

Abstract
Objective
The investment of newly formed endothelial cell tubes with differentiated smooth muscle cells (SMC) is critical for appropriate vessel formation, but the underlying mechanisms remain unknown. We previously showed that depletion of focal adhesion kinase (FAK) in the nkx2.5 expression domain led to aberrant outflow tract (OFT) morphogenesis and strove herein to determine the cell types and mechanisms involved.
Methods and Results
We crossed fakloxp targeted mice with available Cre drivers to deplete FAK in OFT SMC (FAKwnt and FAKnk) or coronary SMC (FAKcSMC). In each case, depletion of FAK led to defective vasculogenesis that was incompatible with post-natal life. Immunohistochemical analysis of the mutant vascular structures revealed that FAK was not required for progenitor cell proliferation, survival, or differentiation into SMC, but was necessary for subsequent SMC recruitment to developing vasculature. Using a novel FAK-null SMC culture model, we found that depletion of FAK did not influence SMC growth or survival, but blocked directional SMC motility and invasion toward the potent endothelial-derived chemokine, PDGFBB. FAK depletion resulted in un-stable lamellipodial protrusions due to defective spatial-temporal activation of the small GTPase, Rac-1 and lack of Rac1-dependent recruitment of cortactin (an actin stabilizing protein) to the leading edge. Moreover, FAK null SMC exhibited a significant reduction in PDGF-stimulated extracellular matrix degradation.
Conclusions
FAK drives PDGFBB-stimulated SMC chemotaxis/invasion and is essential for SMC to appropriately populate the aorticopulmonary septum and the coronary vascular plexus.
Keywords: vasculogenesis, PDGF, FAK, vascular smooth muscle, chemotaxis
Introduction
The investment of newly formed endothelial cell tubes with differentiated smooth muscle cells (SMC) is a very important process during vessel formation and requires intricate regulation of SMC specification, motility, growth, and differentiation. Failure of SMC recruitment to and migration along developing vessels can lead to vascular instability and regression, an event that is likely due in part to the ability of these cells to secrete and organize extracellular matrix-containing basement membranes and elastin. Moreover, mature medial SMC express high levels of contractile genes (i.e. myosin heavy chain (SM-MHC), α-smooth muscle actin (α-SMA), SM22 among others) and their presence is essential for maintaining vessel tone and for providing dynamic control of blood pressure (see 1 for review).
While the precise signaling pathways have yet to be identified, a number of major extrinsic factors have been shown to regulate SMC recruitment and function. TGF-β, which promotes SMC differentiation2-4, and PDGF, which promotes SMC growth and motility, are both important extrinsic regulators of SMC phenotype and genetic ablation of receptors for these genes resulted in defective outflow tract (OFT) and/or coronary vascular morphogenesis 5-7. Extensive evidence indicates that extracellular matrix signaling is also an important regulator of SMC growth and differentiation as deletion of either fibronectin (FN), the α5 integrin FN receptor, or focal adhesion kinase (FAK) (the kinase that mediates α5-dependent signaling) each results in extraembryonic and embryonic vessel defects leading to lethality in the mouse from E8.5 to E10 8-10.
Although a direct role for FAK in vascular smooth muscle growth and development has yet to be examined, our lab recently showed that depletion of FAK from nkx2.5-expressing precursors led to perinatal lethality resulting from a profound sub-aortic ventricular septal defect accompanied (in some neonates) by mal-alignment of the outflow tract including double-outlet right ventricle (DORV) and persistent truncus arteriosus (PTA)11. Thus, conditional inactivation of FAK in nkx2.5-expressing cells phenocopies the most common congenital heart disease that is also a subset of abnormalities associated with Tetralogy of Fallot and the DiGeorge Syndrome 12,13. However, whether these defects arose from the inability of nkx2.5 precursor cells to differentiate into SMC or from a specific defect in nkx2.5-derived SMC remained unclear.
The aim of the present study was to identify the critical FAK-dependent functions during vasculogenesis. Since both wnt-1- and nkx2.5-expressing cells contribute to development of conotruncal septal SMC that divide the OFT into the aorta and pulmonary artery 13, 14, we explored the effects of FAK depletion in both origins using available Cre lines. We show FAK deletion (by homologous recombination) in either embryologic origin results in OFT defects including PTA. Moreover, targeted depletion of FAK in these SMC precursors did not affect progenitor cell proliferation, survival, or differentiation, but resulted in lack of appropriate SMC coverage of the developing vasculature. Similarly, depletion of FAK in a third SMC precursor origin, the wt1-expressing epicardial cells that provide SMC that line coronary vessels, did not affect SMC specification but lead to aberrant recruitment of SMC to the developing coronary vasculature. Collectively, these studies indicate that FAK is essential for a SMC autonomous function during vascular remodeling. Our mechanistic studies in FAK null SMC, revealed that FAK does not influence SMC growth or survival, but functions to promote directional SMC chemotaxis/invasion toward the potent endothelial-derived chemokine, PDGF.
Methods
Experimental Animals
fak+/- mice15 were bred to existing Cre lines including cardiac neural crest (CNC)-specific wnt-1Cre mice obtained from Andrew McMahon14; primary and secondary heart field-specific nkx2.5Cre mice obtained from Robert Schwartz16; and epicardial-specific wt1Cre mice obtained from John Burch17. The resulting fak+/-/Cre+/- mice were bred with fakflox/flox/Rosa26RlacZ/lacZ mice to obtain fakflox/-Cre+/-/Rosa26RlacZ/wt (experimental) or genetic control mice (fakflox/-/Rosa26RlacZ/wt without Cre and fakflox/+ with or without Cre). Mice were housed in the University of North Carolina Animal Care Facility, which is accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International, and all experimental procedures were approved by the University of North Carolina Animal Care and Use Committee.
Statistical Analysis
All quantitative data represent at least 3 separate experiments and are presented as mean±SEM. Means were compared by 2-tailed Student's t test. p< .05 was considered statistically significant.
See Supplemental Data Section for additional experimental procedures.
Results
FAK deletion impairs recruitment of nkx2.5- and wnt-1-derived SMC to the developing aorta and pulmonary artery
We previously showed that conditional deletion of FAK in nkx2.5-expressing cells (FAKnk) resulted in mal-alignment of the outflow tract and incomplete aorticopulmonary septum formation in a percentage of neonates11. Since nkx2.5-derived SMC contribute to the formation of the aortic root and septum, we hypothesized that this phenotype may have resulted from a SMC defect in FAK depleted cells. To further examine FAK's role in outflow tract development, we utilized the wnt-1Cre line to deplete FAK from the CNC-derived SMC that also make a major contribution to this process 14. These well characterized mice generate high efficiency recombination around E9.5 in the CNC-derived cells populating the truncus arteriosis and pharyngeal arch arteries (3, 4, and 6) and in the CNC-derived cells invading the conotruncal cushions and the aorticopulmonary septum by E11.514. While fakflox/-wnt-1Cre/+/Rosa26RlacZ/wt mice (herein referred to as FAKwnt) were present at the appropriate Mendelian ratio at P0 (Table S1), all FAKwnt mice died within a few days after birth. Gross morphology of the cardiac outflow tract revealed aberrant septation and branching of the major outflow vessels in all FAKwnt mice with the majority (95%; N=42/44) exhibiting PTA (Fig 1A). Thus, our data indicate that FAK activity in nkx2.5- and wnt-1-derived cells is required for appropriate septation of the truncus arteriosis.
Figure 1. FAK deletion impairs recruitment of cardiac neural crest-derived SMC to the developing aorta and pulmonary artery.
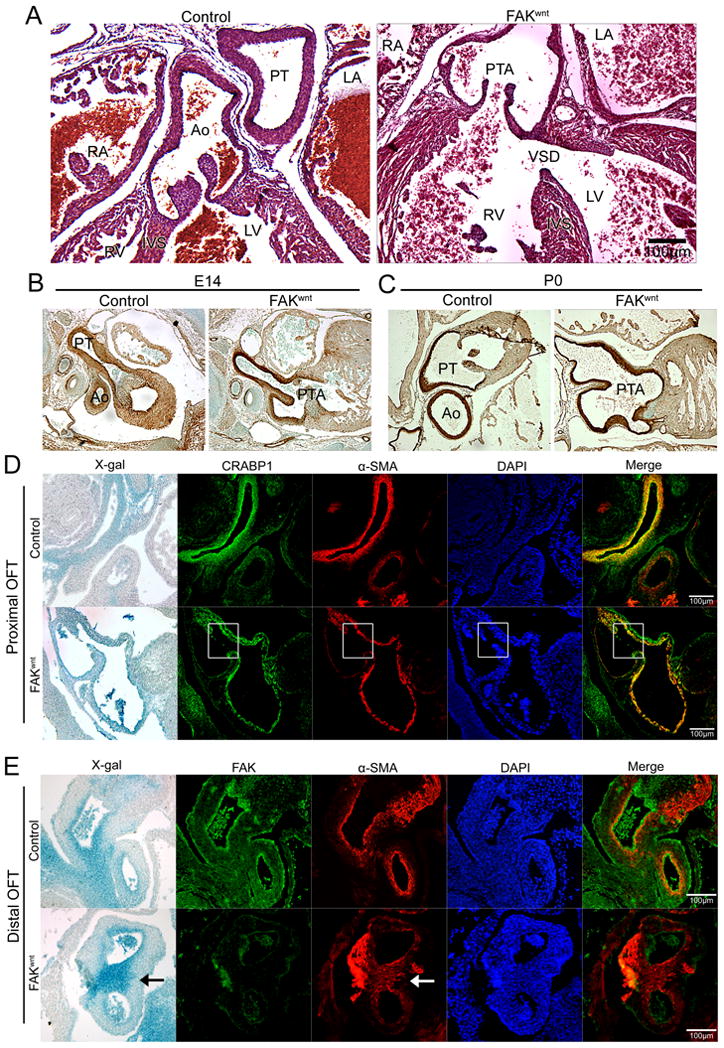
(A) H&E staining of genetic control and FAKwnt mice at postnatal day 0 (P0). FAKwnt outflow tract (OFT) shows persistent truncus arteriosus (PTA) and ventricular septal defect (VSD). LA, left atrium; RA, right atrium; Ao, aorta; PT, pulmonary trunk; IVS, intraventricular septum. (B-C) SM-22 immunostaining (brown) of embryonic day 14 (E14; B) or P0 (C) genetic control and FAKwnt OFT revealed PTA without defect in SMC differentiation. (D) Proximal OFT of genetic control and FAKwnt hearts at E 12 reveals co-localization of α-SMA (red) with the CNC lineage marker CRABP1 (green), indicating that the SMC covering the aorta and pulmonary artery were derived from CNC cells. X-gal staining demarcates wnt-1-derived CNC in the Rosa26RLacZ –positive embryos. Nuclei (blue) were stained with DAPI. Note the lack of SMC coverage (highlighted in white boxes) in the inner walls of the aorta and pulmonary artery in FAKwnt mice. (E) Distal OFT at E 12 stained with X-gal (blue in bright field), FAK (green), and α-SMA (red) reveals FAK-depleted CNC differentiate into SMC (arrows) but fail to appropriately populate the conus. Nuclei (blue) were stained with DAPI. Scale bar = 100μm.
Since the aortic arch defects observed in CNC-targeted Notch-/- mice were associated with impaired SMC differentiation within the truncus18, we tested whether FAK depletion affected this process in our models. Expression of α-SMA in the pharyngeal arch region at E11 was similar between control and FAKwnt mice (Fig S1A). The un-septated ascending outflow tract in FAKwnt mice contained layers of SM-22-positive medial SMC that were comparable to littermate controls from E14 onwards (Fig 1B-C). SMC layers and differentiation in the carotid arteries also appeared to be normal in FAKwnt mice (Fig S1B). These mice were bred to contain the Rosa26RLacZ allele to track progenitors of the Cre-expressing cells, and the neural crest origin of carotids was confirmed by β-galactosidase positive staining (Fig S1C). In addition, as shown in figure S1D, SMC investment of coronary SMC (that were not targeted for FAK depletion) was also similar between genetic control and FAKwnt hearts, indicating that global changes in flow or hemodynamic properties do not account for the SMC investment phenotypes observed in the FAKwnt OFTs.
Aorticopulmonary septation occurs between E11.5 and E12.5 when a wedge of CNC-derived SMC partitions the aortic sac into distinct aortic and pulmonary channels 19. To more closely evaluate this process in FAKwnt mice serial sections were co-stained for α-SMA and CNC lineage markers (CRABP1 or expression of the Rosa26RLacZ allele). As shown in figure 1D, the SMC lining the pulmonary artery and aorta at the level of the proximal OFT, are mainly derived from CNC in control and FAKwnt mice. However, a selective lack of CNC (and CNC-derived SMC) was apparent in the aortic wall juxtaposed to pulmonary artery in the FAKwnt mice (Fig 1D; highlighted in box). FAK deficient CNC cells populated the conotruncal region dorsal to the PTA and properly differentiated into SMC (Fig 1E). However, in FAKwnt mice the SMC in the dorsal conus were more centrally located (Fig 1E; arrow) and did not surround the developing vessels as completely in control mice.
We previously showed that FAK deficiency did not alter proliferation or survival within the affected outflow tracts of FAKnk mice11. Similarly, there was no significant difference in levels of cell death in the outflow tract of E13.5 FAKwnt mice in comparison to controls (27.8 +/- 4.3 versus 29.3 +/- 7.4 per 0.5 mm2 respectively; N= 5/5). The numbers of BrdU-labeled cells in the affected outflow tract region of FAKwnt embryos also did not differ significantly from those in the genetic controls (45.8 +/-3.7 versus 43.6 +/-4.8 per 0.5 mm2 respectively; N=4/4). In support of a lack of major effect on the proliferation and survival of CNC-derived cells, the more distal CNC-derived structures such as the aforementioned carotid arteries (Fig S1B-C) and other neural crest-derived structures including the thymic lobes, thyroid, tongue, and ear appeared to be unaffected by FAK deletion (data not shown). Close evaluation of the cranial cartilage and bones did reveal a modest mandibular truncation in 3/6 FAKwnt neonates, but the crown-rump length and maxilla formation were normal (Fig S1E). Collectively, these studies reveal that CNC functions, population of the developing OFT, and differentiation of wnt-1 lineages into SMC occur normally in FAKwnt mutants. However, in these mice and those in which FAK was depleted in nkx2.5-derived cells, FAK null SMC fail to appropriately invest the inner walls of the developing aorta and pulmonary trunk, indicating that FAK may be necessary for directional SMC movement within the conotruncus.
FAK deletion impairs recruitment of epicardial-derived SMC to the coronary plexus
To provide additional in vivo evidence for our hypothesis, we tested the effects of FAK depletion on the recruitment of epicardial-derived SMC to the intra-myocardial endothelial plexus, a process also dependent on directional motility7. To this end, we intercrossed our FAKflox/flox/Rosa26RlacZ/lacZ mice to an available Wilm's Tumor-1Cre line (wt-1, hereafter referred to as FAKcSMC so as to not be confused with FAKwnt) that induces strong and uniform recombination in the proepicardium by E9.5 and is restricted to the epicardium from E10.5 onward17, 20. Previous studies revealed that wt1-derived cells give rise to the SMC that invest the coronary arteries and veins 7, 20, and we found similar results upon analysis of β-galactosidase expression in P0 wt1Cre/Rosa26RLacZ hearts (Fig S2). We observed Mendelian distribution of FAKcSMC at P0 (Table S1), but all FAKcSMC mice died within a few days after birth by still to be determined mechanisms.
Importantly, immunohistochemical assessment of α-SMA positive cells in P0 hearts, revealed a striking and consistent lack of SMC coverage of the coronary endothelial plexus (Fig 2). All large diameter vessels in FAKcSMC hearts exhibited a thin non-continuous layer of SMC in comparison to comparable genetic control vessels (Fig 2B, arrowheads), while most of the smaller arterioles exhibited a complete lack of associated SMC (Fig 2B, arrows). Quantification of SMC numbers revealed significantly fewer SMC associated with both arteries and arterioles in FAKcSMC hearts relative to genetic controls (Fig 2C,D). In contrast, significantly higher numbers of α-SMA positive cells were observed in the sub-epicardial space in FAKcSMC, compared to genetic control hearts (Fig 2A, arrows, Fig 2E), indicating that the FAK null cells efficiently delaminated from the epicardium and differentiated into SMC, but failed to migrate into the myocardium. Importantly, a reduction in PECAM labeled coronary vasculature was not observed in the FAKcSMC hearts (Fig 3), indicating appropriate formation of the primary plexus. This conclusion is supported by the finding that these hearts did not exhibit signs of hypoplastic growth or non-compaction that are associated with defects in endothelial plexus function (Fig S3A,B)21. As well, no defects in the formation or SMC investment in carotid arteries (not targeted for FAK depletion) were observed in the FAKcSMC neonates, indicating a lack of effect on global hemodynamics (Fig S3C).
Figure 2. FAK deletion impairs recruitment of epicardial-derived SMC to the coronary vessels.

(A) P0 genetic control and FAKcSMC heart stained with FAK (green) and α-SMA (red). Phase image is shown on left. Sub-epicardial α-SMA-stained cells (arrows) are present in FAKcSMC but not genetic control hearts. Scale bar = 40μm. Boxed regions (a'-e') in A are shown at higher magnification in B. (B) FAKcSMC hearts exhibited reduced presence of SMC lining the coronary vasculature, including large vessels (arrowheads) and small arterioles (arrows), in comparison to genetic control vessels. FAK deletion was confirmed by absence of FAK staining in SMC from FAKcSMC hearts (arrowheads). Scale bar = 20μm or 10μm. (C-E) Numbers of SMC lining arteries (diameter > 65μm), arterioles (diameter < 65μm as previously described45), or localized in the sub-epicardium were quantified using Image J. *p < .05; **p< .01.
Figure 3. Endothelial plexus formation in FAKcSMC hearts.

P0 genetic control and FAKcSMC hearts stained with endothelial marker PECAM (green) and α-SMA (red) showing normal endothelial plexus formation with reduced SMC coverage (arrowhead) of the coronary endothelial plexus. Nuclei (blue) were stained with DAPI. Boxed regions (a'-c') in A are shown at higher magnification in B. Scale bar = 40μm (A) and 10μm (B), respectively.
We next performed additional studies to confirm that FAK activity was not required for the proliferation or survival of SMC progenitors, or their differentiation into coronary SMC. As shown in figure S4, there was no statistical difference in the rates of cell proliferation or apoptosis in β-gal labeled wt-1-derived cells within FAKcSMC hearts compared to genetic controls at either E15.5 or P0. Interestingly, by E15.5, many β-gal labeled cells in genetic control hearts had moved into the sub-epicardial zone, while most of the β-gal labeled cells in E15.5 FAKcSMC hearts remained associated with the epicardium (Fig S4A), corroborating our previous findings in the P0 hearts that suggested impaired directional motility of the FAK-depleted SMC (Fig 2B, E). We also analyzed SMC differentiation marker gene expression in quail pro-epicardial organ explants treated with adenoviruses that express GFP or the FAK inhibitor, GFP-FRNK. As shown in figure S5, expression of FRNK attenuated the outward movement of the epicardial explants but did not affect the induction of SMC marker gene expression in these cultures. These studies strongly suggest that the SMC investment defect observed in FAKcSMC coronary vessels did not result from a block in the proliferation, survival or differentiation of wt-1 progenitors, but rather from a lack of SMC recruitment to the endothelial plexus.
FAK deletion impairs SMC chemotaxis but not proliferation or survival
To confirm that FAK plays a significant role in the regulation of SMC chemotaxis, we established a conditional FAK null SMC culture model using cells isolated from fakflox/flox mice. As shown in figure 4A, treatment of fakflox/flox SMC with Cre adenovirus (but not LacZ) resulted in a significant reduction of FAK protein. In excellent agreement with our in vivo studies, FAK deletion had no effect on SMC differentiation marker gene expression (Fig 4A) and importantly, did not alter the expression of FRNK or the FAK homologue, Pyk2 (Fig S6A). Moreover, continuously adherent FAK null cells revealed no major differences in focal adhesion or actin filament organization (Fig S6B).
Figure 4. FAK deletion impairs SMC chemotaxis/invasion toward PDGF and serum.

(A) fakflox/flox SMC were infected with either LacZ or Cre adenovirus for 72 hrs and cell lysates were immunoblotted with antibodies for smooth muscle differentiation markers (SM-MHC and α-SMA). α-tubulin was used as a loading control. Data are representative of at least four experiments. (B-D) Cells treated as above were plated on fibronectin-coated inserts (10 μg/ml; Bio-Coat) in serum-free media using either PDGF-BB (20 ng/ml; B) or 10% serum (C) as the chemoattractant. (D) LacZ- or Cre- infected cells were co-transfected with GFP and either Flag vector control, Flag-FAK, or the inactive mutant Flag-Y397FFAK. Chemotaxis towards PDGF-BB was assessed 24 hours after transfection. Wild-type FAK but not Y397FFAK restored chemotaxis towards PDGF in the Cre expressing cells. Results in B-D are mean±SEM of cells counted in 4 fields from 4 independent experiments. *p< .05; ***p< .001.
Since the strongly chemotactic PDGFs are highly expressed in the conotruncus during outflow tract and coronary vessel morphogenesis, and have been implicated in guiding cells during these critical processes5,22, we hypothesized that the cause of the PTA (in FAKwnt and FAKnk mice) and the impaired coronary vessel formation (in FAKcSMC mice) was due to a defective SMC migratory response to this chemokine. We found that PDGF treatment induced robust FAK activation at the leading edge of SMC (Fig S6C) and that FAK was necessary for PDGF-stimulated SMC chemotaxis as assessed using FN-coated transwells (Fig 4B). Chemotaxis towards serum was also inhibited but this effect was much more modest (Fig 4C). Importantly, ectopic expression of wildtype FAK but not phosphorylation-deficient FAK (Y397FFAK) restored PDGF-stimulated motility in FAK-deficient cells (Fig 4D). Confirming our in vivo results, we detected no significant difference in cell proliferation between control and FAK null SMC grown in 10% serum (data not shown), under serum-free conditions, or following treatment with PDGF or EGF (Fig S6D). Accordingly, PDGF receptor activation (as assessed by staining and Western analysis for pY716PDGFRβ; data not shown), PDGF-stimulated ERK activity (Fig S6E) and cyclin D protein levels (Fig S6F) were similar in Cre- and LacZ- treated cells, indicating that FAK was not required for activation of the major mitogenic Ras/ERK signaling pathway. We also observed no significant difference in cell survival in FAK-depleted SMC as measured by caspase 3/7 activity (data not shown). As well, PDGF-stimulated activation of the pro-survival kinase, AKT was un-changed (data not shown). Collectively, these data indicate that defective aorticopulmonary septation and coronary vasculogenesis observed in our conditional FAK mutants was likely due to a specific defect in PDGF-dependent SMC chemotaxis.
FAK increases lamellipodial stability through Rac1-dependent recruitment of cortactin
Since PDGF-dependent motility involves rapid remodeling of the plasma membrane and underlying actin cytoskeleton, we next explored whether FAK-depleted SMC exhibited a biomechanical defect in one of these processes. To this end, we performed time-lapse imaging of Cre- and LacZ-treated fakflox/flox SMC following PDGF treatment. In control cells, PDGF stimulated extensive membrane ruffling as early as 2.5 minutes that culminated in a formation of a dominant, stable leading edge lamellipodium by 15-20 min (Fig 5A, left). In contrast, PDGF treated FAK null SMC remained un-polarized and rarely formed a distinctive leading edge (Fig 5A, right). Kymographic analysis of lamellipodia dynamics indicated that this difference was mainly due to decreased lamellipodia persistence in FAK null SMC (Fig 5B-D) and that this defect culminated in a significant decrease in leading edge displacement (Fig 5E) and cell speed in 2-D (Fig 5F).
Figure 5. FAK regulates Rac1-dependent cortactin recruitment and lamellipodial stability.

LacZ- or Cre-infected fakflox/flox SMC were treated with 20 ng/ml PDGF-BB and immediately imaged at 5 second intervals for 60 min. (A) Representative images of cells 20 min following PDGF treatment. Cell polarization as shown by formation of leading edge (arrowhead) and trailing edge (arrow) was observed in control but not in FAK-deficient SMC. (B) Representative kymographs of LacZ and Cre-infected cells 0-20 min following PDGF-BB treatment. Ascending slopes represent protrusion (arrows) and descending slopes represent retraction (arrowheads). (C-E) Kymographic analysis of lamellipodial dynamics following PDGF treatment. Calculated protrusion rate (C), lamellipodial persistence (D) and leading edge displacement (E) from 8-10 cells from 4 independent experiments (mean±SEM; *p < .05.) (F) Average 2-D cell speed within 4 hr following PDGF treatment. Results are mean±SEM of 30-60 cells from 4 independent experiments. *p < .05. (G-I) LacZ or Cre infected fakflox/flox cells stained for Rac1 showing dorsal ruffle localization in both control (G, top left) and FAK-deficient (G, top right) cells at 2.5 min following PDGF treatment. At 15 min, leading edge localization of Rac1 (arrows) was observed in control cells, whereas FAK-deficient cells showed un-polarized Rac1 localization (arrowheads). (H, I) Quantification of cells exhibiting Rac1-labeled dorsal ruffles 2.5 min post PDGF treatment (H) and Rac1-labeled dominant leading edge lamellipodia 15 min post PDGF treatment (I). Results are mean±SEM of 150-200 cells from 3 independent experiments. ***p< .001. (J-L) SMC treated as above were stained with anti-cortactin antibodies and processed as described in G-I. Similar dorsal ruffle but not leading edge recruitment of cortactin was observed in FAK-deficient cells. Results are mean±SEM of 200-250 cells from 4 independent experiments. **p< .01. Scale bar = 10μm.
PDGF-stimulated motility is dependent on Rac1, a small GTPase that controls cell protrusion and lamellipodial dynamics. To test whether FAK is necessary for Rac1 activation we first analyzed total Rac1 activation in response to PDGF using the standard GST-PBD pull-down assay. As shown in figure S6G, PDGF robustly and transiently activated Rac1 with peak activity by 2.5 minutes and, somewhat surprisingly, this response was not affected by depletion of FAK. Thus, global activation of Rac1 is FAK-independent. Since recent studies indicate that PDGF induces leading edge localized activation of Rac1 via induction of new integrin-ECM connections23, we explored the idea that FAK might be necessary for restricting localized Rac1 activity to the leading edge. Immunofluorescent staining with a Rac1 antibody revealed that PDGF treatment of wildtype and FAK deficient SMC led to rapid and equivalent accumulation of Rac1 at dorsal ruffles that peaked at 2.5 minutes (Fig 5G top, 5H). By 15 minutes following PDGF-treatment, restricted enrichment of Rac1 at the leading edge was observed in wildtype SMC, but this response was perturbed in FAK null SMC, which exhibited random distribution of membrane-associated Rac1 (Fig 5G bottom, 5I).
To confirm a role for FAK in localized Rac1 activation, we next examined the spatiotemporal distribution of cortactin, an actin binding protein that is recruited to the cell periphery in a Rac1-dependent fashion24. Immunofluorescent staining showed that recruitment of cortactin to leading edge-lamellipodia (Fig 5J bottom, 5L), but not dorsal ruffles (Fig 5J top, 5K) was significantly reduced in FAK null SMC, indicating that FAK is critical for this aspect of PDGF-stimulated cytoskeletal remodeling. These data are consistent with the finding that active FAK accumulates at the leading edge of cells following PDGF treatment (Fig S6C). Importantly, either blocking Rac1 activity with the pharmacological inhibitor, NSC23766 (10 μM, Calbiochem) or treatment with cortactin siRNAs mirrored the effects of depleting FAK on PDGF-stimulated SMC membrane dynamics and chemotaxis (Fig S7). In sum, these studies indicate that FAK plays a critical role in coordinating and stabilizing the protein complexes that are required for Rac1-dependent leading edge protrusions, and hence, directional SMC motility toward the potent endothelial-derived chemokine, PDGF.
FAK is necessary for SMC-mediated extracellular matrix degradation
SMC are known to have a high basal invasive potential25, a function mediated by podosomes, sites of dynamic actin polymerization activity thought to be the topological equivalent of lamellipodia formed during 3-D movement. Interestingly, podosomes are initiated at the inner-face of focal adhesion complexes and require cortactin and its associated Arp 2/3 complex for extension into the underlying matrix and for the recruitment and secretion of matrix-degrading proteases26, 27. To determine whether the chemotactic defect of FAK null SMC in the Boyden chamber assays resulted from impaired directional movement alone or to a combination of impaired motility and invasion, we assessed the capacity of wt and FAK null SMC to degrade ECM. To this end, we cultured LacZ- or Cre- adenovirus pre-treated fakflox/flox SMC on fluorescent matrix (Oregon green 488 gelatin/FN mixture) and treated them with or without PDGF for 90 min. As shown in figure 6a, PDGF induced focal areas of matrix degradation (cell associated dark spots) in wt SMC, but this response was significantly diminished in FAK-deficient SMC. We next evaluated the ability of FAK-containing and FAK null SMC to degrade DQ-gelatin, which fluoresces upon degradation allowing quantification of protease activity via fluorescent spectrophotometry. As shown in figure 6C, significantly lower levels of gelatin degradation occurred in FAK-depleted SMC compared to control SMC, confirming that SMC matrix protease activity is FAK-dependent. In addition, we found that invasion of FAK null SMC into matrigel was significantly impaired in comparison to wt SMC (Fig 6D). These studies indicate that FAK is necessary for both PDGF-stimulated chemotaxis and invasive cell migration; and may explain, at least in part, the selective requirement for FAK in coronary and OFT morphogenesis, as these processes each require SMC to cross tissue/ECM boundaries.
Figure 6. FAK is necessary for SMC-mediated extracellular matrix degradation.

(A) LacZ- or Cre-infected fakflox/flox SMC were plated on Oregon Green 488 gelatin/FN matrix in serum-free medium and treated with vehicle or PDGF-BB for 90 minutes. Cells were fixed and co-stained with anti-FAK antibody and phalloidin. (B) Cells were scored for the presence of degradation puncta (black spots) and data are presented as puncta/per cell normalized to values for the LacZ-infected SMC following PDGF treatment. Data represent mean±SEM of at least 200 cells from 3 independent experiments. *p< .05. (C). LacZ- or Cre-infected fakflox/flox SMC were plated on DQ-gelatin-coated 96-well plate and treated with PDGF-BB for 90 minutes. Fluorescence intensity was monitored at Ex/Em 495/515 nm. Data represent mean±SEM of 3 independent experiments. *p< .05. (D) GFP and LacZ or Cre co-infected fakflox/flox SMC were Cells treated as above were plated on matrigel-coated inserts (10 μg/ml; Bio-Coat) in serum-free media using either PDGF-BB (20 ng/ml) as the chemoattractant. Invading cells were counted by direct fluorescence at 10X magnification. Data represent mean±SEM of 3 independent experiments. ***p< .001.
Discussion
Our studies using three independent Cre lines that induce recombination in SM precursor cells revealed a SMC autonomous role for FAK in vascular development. Depletion of FAK in wnt-1- and nkx2.5-derived cells (i.e. FAKwnt and FAKnk embryos respectively) led to aberrant septation of the great vessels; while depletion of FAK in pro-epicardial wt1 -derivatives (i.e. FAKcSMC embryos) led to defective coronary vascular formation. Concomitantly, the OFT and coronary vessels were distended by P0, likely due to a block in vessel tone normally imparted by SMC and SMC-elaborated basement membrane components. Phenotypic assessment of each mouse model revealed that the FAK-null SMC were specified, but failed to be appropriately recruited to established endothelial tubes. Our studies using FAK-null SMC cultures revealed a selective function for FAK in regulating PDGF-dependent motility, but not growth or survival. Specifically, we found that FAK depletion decreased PDGF-stimulated leading edge persistence, cell migration speed, and directional chemotaxis. Given the fact that blockade of PDGF led to defects in recruitment of vascular smooth muscle cells to the developing vasculature28, 29 and CNC- and epicardial-targeted ablation of PDGFR α/β, led to fully penetrant PTA and defective coronary vasculature respectively5, 7, we speculate that the morphogenesis of these vascular structures involves FAK-dependent SMC motility/chemotaxis induced by endothelial derived PDGFs.
Interestingly, PTA has been associated with a block in SMC differentiation within the truncus of mice harboring null mutations in the BMP receptor, Alk-2 and MRTF-B as well as those with CNC-targeted expression of a dominant negative Notch variant18, 30-32. However, similar to our studies, CNC-targeted ablation of TGFβR2, PDGFBBR α/β, and Gata 6 each led to fully penetrant PTA without a noticeable reduction in SMC number or SMC maturation5, 6, 33. Interestingly, the interrupted arch phenotype observed in CNC-specific FAK knockout mice recently reported by Vallejo-Illarramemdi et al. was attributed to impaired SMC differentiation of a subset of CNC (within the aortic arch), although the authors observed normal differentiation of CNC cells into SMC within the aorticopulmonary septum 34. Thus, the underlying cause for impaired septation was not identified in the aforementioned manuscript. As we did, this group observed normal migration and growth of CNC in FAKwnt embryos as well as normal differentiation of neural crest cells into SMC within the aorticopulmonary septum. These findings coupled with our mechanistic studies in FAK null SMC suggest that precise regulation of SMC phenotype within the walls of the aortic sac is necessary for subsequent formation of the aorticopulmonary septum. Since PDGF 29 and now FAK have been implicated in the promotion of SMC chemotaxis, it is tempting to speculate that the final septation event may be due to coordinated movement of CNC-derived SMC to the conus. Mis-localization of these cells could therefore lead to impaired coverage of the outflow tract vessels and impaired force generation within the conus that could impact the dynamic morphogenetic movements during septogenesis.
Because formation of the outflow tract is extremely complex, and involves multiple cell types with interconnected functions, we chose a separate in vivo model (coronary vasculogenesis) to confirm a role for FAK in SMC chemotaxis. The epicardium contains progenitor cells, the major source of SMC that will eventually line the coronary vessels. Elegant lineage tracing studies have shown that SMC precursors are specified in the pro-epicardium prior to spreading of this epithelial tissue around the heart 35-37. In response to signals from the myocardium, these specified cells undergo an epithelial to mesenchyme transition (EMT), delaminate from the epicardium, and move into the sub-epicardial space. Epicardial-derived mesenchymal cells (EPDCs) will eventually be induced to migrate into the myocardium and to differentiate into definitive cell types; predominantly the SMC that cover the primary endothelial plexus but also some endothelial and myocardial cells20. The endothelial primary plexus originates from the atrial-ventricular groove at the base of the heart and gradually extends to the apex between E11.5-E13.5 in response to prior wave-like secretion of tropic factors from the myocardium (VEGFA and B) and epicardium (sonic hedgehog)21 an event that is necessary to support mid-gestational growth. Since the investment of the coronary plexus with mature SMC occurs in a similar (albeit delayed) spatial-temporal pattern, it has been postulated that endothelial-derived factors initiate the recruitment of epicardial-derived SMC. While the spatial-temporal coupling of EPDC differentiation and migration has not been well characterized, the possibility that SMC differentiation may occur prior to chemotaxis/invasion towards the endothelial plexus is supported by the recent finding that cardiac-restricted depletion of the chemoattractant, thymosin β-4 resulted in robust α-SMA stained cells that aberrantly lined the epicardium (at E14.5 in mouse), but failed to invade the myocardium38.
The following findings from our studies support the hypothesis that activation of FAK in SMC by endothelial-derived signals regulates coronary SMC recruitment to (and along) the developing arterioles: 1) defects were observed in SMC coverage of arterioles in FAKcSMC hearts but not in the formation or function of the endothelial plexus (as assessed by PECAM staining and appropriate mid-gestational heart growth); 2) no significant difference was observed in either the rates of proliferation or apoptosis in FAK-null precursor cells, and 3) significantly higher numbers of α-SMA-positive cells were apparent in the sub-epicardial space (as opposed to along the endothelial plexus) of the conditional mutants at P0, indicating that FAK was not essential for epicardial-mesenchymal transition, but was necessary for movement into the myocardium. These findings coupled with our in vitro evidence that FAK is necessary for chemotaxis/invasion towards the major endothelial-derived chemo-attractant, PDGFBB, indicate that lack of directional motility is likely causal for the SMC investment phenotypes observed in the SMC-selective FAK knock-outs. However, it is formally possible that defects in other parameters not evaluated herein could contribute to the defects observed.
Our mechanistic studies revealed that depletion of FAK markedly impaired PDGF-stimulated formation of a stable leading edge lamellipodium, the hallmark of polarized movement. This defect was accompanied by reduced leading edge recruitment of cortactin, a branch filament stabilizer that is essential for lamellipodial persistence and polarized motility 39. Moreover, cortactin knock-down was shown to affect lammellipodial activity and directional chemotaxis in exactly the same way as depletion of FAK, suggesting that the functions of these two proteins are interrelated. Interestingly, we found that formation of circular dorsal ruffles in response to PDGF does not require Rac1 activation, cortactin, or FAK. While the function of these transient dorsal ruffles is not completely understood there is considerable support for the idea that they are formed as a prelude to leading edge lamella and are important for recruitment and recycling of membrane and actin polymerizing/depolymerizing agents to the presumptive leading edge. Indeed, dorsal ruffles contain many of the same actin regulating components as leading edge lamella including (among others) ARP2/3, WASP1/2, WAVE, dynamin, and cofilin 40. We surmise that FAK regulates leading edge and not dorsal ruffle formation because FAK is essential for the spatial re-distribution of active Rac1 from these complexes to the leading edge.
Since FAK is one of the first proteins recruited to nacient focal complexes, we surmise that FAK initiates recruitment of active Rac1 to these newly formed sites via multifunctional protein complex formation. FAK associates with a number of adapter molecules through well defined protein interaction sites that could serve as a Rac1 binding platforms. FAK-dependent recruitment of Rac1 could proceed through FAK/(CAS or paxillin)/Crk/DOCK 180 complex formation 41, 42 or a paxillin/PIX/COOL complex 43. Whether FAK-dependent activation of Rac1 at the leading edge is linked to the impaired spatial-temporal leading edge specific re-distribution (and/or phosphorylation) of these or other FAK substrates are important questions for future studies.
There are a few possible explanations for why FAK might be essential for SMC investment of the conotruncus and coronary vasculature, but not for the recruitment of SMC to the distal vasculature (i.e. carotid arteries). First, there may be cell type specific differences in the factors that drive chemotaxis or regional differences in the levels and locale of these so-called motogens. In this regard, it is interesting to note that SMC chemotaxis toward PDGF was exquisitely sensitive to depletion of FAK, while chemotaxis toward serum was only modestly decreased in FAK null SMC (indicating that there are likely FAK-dependent and FAK-independent SMC motogens). The findings that depletion of PDGF receptors from CNC- or proepicardial cells phenocopies the outflow tract and coronary vessel defects observed in our FAK-depleted models and that PDGFs are expressed in high levels within the conotruncus 5-7, supports the possibility that PDGF and FAK may work in concert to spatially regulate SMC motility in these regions. The second (not mutually exclusive) possibility is that a SMC invasive phenotype (that requires FAK activity) is required for SMC to cross tissue boundaries during both septal formation and coronary vasculogenesis (but not carotid vasculogenesis). Indeed, it may be possible that SMC coverage of carotid vessels occurs by a sheet-like movement/expansion of SMC along the continuously remodeling SMC-lined pharyngeal arches. In support of the possibility that FAK promotes an invasive phenotype, we showed that FAK is necessary for SMC podosome-mediated matrix degradation, and others have recently reported that FAK is essential for invadopodia-mediated matrix degradation in colon cancer cells44. Lack of appropriate cortactin recruitment could also be causal for the impaired invasion observed in the FAK null SMC, since cortactin has been shown to be necessary for regulating the localized recruitment and secretion of matrix metalloproteinases from lamellipodial-like membrane protrusions26. Future studies will be necessary to more fully understand the requirement for SMC invasion versus chemotaxis during the formation of various vascular beds.
In conclusion, our studies indicate that FAK plays a SMC autonomous role in aorticopulmonary septum and coronary vessel formation. Our studies in cultured SMC indicate that FAK activity is critical for SMC chemotaxis/invasion toward PDGF, a potent chemoattractant elaborated from the endothelium. Thus, we surmise that defective PDGF-dependent SMC recruitment leads to improper morphogenesis of FAK null vessels. We found that FAK functions to induce directional SMC motility by regulating the spatial and temporal locale of Rac1-dependent processes, including leading edge recruitment of the actin modifier, cortactin, which is necessary to stabilize lamellipodia and to form productive podosomes. It will be of future interest to evaluate the extent to which FAK may play a role in the pathogenesis of microaneurysms and other vascular diseases associated with defective recruitment of PDGF-Rβ positive progenitors 28.
Supplementary Material
Acknowledgments
Sources of Funding: This work was supported in part by grants from NIH-NHLBI (HL-081844 and HL-071054) and the American Heart Association (AHA. 0355776U) to J.M.T, NIH-NHLBI (HL070953) and AHA (0555476U) to C.P.M and NIH HL-19242 and HL-93594 to M.W.M. L.S.S was supported by an American Heart pre-doctoral fellowship (0515329U).
Mouse Line Abbreviations
- FAKnk
fakflox/- nkx2.5Cre/+
- FAKwnt
fakflox/- wnt-1Cre/+ / Rosa26RlacZ/wt
- FAKcSMC
fakflox/- wt-1Cre/+ / Rosa26RlacZ/wt
Footnotes
Disclosures: None.
References
- 1.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 2.Ninomiya K, Takahashi A, Fujioka Y, Ishikawa Y, Yokoyama M. Transforming growth factor-beta signaling enhances transdifferentiation of macrophages into smooth muscle-like cells. Hypertens Res. 2006;29:269–276. doi: 10.1291/hypres.29.269. [DOI] [PubMed] [Google Scholar]
- 3.Sinha S, Hoofnagle MH, Kingston PA, McCanna ME, Owens GK. Transforming growth factor-beta1 signaling contributes to development of smooth muscle cells from embryonic stem cells. Am J Physiol Cell Physiol. 2004;287:C1560–1568. doi: 10.1152/ajpcell.00221.2004. [DOI] [PubMed] [Google Scholar]
- 4.Lee WC, Rubin JP, Marra KG. Regulation of alpha-smooth muscle actin protein expression in adipose-derived stem cells. Cells Tissues Organs. 2006;183:80–86. doi: 10.1159/000095512. [DOI] [PubMed] [Google Scholar]
- 5.Richarte AM, Mead HB, Tallquist MD. Cooperation between the PDGF receptors in cardiac neural crest cell migration. Dev Biol. 2007;306:785–796. doi: 10.1016/j.ydbio.2007.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Choudhary B, Ito Y, Makita T, Sasaki T, Chai Y, Sucov HM. Cardiovascular malformations with normal smooth muscle differentiation in neural crest-specific type II TGFbeta receptor (Tgfbr2) mutant mice. Dev Biol. 2006;289:420–429. doi: 10.1016/j.ydbio.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 7.Mellgren AM, Smith CL, Olsen GS, Eskiocak B, Zhou B, Kazi MN, Ruiz FR, Pu WT, Tallquist MD. Platelet-derived growth factor receptor beta signaling is required for efficient epicardial cell migration and development of two distinct coronary vascular smooth muscle cell populations. Circ Res. 2008;103:1393–1401. doi: 10.1161/CIRCRESAHA.108.176768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bouvard D, Brakebusch C, Gustafsson E, Aszodi A, Bengtsson T, Berna A, Fassler R. Functional consequences of integrin gene mutations in mice. Circ Res. 2001;89:211–223. doi: 10.1161/hh1501.094874. [DOI] [PubMed] [Google Scholar]
- 9.George EL, Baldwin HS, Hynes RO. Fibronectins are essential for heart and blood vessel morphogenesis but are dispensable for initial specification of precursor cells. Blood. 1997;90:3073–3081. [PubMed] [Google Scholar]
- 10.Ilic D, Kovacic B, McDonagh S, Jin F, Baumbusch C, Gardner DG, Damsky CH. Focal adhesion kinase is required for blood vessel morphogenesis. Circ Res. 2003;92:300–307. doi: 10.1161/01.res.0000055016.36679.23. [DOI] [PubMed] [Google Scholar]
- 11.Hakim ZS, DiMichele LA, Doherty JT, Homeister JW, Beggs HE, Reichardt LF, Schwartz RJ, Brackhan J, Smithies O, Mack CP, Taylor JM. Conditional deletion of focal adhesion kinase leads to defects in ventricular septation and outflow tract alignment. Mol Cell Biol. 2007;27:5352–5364. doi: 10.1128/MCB.00068-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wurdak H, Ittner LM, Sommer L. DiGeorge syndrome and pharyngeal apparatus development. Bioessays. 2006;28:1078–1086. doi: 10.1002/bies.20484. [DOI] [PubMed] [Google Scholar]
- 13.Waldo KL, Hutson MR, Ward CC, Zdanowicz M, Stadt HA, Kumiski D, Abu-Issa R, Kirby ML. Secondary heart field contributes myocardium and smooth muscle to the arterial pole of the developing heart. Dev Biol. 2005;281:78–90. doi: 10.1016/j.ydbio.2005.02.012. [DOI] [PubMed] [Google Scholar]
- 14.Jiang X, Rowitch DH, Soriano P, McMahon AP, Sucov HM. Fate of the mammalian cardiac neural crest. Development. 2000;127:1607–1616. doi: 10.1242/dev.127.8.1607. [DOI] [PubMed] [Google Scholar]
- 15.Beggs HE, Schahin-Reed D, Zang K, Goebbels S, Nave KA, Gorski J, Jones KR, Sretavan D, Reichardt LF. FAK deficiency in cells contributing to the basal lamina results in cortical abnormalities resembling congenital muscular dystrophies. Neuron. 2003;40:501–514. doi: 10.1016/s0896-6273(03)00666-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moses KA, DeMayo F, Braun RM, Reecy JL, Schwartz RJ. Embryonic expression of an Nkx2-5/Cre gene using ROSA26 reporter mice. Genesis. 2001;31:176–180. doi: 10.1002/gene.10022. [DOI] [PubMed] [Google Scholar]
- 17.Norden J, Grieskamp T, Lausch E, van Wijk B, van den Hoff MJ, Englert C, Petry M, Mommersteeg MT, Christoffels VM, Niederreither K, Kispert A. Wt1 and retinoic acid signaling in the subcoelomic mesenchyme control the development of the pleuropericardial membranes and the sinus horns. Circ Res. 2010;106:1212–1220. doi: 10.1161/CIRCRESAHA.110.217455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.High FA, Zhang M, Proweller A, Tu L, Parmacek MS, Pear WS, Epstein JA. An essential role for Notch in neural crest during cardiovascular development and smooth muscle differentiation. J Clin Invest. 2007;117:353–363. doi: 10.1172/JCI30070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Waldo KL, Hutson MR, Stadt HA, Zdanowicz M, Zdanowicz J, Kirby ML. Cardiac neural crest is necessary for normal addition of the myocardium to the arterial pole from the secondary heart field. Dev Biol. 2005;281:66–77. doi: 10.1016/j.ydbio.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 20.Zhou B, Ma Q, Rajagopal S, Wu SM, Domian I, Rivera-Feliciano J, Jiang D, von Gise A, Ikeda S, Chien KR, Pu WT. Epicardial progenitors contribute to the cardiomyocyte lineage in the developing heart. Nature. 2008;454:109–113. doi: 10.1038/nature07060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lavine KJ, Ornitz DM. Shared circuitry: developmental signaling cascades regulate both embryonic and adult coronary vasculature. Circ Res. 2009;104:159–169. doi: 10.1161/CIRCRESAHA.108.191239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Van Den Akker NM, Lie-Venema H, Maas S, Eralp I, DeRuiter MC, Poelmann RE, Gittenberger-De Groot AC. Platelet-derived growth factors in the developing avian heart and maturating coronary vasculature. Dev Dyn. 2005;233:1579–1588. doi: 10.1002/dvdy.20476. [DOI] [PubMed] [Google Scholar]
- 23.del Pozo MA, Price LS, Alderson NB, Ren XD, Schwartz MA. Adhesion to the extracellular matrix regulates the coupling of the small GTPase Rac to its effector PAK. Embo J. 2000;19:2008–2014. doi: 10.1093/emboj/19.9.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Weed SA, Du Y, Parsons JT. Translocation of cortactin to the cell periphery is mediated by the small GTPase Rac1. J Cell Sci. 1998;111:2433–2443. doi: 10.1242/jcs.111.16.2433. [DOI] [PubMed] [Google Scholar]
- 25.Furmaniak-Kazmierczak E, Crawley SW, Carter RL, Maurice DH, Cote GP. Formation of extracellular matrix-digesting invadopodia by primary aortic smooth muscle cells. Circ Res. 2007;100:1328–1336. doi: 10.1161/CIRCRESAHA.106.147744. [DOI] [PubMed] [Google Scholar]
- 26.Clark ES, Weaver AM. A new role for cortactin in invadopodia: regulation of protease secretion. Eur J Cell Biol. 2008;87:581–590. doi: 10.1016/j.ejcb.2008.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ayala I, Baldassarre M, Giacchetti G, Caldieri G, Tete S, Luini A, Buccione R. Multiple regulatory inputs converge on cortactin to control invadopodia biogenesis and extracellular matrix degradation. J Cell Sci. 2008;121:369–378. doi: 10.1242/jcs.008037. [DOI] [PubMed] [Google Scholar]
- 28.Lindahl P, Johansson BR, Leveen P, Betsholtz C. Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science. 1997;277:242–245. doi: 10.1126/science.277.5323.242. [DOI] [PubMed] [Google Scholar]
- 29.Stratman AN, Schwindt AE, Malotte KM, Davis GE. Endothelial-derived PDGF-BB and HB-EGF coordinately regulate pericyte recruitment during vasculogenic tube assembly and stabilization. Blood. 2010;116:4720–4730. doi: 10.1182/blood-2010-05-286872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kaartinen V, Dudas M, Nagy A, Sridurongrit S, Lu MM, Epstein JA. Cardiac outflow tract defects in mice lacking ALK2 in neural crest cells. Development. 2004;131:3481–3490. doi: 10.1242/dev.01214. [DOI] [PubMed] [Google Scholar]
- 31.Oh J, Richardson JA, Olson EN. Requirement of myocardin-related transcription factor-B for remodeling of branchial arch arteries and smooth muscle differentiation. Proc Natl Acad Sci U S A. 2005;102:15122–15127. doi: 10.1073/pnas.0507346102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li J, Zhu X, Chen M, Cheng L, Zhou D, Lu MM, Du K, Epstein JA, Parmacek MS. Myocardin-related transcription factor B is required in cardiac neural crest for smooth muscle differentiation and cardiovascular development. Proc Natl Acad Sci U S A. 2005;102:8916–8921. doi: 10.1073/pnas.0503741102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lepore JJ, Mericko PA, Cheng L, Lu MM, Morrisey EE, Parmacek MS. GATA-6 regulates semaphorin 3C and is required in cardiac neural crest for cardiovascular morphogenesis. J Clin Invest. 2006;116:929–939. doi: 10.1172/JCI27363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vallejo-Illarramendi A, Zang K, Reichardt LF. Focal adhesion kinase is required for neural crest cell morphogenesis during mouse cardiovascular development. J Clin Invest. 2009;119:2218–2230. doi: 10.1172/JCI38194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ratajska A, Czarnowska E, Ciszek B. Embryonic development of the proepicardium and coronary vessels. Int J Dev Biol. 2008;52:229–236. doi: 10.1387/ijdb.072340ar. [DOI] [PubMed] [Google Scholar]
- 36.Majesky MW. Developmental basis of vascular smooth muscle diversity. Arterioscler Thromb Vasc Biol. 2007;27:1248–1258. doi: 10.1161/ATVBAHA.107.141069. [DOI] [PubMed] [Google Scholar]
- 37.Mikawa T, Fischman DA. Retroviral analysis of cardiac morphogenesis: discontinuous formation of coronary vessels. Proc Natl Acad Sci U S A. 1992;89:9504–9508. doi: 10.1073/pnas.89.20.9504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Smart N, Risebro CA, Melville AA, Moses K, Schwartz RJ, Chien KR, Riley PR. Thymosin beta4 induces adult epicardial progenitor mobilization and neovascularization. Nature. 2007;445:177–182. doi: 10.1038/nature05383. [DOI] [PubMed] [Google Scholar]
- 39.Bryce NS, Clark ES, Leysath JL, Currie JD, Webb DJ, Weaver AM. Cortactin promotes cell motility by enhancing lamellipodial persistence. Curr Biol. 2005;15:1276–1285. doi: 10.1016/j.cub.2005.06.043. [DOI] [PubMed] [Google Scholar]
- 40.Buccione R, Orth JD, McNiven MA. Foot and mouth: podosomes, invadopodia and circular dorsal ruffles. Nat Rev Mol Cell Biol. 2004;5:647–657. doi: 10.1038/nrm1436. [DOI] [PubMed] [Google Scholar]
- 41.Schaller MD, Parsons JT. pp125FAK-dependent tyrosine phosphorylation of paxillin creates a high- affinity binding site for Crk. Mol Cell Biol. 1995;15:2635–2645. doi: 10.1128/mcb.15.5.2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kiyokawa E, Hashimoto Y, Kurata T, Sugimura H, Matsuda M. Evidence that DOCK180 up-regulates signals from the CrkII-p130(Cas) complex. J Biol Chem. 1998;273:24479–24484. doi: 10.1074/jbc.273.38.24479. [DOI] [PubMed] [Google Scholar]
- 43.Manser E, Loo TH, Koh CG, Zhao ZS, Chen XQ, Tan L, Tan I, Leung T, Lim L. PAK kinases are directly coupled to the PIX family of nucleotide exchange factors. Mol Cell. 1998;1:183–192. doi: 10.1016/s1097-2765(00)80019-2. [DOI] [PubMed] [Google Scholar]
- 44.Yu HG, Nam JO, Miller NL, Tanjoni I, Walsh C, Shi L, Kim L, Chen XL, Tomar A, Lim ST, Schlaepfer DD. p190RhoGEF (Rgnef) promotes colon carcinoma tumor progression via interaction with focal adhesion kinase. Cancer Res. 2011;71:360–370. doi: 10.1158/0008-5472.CAN-10-2894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Geary GG, Krause DN, Duckles SP. Estrogen reduces mouse cerebral artery tone through endothelial NOS- and cyclooxygenase-dependent mechanisms. Am J Physiol Heart Circ Physiol. 2000;279:H511–519. doi: 10.1152/ajpheart.2000.279.2.H511. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.