duration to 5 s the PPR had returned to 1 (PPR20ms 0.44 ± 0.09, n = 5; PPR50ms 0.58 ± 0.03, n = 20; PPR100ms 0.62 ± 0.04, n = 18; PPR200ms 0.70 ± 0.02, n = 19; PPR500ms 0.73 ± 0.02, n = 17; PPR1000ms 0.84 ± 0.04, n = 5; PPR2000ms 0.94 ± 0.04, n = 6; PPR5000ms 1.00 ± 0.05, n = 5; Fig. 2A and B).
Figure 2. Short-term synaptic dynamics of AMPA receptor-mediated eEPSCs.
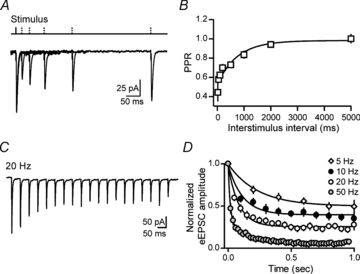
A, sample traces of AMPA eEPSCs elicited by paired stimuli with increasing interstimulus intervals. B, summary graph of paired-pulse ratios (PPR) plotted against interstimulus intervals. C, sample eEPSCs elicited by trains of stimulation (20 Hz, 1 s). D, summary graph of normalized eEPSC amplitudes during trains of stimuli evoked at varying frequencies (5–50 Hz).
To extend this analysis, the responses to longer trains of stimulation were evaluated. When the synapses were stimulated with a 1 s train of varying frequencies (5–50 Hz) we observed a frequency-dependent depression in eEPSC amplitude until a near steady state was achieved (Fig. 2C). The majority of depression occurred within the first few stimulations and as a percentage of the first eEPSC, the synaptic response depressed to 6.5 ± 3.5% at 50 Hz (eEPSC51/eEPSC1, n = 8), 22.9 ± 2.7% at 20 Hz (eEPSC21/eEPSC1, n = 12), 36.6 ± 3.5% at 10 Hz (eEPSC11/eEPSC1, n = 12) and 49.6 ± 4.3% at 5 Hz (eEPSC6/eEPSC1, n = 13) (Fig. 2D). The time constants of depression at 50 Hz, 20 Hz, 10 Hz, 5 Hz were 15.0 ms, 86.2 ms, 115.3 ms and 234.7 ms, respectively. In order to determine whether this depression may be the result of glutamate feedback at presynaptic metabotropic glutamate receptors (mGluRs) (Takahashi et al. 1996; von Gersdorff et al. 1997), we first conducted experiments to determine the identity of the mGluRs that may be present at presynaptic terminals. Group III mGluRs powerfully regulate glutamate release at excitatory terminals onto magnocellular neurosecretory cells in PVN (Gordon & Bains, 2003; Kuzmiski et al. 2009). Bath application of the group III mGluR agonist, l AP4 elicited a robust depression (59.2 ± 4.6% at 50 μm, n = 4). Next, we examined the effects of the mGluR antagonist MAP4 on synaptic depression during a 20 Hz train. Synaptic depression was unaffected by MAP4 (eEPSC21/eEPSC1 30.3 ± 8.7%; MAP4 250 μm, n = 7). This indicates that glutamate autoreceptors probably do not play a major role in synaptic depression induced by high frequency trains.
AMPA receptor desensitization does not contribute to synaptic depression
Both presynaptic and postsynaptic mechanisms can contribute to short-term depression of paired-pulse responses. AMPA receptors activated by glutamate undergo rapid desensitization and this can contribute to paired-pulse depression (Trussell & Fischbach, 1989; Trussell et al. 1993; Isaacson & Walmsley, 1996; Rozov et al. 2001; Xu-Friedman & Regehr, 2003). To investigate the potential role of postsynaptic AMPA receptor desensitization in paired-pulse depression we analysed the effects of cyclothiazide (100 μm; CTZ) and aniracetam (2 mm; ANI), two structurally unrelated compounds that slow AMPA receptor desensitization (Vyklicky et al. 1991; Isaacson & Nicoll, 1991; Trussell et al. 1993; Otis et al. 1996). Both CTZ and ANI increased the decay time of eEPSCs (control: 4.6 ± 0.4 ms, n = 16; CTZ: 11.0 ± 1.3 ms, n = 9, P < 0.01; ANI: 10.3 ± 1.7 ms, n = 7, P < 0.01 ANOVA, Kruskal–Wallis; Fig. 3A and B) and increased the amplitude of eEPSCs (CTZ: 127.5 ± 10.7% of control, n = 9, P = 0.006; ANI: 138.5 ± 13.1% of control, n = 7, P = 0.02 Wilcoxon signed rank; Fig. 3A–C). These results suggest that a portion of AMPA receptors at the synapse are desensitized by glutamate release during a single stimulation. However, CTZ and ANI failed to affect PPR suggesting that AMPA receptor desensitization does not significantly contribute to paired-pulse depression in PNCs (PPR50ms 0.60 ± 0.03, P = 0.69; PPR100ms 0.65 ± 0.04, P = 0.57; PPR200ms 0.63 ± 0.03, P = 0.39; PPR500ms 0.82 ± 0.08, P = 0.09; PPR5000ms 1.03 ± 0.01, P = 0.66, n = 6; Fig. 3D).
Figure 3. AMPA receptor desensitization does not contribute to short-term depression.
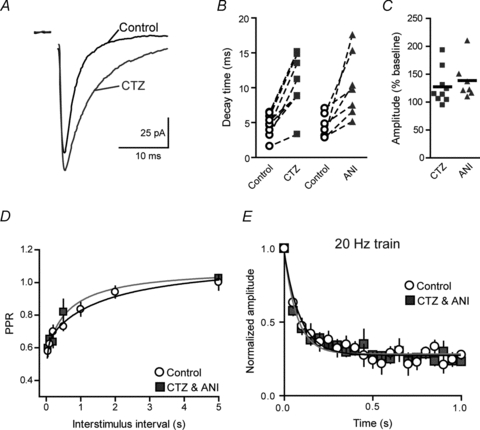
A, sample traces of AMPA receptor eEPSCs in control and after application of the AMPA receptor desensitization inhibitor, cyclothiazide (100 μm; CTZ). B, summary graph of changes in eEPSC decay time following application of either CTZ or aniracetam (2 mm, ANI). C, summary graph of percentage change from baseline in eEPSC amplitude after application of either CTZ or ANI. D, summary graph of PPRs plotted against interstimulus intervals in control and after application of CTZ or ANI. E, summary graph of normalized eEPSC amplitudes during trains of stimulation evoked at 20 Hz in control or CTZ/ANI.
Next we determined whether AMPA receptor desensitization contributes to use-dependent synaptic depression during a stimulation train at various frequencies. If desensitization played a role in the synaptic depression during train of stimulations, we would expect that the depression should be reduced in the presence of CTZ and ANI. However, we found that neither CTZ nor ANI significantly affected the relative amplitude of the last eEPSC in the train compared to control (20 Hz, eEPSC21/eEPSC1 29.9 ± 7.1%, n = 5, P = 0.27, Fig. 3E; 10 Hz, eEPSC11/eEPSC1 38.6 ± 6.2%, n = 6, P = 0.92; 5 Hz, eEPSC6/eEPSC1 56 ± 3.1%, n = 6, P = 0.36). Since there was no difference between CTZ and ANI, the data were pooled. The time constant of depression during CTZ application at 20 Hz, 10 Hz and 5 Hz were 85.3 ± 4.2 ms, 137.9 ± 7.5 ms and 141.4 ± 6.4 ms, respectively.
Inhibitors of AMPA receptor desensitization are known to have presynaptic effects (Bellingham & Walmsley, 1999; Ishikawa & Takahashi, 2001). We previously showed that AMPA receptors are not saturated under our recording conditions (Kuzmiski et al. 2010), as bath application of the low-affinity, rapidly dissociating competitive antagonist of AMPA receptors, γ-d-glutamylglycine (γ-DGG; 2 mm) reduced eEPSC amplitude (52.1 ± 6.4% of baseline, n = 7) without altering PPR (97.8 ± 3.7% of baseline, P = 0.23, Fig. 4A and C). As a control for voltage escape, a low dose of the slowly unbinding competitive antagonist DNQX (100 nm) was applied. DNQX inhibited eEPSC amplitude to a similar extent (57.3 ± 6.7% of baseline, n = 4) and did not alter PPR (102.7 ± 3.1% of baseline, P = 0.31, Fig. 4C). Therefore, we also used γ-DGG to further investigate a role for AMPA receptor desensitization in use-dependent synaptic depression. The fast off-rate of γ-DGG minimizes desensitization in the range of 90% by allowing re-equilibration of blocked and unblocked receptors between stimuli (Wong et al. 2003). However, consistent with CTZ and ANI, there was no change in synaptic depression in the presence of γ-DGG (20 Hz, eEPSC21/eEPSC1 27.0 ± 6.0%, n = 5, P = 0.81 vs. control, Fig. 4A and B; 10 Hz, eEPSC11/eEPSC1 42.1 ± 7.5%, n = 5, P = 1; 5 Hz, eEPSC6/eEPSC1 51.0 ± 9.7%, n = 5, P = 0.81). Taken together, these data suggest that desensitization of AMPARs does not contribute to short-term depression which is probably due to a presynaptic mechanism (Forsythe et al. 1998; Dittman & Regehr, 1998; Bellingham & Walmsley, 1999; Xu & Wu, 2005).
Figure 4. Short-term depression is not relieved by γDGG.
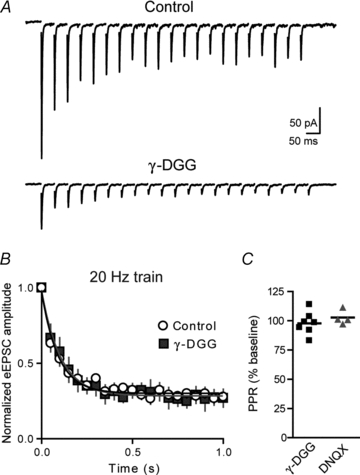
A, sample eEPSCs elicited by trains of stimulation (20 Hz) in control and after application of the low affinity, competitive AMPA receptor antagonist γ-DGG (2 mm). B, summary graph of normalized eEPSC amplitudes during trains of stimulation evoked at 20 Hz in control and after application of γ-DGG. C, summary graph of changes in PPR after application of either γ-DGG or DNQX (100 nm).
Variance–mean analysis
To further study the synaptic properties and mechanisms underlying the synaptic depression at glutamate synapses onto PNCs, we characterized the transmitter release properties using a non-stationary variance–mean analysis of eEPSCs at 4 mm Ca2+o (Meyer et al. 2001; Scheuss et al. 2002; Clements, 2003). An estimate of the release probability (Pr), number of release sites (N) and the response to release of a quantum of transmitter at one release site (q), was obtained by using a protocol consisting of trains of five stimuli at 50 Hz repeated every 10 s (Fig. 5A). This duration of interval between train repetitions allowed for recovery from depression as assessed by fitting a straight line to the amplitudes of the first stimulus in the train plotted against repetition number (Fig. 5B). For each of the five stimuli within the 50 Hz trains, the mean eEPSC amplitude and the variance of the eEPSC amplitudes was determined by summing over 50 consecutive trains. The relationship between variance (V) and mean (M) can be described by the following equation: V = qM–M2/N. eEPSCs recorded in 4 mm Ca2+o displayed strong depression with the amplitude of the fifth eEPSC in the train depressed to 25.3 ± 3.8% of the first eESPC (n = 11). The average V and M relationship of eEPSCs in the trains could be fitted with a parabolic function with all five responses lying along the parabola (r2 = 0.92, Fig. 5C). The finding that the fourth and fifth responses do not deviate from the parabola suggests that quantal size does not change late in the train and is consistent with our previous assertion that desensitization does not contribute to synaptic depression in PNCs. The estimates of q based on the initial slope were 16.7 pA, N was estimated as 23 release sites and at 4 mm Ca2+o the Pr was 0.6.
Figure 5. Variance–mean analysis of glutamate synapses.
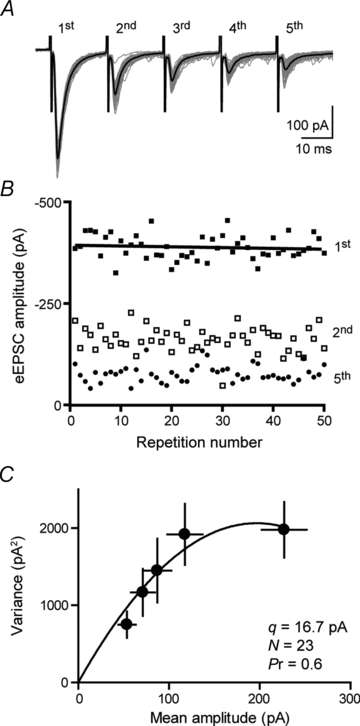
A, sample traces of five eEPSCs in a 50 Hz stimulation train evoked every 10 s in 4 mm Ca2+o. The thick black line indicates average eEPSC. B, amplitudes of first, second and fifth eESPCs in a train (for better visibility) plotted versus repetition number (50 repetitions). A regression line was fitted to the amplitudes of the first eEPSCs in the trains. C, eESPC amplitude variance–mean plot from all neurons (n = 11). Parabolic fit was constrained to pass through the origin.
Recovery from depletion and estimate of the readily releasable pool
Following high frequency activity, the readily releasable pool is depleted and has to be refilled rapidly to ensure synaptic fidelity (Wu & Borst, 1999; Sakaba & Neher, 2001). To determine the time course of recovery from synaptic depression, we delivered a 50 Hz train (1 s) and then gave a test pulse at different intervals following each train. Recovery from synaptic depression follows a slow time course that can be fitted with a single-exponential function (τ = 1.54 ± 0.13 s, n = 9, Fig. 6A and B). Following a 50 Hz stimulation train, the eEPSC amplitudes recovered to their initial values within 5 s.
Figure 6. Recovery from depletion and size of the RRP.
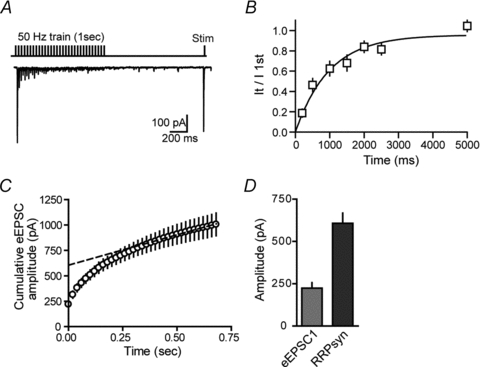
A, sample experiment illustrating recovery after a conditioning train of 50 pulses (50 Hz, 1 s). Test pulses were given at various intervals following the conditioning train. B, summary graph of the test eEPSC current amplitude normalized to the first eEPSC amplitude in the train. C, cumulative amplitude plot of the eEPSCs during a 50 Hz stimulation train. D, summary graph of the average amplitude of the first eEPSC in the train (eEPSC1) and the average size of the RRP of synchronous release (RRPsyn).
Next we analysed the cumulative amplitude profile during high-frequency stimulation trains (50 Hz) to estimate the size of the readily releasable pool (RRP). We used 2.5 mm Ca2+o since under these conditions AMPA receptors are not saturated and desensitization does not contribute to synaptic depression during a train. Estimates of the RRP size depend on complete and rapid depletion of the pool, as well as a lack of AMPA receptor desensitization/saturation. In response to 50 Hz stimulation, there was a rapid depression of eEPSCs during the train followed by a near steady-state amplitude (Fig. 2D). The cumulative amplitude profile of repeated eEPSCs showed a rapid rise followed by a slower linear increase (Fig. 6C). The slow linear rise is attributable to the equilibrium between the release-induced depletion and the constant replenishment of the RRP (Schneggenburger et al. 1999). This assumes that depression is largely caused by a transient decrease in the readily releasable quanta. Back-extrapolation of the linear portion to time 0 (y-intercept of the linear fit) provides an estimate of the size of the RRP of synchronous release (RRPsyn; 607.5 ± 60.3 pA, n = 8, Fig. 6C and D). The ratio between the first eEPSC in the train and the RRPsyn gave an estimate of the synaptic vesicle release probability (Pr,ves; 0.36 ± 0.02). Finally, the number of quanta forming the RRPsyn (Nq), estimated by dividing the RRPsyn by the average quantal amplitude was calculated to be 38 ± 4. Since estimates of the RRPsyn depend on a complete and rapid depletion of the pool and a stimulation frequency of 50 Hz may not be sufficiently fast to deplete the entire pool, we reconfirmed the estimates of RRPsyn, Pr,ves and Nq with high frequency trains. At 100 Hz, there was not a significant difference in RRPsyn (470.9 ± 62.2 pA; P = 0.17, n = 15), Pr,ves (0.40 ± 0.04; P = 0.49), and Nq (31 ± 4; P = 0.21) when compared to the estimates at 50 Hz stimulation.
Physiological consequences of synaptic depression
Activity-dependent synaptic depression may play a crucial role in determining the rate at which synaptic information (the conversion of EPSPs to spikes) is transferred at the synapse. Based on the observations above, one plausible scenario is that EPSP–spike coupling is weaker during high frequency activity. To test this hypothesis, we conducted current clamp experiments in which we delivered 1 s trains at frequencies of 5, 10, 20 or 50 Hz and assessed postsynaptic spiking in response (Fig. 7A). Ten trains were delivered for each frequency and trains were repeated in six cells. We ensured that the first EPSP in each train always generated a spike. An increase in frequency was accompanied by an increase in the total number of spikes during the train (5 Hz: 3.0 ± 0.4, 10 Hz: 3.6 ± 0.6, 20 Hz: 5.2 ± 0.6, 50 Hz: 7.1 ± 1.1, Fig. 7B). However, the probability of an EPSP generating a spike during the course of the train decreased at 5 Hz, and was further reduced at higher frequencies (Fig. 7C). We used these observations to calculate the spike efficiency during each of the trains. This calculation is simply the total number of spikes in a 1 s train divided by the number of presynaptic stimuli. Here again, we find a frequency-dependent decrease in spike-efficiency (5 Hz: 0.51 ± 0.1, 10 Hz: 0.28 ± 0.06, 20 Hz: 0.22 ± 0.03, 50 Hz: 0.13 ± 0.02, Fig. 7D). These data suggest that information transfer at these synapses occurs more reliably during lower rates of synaptic activity.
Figure 7. EPSP–spike coupling during trains of synaptic stimulation.
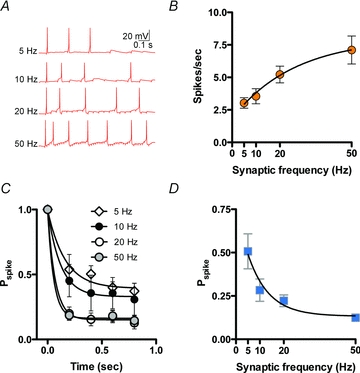
A, sample current clamp recordings of spikes generated by trains of synaptic stimulation at various frequencies. B, summary graph of the number of spikes generated by trains of synaptic stimulation. C, summary graph of the probability of an EPSP generating a spike (Pspike) during the course of a train of synaptic stimulation. D, summary graph of the spike efficiency during EPSP trains at various frequencies.
Discussion
A detailed understanding of the basic properties of synaptic transmission in the PVN is a requirement for the comprehension of the regulation of the neuroendocrine system in both physiological and pathophysiological conditions. In the present study, we investigated the short-term dynamics of glutamate synapses onto PNCs in the PVN. Glutamatergic synapses onto PNCs generally displayed a robust paired-pulse depression indicative of a relatively high Pr at presynaptic terminals. During trains of synaptic stimuli, eEPSCs exhibited a robust frequency-dependent depression that was most likely attributable to vesicle depletion or activity-dependent decrease in Pr and not AMPAR desensitization. This short-term depression contributes to a reduction in the efficiency with which high frequency synaptic inputs generate spikes in PNCs. Thus, these synapses effectively operate as low-pass filters in basal conditions.
Glutamatergic terminals onto the PVN show a relatively high Pr and repetitive stimulation induces a short-term depression of synaptic responses that is dependent on the frequency of stimulation. Both presynaptic and postsynaptic mechanisms may contribute to this short-term synaptic plasticity. Depression of synaptic strength during trains can arise from depletion of neurotransmitter from the presynaptic terminal (Varela et al. 1997; Forsythe et al. 1998; Staley et al. 1998; Dittman & Regehr, 1998; Bellingham & Walmsley, 1999; Foster & Regehr, 2004; Xu & Wu, 2005), activity-dependent decrease in Pr (Silver et al. 1998; Wu & Borst, 1999; Sakaba & Neher, 2001), and the desensitization of postsynaptic AMPARs (Trussell & Fischbach, 1989; Trussell et al. 1993; Isaacson & Walmsley, 1996; Rozov et al. 2001; Xu-Friedman & Regehr, 2003). Our results indicate that paired-pulse depression at glutamate synapses onto PNCs only involves a presynaptic mechanism. PPRs and trains of synaptic stimuli were not significantly altered by preventing AMPAR desensitization with either CTZ or ANI, despite the observation that inhibiting AMPAR desensitization increased the decay time and amplitude of AMPAR-mediated EPSCs indicating that AMPARs are desensitized under basal release conditions. The finding that CTZ failed to alter PPRs also excludes the possibility that CTZ acts at the presynaptic terminal to affect K+ currents (Ishikawa & Takahashi, 2001). Secondly, depression during trains of eEPSCs in the presence of γ-DGG was not significantly different from control trains. γ-DGG prevents desensitization of AMPARs by protecting a population of receptors from exposure to glutamate (Wong et al. 2003; Crowley et al. 2007). Since AMPARs are not saturated under basal release conditions (Kuzmiski et al. 2010) this is an effective tool for studying desensitization. Finally, variance–mean analysis of the eEPSCs in a train failed to demonstrate a significant deviation of the latter (3rd to 5th) eEPSCs in the train from the parabola indicating that quantal size was not reduced late in the depressing train. A reduction in quantal size late in the train reflects a use-dependent reduction in postsynaptic responsiveness due to AMPAR desensitization or saturation (Scheuss et al. 2002). Thus, our data point to a presynaptic mechanism for the use-dependent depression of eEPSC amplitude during trains of synaptic stimuli. Potential mechanisms for this presynaptic depression include vesicle pool depletion (von Gersdorff & Matthews, 1997; Schneggenburger et al. 1999; Foster & Regehr, 2004), reduction in Ca2+ influx (Forsythe et al. 1998) or activity-dependent inactivation of release machinery (Kraushaar & Jonas, 2000). Our observations provide little evidence to support a role for mGluR autoreceptors (Takahashi et al. 1996; von Gersdorff et al. 1997) under our experimental conditions.
The short-term plasticity of glutamatergic synapses onto PNCs is relatively homogeneous with the majority of neurons displaying paired-pulse depression (Wamsteeker et al. 2010; Kuzmiski et al. 2010). This is in spite of the potential for significant heterogeneity in target cell phenotype. Neurons recorded in this study were electrophysiologically identified PNCs (Tasker & Dudek, 1991; Stern, 2001; Luther et al. 2002) and there were no discernable differences among them in terms of postsynaptic spiking characteristics, current–voltage relationships and synaptic properties. Interestingly, despite the relative homogeneity in electrophysiological properties, single cell RT-PCR work has demonstrated that there is a notable diversity in the expression of mRNA for peptide transmitters and PNCs often express more than one neuropeptide that may include vasopressin, corticotrophin-releasing hormone and thyrotrophin-releasing hormone (Price et al. 2008, 2009). Thus, it is likely that glutamatergic synapses onto phenotypically diverse PNCs display remarkably similar short-term plasticity. In addition to postsynaptic phenotype heterogeneity, glutamatergic afferents probably originate in several anatomically distinct areas including limbic structures, hypothalamic regions such as the suprachiasmatic nucleus, dorsomedial hypothalamus, anterior hypothalamic nucleus as well as locally from within the PVN (Hermes et al. 1996; Boudaba et al. 1997; Csáki et al. 2000). This raises the possibility that the homogeneity in short-term plasticity is dependent on the postsynaptic PNC and not the originating neuron, or that the majority of synapses activated during stimulation are from a homogeneous afferent population such as a local peri-paraventricular glutamatergic population (Boudaba et al. 1997). The short-term depression we have described here is similar to that described in other brain regions in terms of its underlying presynaptic cause.
Short-term synaptic plasticity contributes to a dramatic change in the activity of the postsynaptic neuron. Our data demonstrate that PNCs have a moderately high initial Pr and depression is the dominant form of short-term plasticity during trains or bursts of stimulation. This use-dependent depression ensures that these synapses function as low-pass filters and are most effective at ensuring faithful postsynaptic spiking at lower frequencies in response to the first few EPSPs. Additional postsynaptic mechanisms such as the membrane time constant and the duration/magnitude of spike afterhyperpolarization may act in tandem with synaptic depression to curtail information transfer at high rates of activity. Together, these mechanisms may act as an important form of gain control to ensure the appropriate release of peptide hormones. Following acute stress, synapses that undergo bursts of high-frequency stimulation display an increase in initial Pr that is mediated, at least in part, by multivesicular release and an enhanced paired-pulse depression (Kuzmiski et al. 2010). This suggests that acute stress can increase synaptic gain that may enhance the fidelity of spikes early in a burst thereby facilitating the release of hormones. Furthermore, the pronounced synaptic depression at these synapses may be an important regulatory mechanism that optimizes postsynaptic excitation following stress when GABAergic synaptic inhibition becomes depolarizing (Hewitt et al. 2009). Therefore, short-term synaptic dynamics provide an important means for shaping an appropriate neuroendocrine response.
Glossary
Abbreviations
- ACSF
artificial cerebrospinal fluid
- AMPAR
AMPA receptor
- ANI
aniracetam
- CTZ
cyclothiazide
- eEPSCs
evoked EPSCs
- HPA
hypothalamic-pituitary-adrenal
- mEPSCs
miniature EPSCs
- mGluRs
metabotropic glutamate receptors
- NMDAR
NMDA receptor
- PNCs
parvocellular neurosecretory cells
- PPR
paired-pulse ratio
- PVN
paraventricular nucleus
- RRP
readily releasable pool
- RRPsyn
RRP of synchronous release
- sEPSCs
spontaneous EPSCs
- V–M
variance–mean
Author contributions
The experiments were carried out at the University of Calgary. J.B.K. and J.S.B. were responsible for the design and conception of the experiments. V.M., J.B.K. and D.V.B. were responsible for collection, analysis and interpretation of data. J.S.B, J.B.K. and V.M. drafted the article and revised it for important intellectual content. All authors approved the final version of the manuscript.
Author's present address
V. Marty: University of California, Los Angeles, Oral Biology and Medicine, 10833 Le Conte Avenue, Los Angeles, CA 90095-1668, USA.
References
- Bellingham MC, Walmsley B. A novel presynaptic inhibitory mechanism underlies paired pulse depression at a fast central synapse. Neuron. 1999;23:159–170. doi: 10.1016/s0896-6273(00)80762-x. [DOI] [PubMed] [Google Scholar]
- Boudaba C, Schrader LA, Tasker JG. Physiological evidence for local excitatory synaptic circuits in the rat hypothalamus. J Neurophysiol. 1997;77:3396–3400. doi: 10.1152/jn.1997.77.6.3396. [DOI] [PubMed] [Google Scholar]
- Clements JD. Variance–mean analysis: a simple and reliable approach for investigating synaptic transmission and modulation. J Neurosci Methods. 2003;130:115–125. doi: 10.1016/j.jneumeth.2003.09.019. [DOI] [PubMed] [Google Scholar]
- Cole RL, Sawchenko PE. Neurotransmitter regulation of cellular activation and neuropeptide gene expression in the paraventricular nucleus of the hypothalamus. J Neurosci. 2002;22:959–969. doi: 10.1523/JNEUROSCI.22-03-00959.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley JJ, Carter AG, Regehr WG. Fast vesicle replenishment and rapid recovery from desensitization at a single synaptic release site. J Neurosci. 2007;27:5448–5460. doi: 10.1523/JNEUROSCI.1186-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csáki A, Kocsis K, Halász B, Kiss J. Localization of glutamatergic/aspartatergic neurons projecting to the hypothalamic paraventricular nucleus studied by retrograde transport of [3H]d-aspartate autoradiography. Neuroscience. 2000;101:637–655. doi: 10.1016/s0306-4522(00)00411-5. [DOI] [PubMed] [Google Scholar]
- Darlington DN, Miyamoto M, Keil LC, Dallman MF. Paraventricular stimulation with glutamate elicits bradycardia and pituitary responses. Am J Physiol Regul Integr Comp Physiol. 1989;256:R112–R119. doi: 10.1152/ajpregu.1989.256.1.R112. [DOI] [PubMed] [Google Scholar]
- Decavel C, Van den Pol AN. GABA: a dominant neurotransmitter in the hypothalamus. J Comp Neurol. 1990;302:1019–1037. doi: 10.1002/cne.903020423. [DOI] [PubMed] [Google Scholar]
- Dittman JS, Regehr WG. Calcium dependence and recovery kinetics of presynaptic depression at the climbing fibre to Purkinje cell synapse. J Neurosci. 1998;18:6147–6162. doi: 10.1523/JNEUROSCI.18-16-06147.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsythe ID, Tsujimoto T, Barnes-Davies M, Cuttle MF, Takahashi T. Inactivation of presynaptic calcium current contributes to synaptic depression at a fast central synapse. Neuron. 1998;20:797–807. doi: 10.1016/s0896-6273(00)81017-x. [DOI] [PubMed] [Google Scholar]
- Foster KA, Regehr WG. Variance-mean analysis in the presence of a rapid antagonist indicates vesicle depletion underlies depression at the climbing fibre synapse. Neuron. 2004;43:119–131. doi: 10.1016/j.neuron.2004.06.022. [DOI] [PubMed] [Google Scholar]
- Frerking M, Wilson M. Effects of variance in mini amplitude on stimulus-evoked release: a comparison of two models. Biophys J. 1996;70:2078–2091. doi: 10.1016/S0006-3495(96)79774-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon GRJ, Bains JS. Priming of excitatory synapses by α1 adrenoceptor-mediated inhibition of group III metabotropic glutamate receptors. J Neurosci. 2003;23:6223–6231. doi: 10.1523/JNEUROSCI.23-15-06223.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermes ML, Coderre EM, Buijs RM, Renaud LP. GABA and glutamate mediate rapid neurotransmission from suprachiasmatic nucleus to hypothalamic paraventricular nucleus in rat. J Physiol. 1996;496:749–757. doi: 10.1113/jphysiol.1996.sp021724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewitt SA, Wamsteeker JI, Kurz EU, Bains JS. Altered chloride homeostasis removes synaptic inhibitory constraint of the stress axis. Nat Neurosci. 2009;12:438–443. doi: 10.1038/nn.2274. [DOI] [PubMed] [Google Scholar]
- Isaacson JS, Nicoll RA. Aniracetam reduces glutamate receptor desensitization and slows the decay of fast excitatory synaptic currents in the hippocampus. Proc Natl Acad Sci U S A. 1991;88:10936–10940. doi: 10.1073/pnas.88.23.10936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isaacson JS, Walmsley B. Amplitude and time course of spontaneous and evoked excitatory postsynaptic currents in bushy cells of the anteroventral cochlear nucleus. J Neurophysiol. 1996;76:1566–1571. doi: 10.1152/jn.1996.76.3.1566. [DOI] [PubMed] [Google Scholar]
- Ishikawa T, Takahashi T. Mechanisms underlying presynaptic facilitatory effect of cyclothiazide at the calyx of Held of juvenile rats. J Physiol. 2001;533:423–431. doi: 10.1111/j.1469-7793.2001.0423a.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jezová D, Tokarev D, Rusnák M. Endogenous excitatory amino acids are involved in stress-induced adrenocorticotropin and catecholamine release. Neuroendocrinology. 1995;62:326–332. doi: 10.1159/000127021. [DOI] [PubMed] [Google Scholar]
- Kraushaar U, Jonas P. Efficacy and stability of quantal GABA release at a hippocampal interneuron–principal neuron synapse. J Neurosci. 2000;20:5594–5607. doi: 10.1523/JNEUROSCI.20-15-05594.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzmiski JB, Marty V, Baimoukhametova DV, Bains JS. Stress-induced priming of glutamate synapses unmasks associative short-term plasticity. Nat Neurosci. 2010;13:1257–1264. doi: 10.1038/nn.2629. [DOI] [PubMed] [Google Scholar]
- Kuzmiski JB, Pittman QJ, Bains JS. Metaplasticity of hypothalamic synapses following in vivo challenge. Neuron. 2009;62:839–849. doi: 10.1016/j.neuron.2009.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luther JA, Daftary SS, Boudaba C, Gould GC, Halmos KC, Tasker JG. neurosecretory and non-neurosecretory parvocellular neurones of the hypothalamic paraventricular nucleus express distinct electrophysiological properties. J Neuroendocrinol. 2002;14:929–932. doi: 10.1046/j.1365-2826.2002.00867.x. [DOI] [PubMed] [Google Scholar]
- Makara GB, Stark E. Effect of intraventricular glutamate on ACTH release. Neuroendocrinology. 1975;18:213–216. doi: 10.1159/000122400. [DOI] [PubMed] [Google Scholar]
- Meyer AC, Neher E, Schneggenburger R. Estimation of quantal size and number of functional active zones at the calyx of Held synapse by nonstationary EPSC variance analysis. J Neurosci. 2001;21:7889–7900. doi: 10.1523/JNEUROSCI.21-20-07889.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otis T, Zhang S, Trussell LO. Direct measurement of AMPA receptor desensitization induced by glutamatergic synaptic transmission. J Neurosci. 1996;16:7496–7504. doi: 10.1523/JNEUROSCI.16-23-07496.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price CJ, Hoyda TD, Samson WK, Ferguson AV. Nesfatin 1 influences the excitability of paraventricular nucleus neurones. J Neuroendocrinol. 2008;20:245–250. doi: 10.1111/j.1365-2826.2007.01641.x. [DOI] [PubMed] [Google Scholar]
- Price CJ, Samson WK, Ferguson AV. Neuropeptide W has cell phenotype-specific effects on the excitability of different subpopulations of paraventricular nucleus neurones. J Neuroendocrinol. 2009;21:850–857. doi: 10.1111/j.1365-2826.2009.01904.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roland BL, Sawchenko PE. Local origins of some GABAergic projections to the paraventricular and supraoptic nuclei of the hypothalamus in the rat. J Comp Neurol. 1993;332:123–143. doi: 10.1002/cne.903320109. [DOI] [PubMed] [Google Scholar]
- Rozov A, Jerecic J, Sakmann B, Burnashev N. AMPA receptor channels with long-lasting desensitization in bipolar interneurons contribute to synaptic depression in a novel feedback circuit in layer 2/3 of rat neocortex. J Neurosci. 2001;21:8062–8071. doi: 10.1523/JNEUROSCI.21-20-08062.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaba T, Neher E. Quantitative relationship between transmitter release and calcium current at the calyx of Held synapse. J Neurosci. 2001;21:462–476. doi: 10.1523/JNEUROSCI.21-02-00462.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuss V, Schneggenburger R, Neher E. Separation of presynaptic and postsynaptic contributions to depression by covariance analysis of successive EPSCs at the calyx of Held synapse. J Neurosci. 2002;22:728–739. doi: 10.1523/JNEUROSCI.22-03-00728.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneggenburger R, Meyer AC, Neher E. Released fraction and total size of a pool of immediately available transmitter quanta at a calyx synapse. Neuron. 1999;23:399–409. doi: 10.1016/s0896-6273(00)80789-8. [DOI] [PubMed] [Google Scholar]
- Silver RA, Momiyama A, Cull-Candy SG. Locus of frequency-dependent depression identified with multiple-probability fluctuation analysis at rat climbing fibre–Purkinje cell synapses. J Physiol. 1998;510:881–902. doi: 10.1111/j.1469-7793.1998.881bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staley KJ, Longacher M, Bains JS, Yee A. Presynaptic modulation of CA3 network activity. Nat Neurosci. 1998;1:201–209. doi: 10.1038/651. [DOI] [PubMed] [Google Scholar]
- Stern JE. Electrophysiological and morphological properties of pre-autonomic neurones in the rat hypothalamic paraventricular nucleus. J Physiol. 2001;537:161–177. doi: 10.1111/j.1469-7793.2001.0161k.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Forsythe ID, Tsujimoto T, Barnes-Davies M, Onodera K. Presynaptic calcium current modulation by a metabotropic glutamate receptor. Science. 1996;274:594–597. doi: 10.1126/science.274.5287.594. [DOI] [PubMed] [Google Scholar]
- Tasker JG, Dudek FE. Electrophysiological properties of neurones in the region of the paraventricular nucleus in slices of rat hypothalamus. J Physiol. 1991;434:271–293. doi: 10.1113/jphysiol.1991.sp018469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trussell LO, Fischbach GD. Glutamate receptor desensitization and its role in synaptic transmission. Neuron. 1989;3:209–218. doi: 10.1016/0896-6273(89)90034-2. [DOI] [PubMed] [Google Scholar]
- Trussell LO, Zhang S, Raman IM. Desensitization of AMPA receptors upon multiquantal neurotransmitter release. Neuron. 1993;10:1185–1196. doi: 10.1016/0896-6273(93)90066-z. [DOI] [PubMed] [Google Scholar]
- Ulrich-Lai YM, Herman JP. Neural regulation of endocrine and autonomic stress responses. Nat Rev Neurosci. 2009;10:397–409. doi: 10.1038/nrn2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Pol AN, Wuarin JP, Dudek FE. Glutamate, the dominant excitatory transmitter in neuroendocrine regulation. Science. 1990;250:1276–1278. doi: 10.1126/science.1978759. [DOI] [PubMed] [Google Scholar]
- Varela JA, Sen K, Gibson J, Fost J, Abbott LF, Nelson SB. A quantitative description of short-term plasticity at excitatory synapses in layer 2/3 of rat primary visual cortex. J Neurosci. 1997;17:7926–7940. doi: 10.1523/JNEUROSCI.17-20-07926.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Gersdorff H, Matthews G. Depletion and replenishment of vesicle pools at a ribbon-type synaptic terminal. J Neurosci. 1997;17:1919–1927. doi: 10.1523/JNEUROSCI.17-06-01919.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Gersdorff H, Schneggenburger R, Weis S, Neher E. Presynaptic depression at a calyx synapse: the small contribution of metabotropic glutamate receptors. J Neurosci. 1997;17:8137–8146. doi: 10.1523/JNEUROSCI.17-21-08137.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyklicky L, Patneau DK, Mayer ML. Modulation of excitatory synaptic transmission by drugs that reduce desensitization at AMPA/kainate receptors. Neuron. 1991;7:971–984. doi: 10.1016/0896-6273(91)90342-w. [DOI] [PubMed] [Google Scholar]
- Wamsteeker JI, Kuzmiski JB, Bains JS. Repeated stress impairs endocannabinoid signalling in the paraventricular nucleus of the hypothalamus. J Neurosci. 2010;30:11188–11196. doi: 10.1523/JNEUROSCI.1046-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong AYC, Graham BP, Billups B, Forsythe ID. Distinguishing between presynaptic and postsynaptic mechanisms of short-term depression during action potential trains. J Neurosci. 2003;23:4868–4877. doi: 10.1523/JNEUROSCI.23-12-04868.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LG, Borst JG. The reduced release probability of releasable vesicles during recovery from short-term synaptic depression. Neuron. 1999;23:821–832. doi: 10.1016/s0896-6273(01)80039-8. [DOI] [PubMed] [Google Scholar]
- Xu J, Wu L-G. The decrease in the presynaptic calcium current is a major cause of short-term depression at a calyx-type synapse. Neuron. 2005;46:633–645. doi: 10.1016/j.neuron.2005.03.024. [DOI] [PubMed] [Google Scholar]
- Xu-Friedman MA, Regehr WG. Ultrastructural contributions to desensitization at cerebellar mossy fibre to granule cell synapses. J Neurosci. 2003;23:2182–2192. doi: 10.1523/JNEUROSCI.23-06-02182.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegler DR, Herman JP. Local integration of glutamate signalling in the hypothalamic paraventricular region: regulation of glucocorticoid stress responses. Endocrinology. 2000;141:4801–4804. doi: 10.1210/endo.141.12.7949. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]