

Abstract
The in vitro expression and electrophysiological recording of recombinant voltage-gated ion channels in cultured human embryonic kidney cells (HEK-293T) is a ubiquitous research strategy. HEK-293T cells must be plated onto glass coverslips at low enough density so that they are not in contact with each other in order to allow for electrophysiological recording without confounding effects due to contact with adjacent cells. Transfected channels must also express with high efficiency at the plasma membrane for whole-cell patch clamp recording of detectable currents above noise levels. Heterologous ion channels often require long incubation periods at 28°C after transfection in order to achieve adequate membrane expression, but there are increasing losses of cell-coverslip adhesion and membrane stability at this temperature. To circumvent this problem, we developed an optimized strategy to transfect and plate HEK-293T cells. This method requires that cells be transfected at a relatively high confluency, and incubated at 28°C for varying incubation periods post-transfection to allow for adequate ion channel protein expression. Transfected cells are then plated onto glass coverslips and incubated at 37°C for several hours, which allows for rigid cell attachment to the coverslips and membrane restabilization. Cells can be recorded shortly after plating, or can be transferred to 28°C for further incubation. We find that the initial incubation at 28°C, after transfection but before plating, is key for the efficient expression of heterologous ion channels that normally do not express well at the plasma membrane. Positively transfected, cultured cells are identified by co-expressed eGFP or eGFP expressed from a bicistronic vector (e.g. pIRES2-EGFP) containing the recombinant ion channel cDNA just upstream of an internal ribosome entry site and an eGFP coding sequence. Whole-cell patch clamp recording requires specialized equipment, plus the crafting of polished recording electrodes and L-shaped ground electrodes from borosilicate glass. Drug delivery to study the pharmacology of ion channels can be achieved by directly micropipetting drugs into the recording dish, or by using microperfusion or gravity flow systems that produce uninterrupted streams of drug solution over recorded cells.
Protocol
1. Cell Culture
Human embryonic kidney 293T cells (HEK-293T, Invitrogen) are grown in adherent monolayers in vented 25cm2 flasks (tissue culture treated; Cellstar; greiner bio-one) containing 6mL of Dulbecco's Modified Eagles Medium (DMEM; Sigma Aldrich) supplemented with 10% v/v fetal bovine serum (FBS; Sigma Aldrich), 1% sodium pyruvate, and 250 μg/mL penicillin/streptomycin at 37°C with 5% CO2). Cells are incubated in CO2 incubators (5% CO2) set to either 37°C or 28°C. All work with cells is done in a laminar flow hood.
Cells are typically split bi-weekly, from ~90% confluency, in a 1:8 dilution on Mondays, and 1:12 on Thursdays. (See Figure 1 for images of HEK-293T cells plated at different densities)
For splitting, media is removed from flasks of cells by aspiration, and cells are detached by applying 1mL of Trypsin (Sigma Aldrich) solution heated to 37°C to the inverted flask. The flask is subsequently inverted and gently rocked by hand such that the trypsin solution moves over the attached cells several times. The trypsin is then quickly removed by aspiration and 250 μL of trypsin is micropipetted into the flask which is once again rocked to move the trypsin solution over the cells.
Flasks are then incubated in a CO2 incubator set to 37°C for 3-5 minutes, and a pipette is used to suspend the cells in 4mL of warm supplemented DMEM. Incomplete trypsinization and suspension will cause clumps of cells and could cause cells to grow upwards and not in the intended monolayer. Over-trypsinization may damage the cells.
For a 1:8 split, add 500μL of suspended cells are added to a new flask containing 5.5mL of DMEM; for a 1:12 split (see Figure 1), 333μL of cells are added to a new flask containing 5.7mL of DMEM. Flasks are gently mixed then transferred to a 37°C incubator. During the 3-4h incubation at 37°C after splitting, the cells will sediment and adhere to the flask. The cells initially look round but will flatten and have extensions (pseudopodia) with firmer attachment (Figure 1). Cells should grow in an adherent monolayer in a predictable manner. Should cell growth and appearance be atypical or inconsistent, poor technique or contamination (bacteria/mycoplasma) should be considered. The key to successful tissue culture is consistency.
2. Transfection of HEK-293T with recombinant ion channel cDNAs
Here, cDNAs from various ion channels are cloned into the pIRES2-EGFP plasmid (BD Biosciences Clontech), which allows for the identification of positively transfected cells via enhanced green fluorescent protein (eGFP) fluorescence. The eGFP is produced in transfected cells from the same transcript as the cloned insert cDNA, through translation initiated at an internal ribosome entry site just downstream of the insert cDNA and upstream of the eGFP coding sequence (Figure 2). See Figure 3 for images of fluorescent HEK-293T, positively transfected with calcium channel subunits in pIRES2-EGFP constructs.
Prior to transfection, a fully confluent flask is split 1:4 into a new flask, and the cells are allowed to attach at 37°C in a CO2 incubator for 3-4 hrs (see Figure 1).
After cells are attached, a standard calcium phosphate transfection strategy (Cold Spring Harbor protocols) is used to introduce constructs containing ion channel and accessory subunit cDNAs into the HEK cells. Typically, a total of 12μg of DNA in 600 μL of transfection solution ise used per flask/transfection.
After the transfection solution is pipetted over the cells drop-wise, the flask is placed into a 37°C incubator over night.
Following transfection, cells are carefully washed three times with warm media or phosphate buffered saline (4-5 mL) by pipetting the wash solution into an inverted flask, then slowly turning the flask upright and rocking the flask so that the wash solution moves over the cells. If this is not done carefully the cells could detatch and few will be left after the three wash steps. The wash is repeated two more times, then 6 mL of media are added to the flask and it is incubated at 37°C for 2 hrs, then transferred to a 28°C incubator.
Successful transfection of pIRES2-EGFP constructs can be confirmed 1 to 3 days after transfection using an epifluorescence microscope. Cells that fluoresce green when exposed to UV light are positively-transfected and should express the heterologous ion channel and eGFP proteins.
3. Plating transfected HEK293T cells in preparation for electrophysiological recording
Different ion channels express with different efficiencies at the cell membrane of HEK cells, and as such, the remainder of this protocol requires some optimization to maximize channel surface expression allowing for successful electrophysiological recording. Expression of heterologous ion channels in HEK cells, particularly of those from non-mammalian sources, may present some challenges including (1) large protein sizes requiring long times to translate and localize to the plasma membrane (2) absence or dissimilarities of folding and targeting motifs on the channel protein necessary in a mammalian cell line (3) differences in codon usage frequencies from species to species. All of these factors combined can result in a requirement for long incubation times at 28°C, which ensures effective protein expression and surface localization without cell division (which would dilute out the heterologous cDNAs and proteins). Unfortunately, incubation for prolonged periods at 28°C leads to cell detachment and poor membrane stability making electrophysiological recording difficult. We decided to modify conventional transfection strategies (reviewed in Thomas and Smart 2005) in order to minimize incubation at 28°C prior to recording and to maximize surface expression. Our method requires that cells be transfected, incubated for a period of 3-7 days at 28°C, then cells are detached by trypsinization and replated onto coverslips and incubated at 37°C which restabilizes membranes and allows for strong attachment of cells onto coverslips. Cells are then able to be recorded right away, or can be incubated for additional periods of time at 28°C. We find the replating of cells onto coverslips at this later stage in our method to be absolutely essential for producing coverslips with high numbers of channel-expressing, separate attached cells.
We present two alternative strategies, one that we prefer to use for ion channels and ion channel complexes that do not contain large extracellular domains that may be vulnerable to trypsin digestion (e.g. Cav3 voltage-gated calcium channels; Senatore and Spafford 2010), and one that we use for ion channels that do contain large extracellular domains (e.g. L-type voltage-gated calcium channels and their accessory subunits; Senatore, et al. 2010).
- Cells are incubated at 28°C for 2-6 days to prevent cell division and to allow for accumulation of translated protein. This incubation time must be determined empirically for each ion channel, since surface expression is dependent on intrinsic factors that dictate how the channels' nascent mRNA and polypeptide sequences interact with the translational machinery and the secretory pathway, respectively.
- Prior to plating, circular glass coverslips (Circles No. 1 - 0.13 to 0.17mm thick; Size: 12 mm, Fisher Scientific Canada, Ottawa, Ontario) are coated with Poly-L-lysine (Sigma) as per manufacturer's instructions.
- Media is removed by aspiration from transfected cells, and 1 mL of 37°C trypsin solution (Sigma) is micropipetted into an inverted flask (cell side up). The flask is then gently inverted to the upright position to allow the trypsin to run over the cells, and the flask is then re-inverted and the trypsin removed by aspiration. 250 μL of warm trypsin is then added directly over the cells and the flask is incubated at 37°C for 3-5 minutes until the cells detatch.
- Meanwhile, poly-L-lysine-coated coverslips are placed into 6mm culture dishes (3-8 per dish) and 5 mL of warm culture media is added to each dish.
- Trypsinized cells are resuspended with 4mL of warm culture media by pipetting up and down ~6 times, then 250-500μL of resuspended cells are micropipetted into the culture dishes containing coverslips. Prior to addition of cells, the tip of the micropipette is used to push down on the coverslips to ensure that they are at the bottom of the dish.
- The cells are then incubated at 37°C for 3hrs to allow them to adhere to the coverslips, and then they are transferred to 28°C. At this point, the cells are ready for electrophysiological recording, which is ideally done with separate-attached cells (Fig.3 illustrates transfected cells before and after plating onto coverslips). Leaving the cells for too long at 37°C will result in cell division.
- Preparation of cells expressing channels/channel complexes with large extracellular domains is carried out in much the same way as indicated above, with the exception that coverslips are incubated at 28°C for an additional 2-4 days before electrophysiological recording is carried out. This allows for a re-accumulation of channels/channel complexes at the cell membrane after trypsinization.
4. Electrophysiological recording of recombinant channels in HEK-293T cells
Several comprehensive resources are available that describe the general concepts underlying electrophysiological recording (e.g. Ogden and Stanfield 1987; Molleman 2003).
4.1 Electrophysiology rig
A typical electrophysiology rig (Figure 4) includes an amplifier (e.g. Axopatch 200B or Multiclamp 700B amplifier; Axon Instruments, Union City, CA), a PC computer equipped with a Digidata 1440A analog-to-digital converter interface in conjunction with pClamp10.1 software (Molecular Devices, Sunnyvale, California), motorized, dual pipette manipulators (MPC-385-2, Sutter Instrument Company), an epifluorescence microscope (Axiovert 40 CFL, Zeiss Canada, Toronto, Ontario) and perfusion systems (Figures 4 and 5). Vibrations are limited by mounting the equipment on a TMC 63-500 series vibration isolation table (Technical Manufacturing Corporation, Peabody. MA). Stray electrical noise is limited using a 40" tall Type II Faraday Cage (TMC) surrounding the electrical equipment mounted on the vibration isolation table. Electronic control systems (such as computer & monitor, amplifier, analog-to-digital converter, motorized manipulator and Valvelink perfusion control system) are mounted outside of the Faraday cage to a free-standing metal electrical rack (Hammond Manufacturing, Guelph Ontario). All electrical equipment is grounded to a copper distributor and plugged into a Medical Grade Isolation Tranformer (1800W, Tripp Lite UL60601-1) which provides line isolation, a consistent ground and surge suppression.
4.2 Patch pipettes
Borosilicate glass capillary tubes with filament (O.D. 1.5mm, O.D. 0.86mm, 15cm length; BF150-86-15; Sutter Instrument Company) are cut in half and inserted into a Flaming/Brown Micropipette Puller Model P-97 (Sutter Instrument Company). A pipette-puller program is optimized to consistently generate pipettes with resistances between 2 to 5MΩ from electrode to ground (after fire polishing; Fig.6) Pipettes are individually fire polished to smooth the pipette tip surfaces which allows for tight seals against cell membranes (Micro Forge MF-830; Narishige, Japan; Figure 6). Micropipettes are mounted on the headstage with a specialized holder (1-HL-U; Molecular Devices, Sunnyvale California).
4.3 Ground electrodes
The borosilicate glass tubes used to make patch pipettes are also used to make reference electrodes. The 15cm long tubes are cut into 3 pieces. With 2 fine-tipped forceps, the tubes are held in a Bunsen burner flame until the glass becomes malleable and the tubing can be bent to an "L" or "hockey stick" shape with a 90o angle. While the glass tubes are still hot, one end is immediately immersed in a hot 3M CsCl and 1.5% agarose solution which is drawn into the tube by capillary action. Once a reference electrode is completely filled with solution, it is placed in a beaker of cold water. After all of the reference electrodes are filled, they are individually wiped with Kimwipes to remove extra agarose from the outside and are submerged in a 3M CsCl solution. For electrophysiological recording, the reference electrodes are held in a Microelectrode Holder Half-Cells (Holder 90 F Pellet 1.5mm; #MEH3RF15; World Precision Instruments Inc., Sarasota Florida) filled with 3M CsCl solution. (See Figure 7 for illustration of a ground electrode in bath solution)
4.4 Electrophysiology recording solutions
Different types of voltage-gated ion channels require different recording solutions depending on ion permeability/selectivity. For voltage-gated calcium channels, cells are typically bathed in external solutions containing either barium or calcium at various concentrations. For example, electrophysiological recording of the invertebrate Cav3 voltage-gated calcium channel (LCav3) can be performed using an external solution containing 5mM CaCl2, 166mM tetraethylammonium (TEA-Cl), 10mM HEPES with a pH adjusted to 7.4 with TEA-OH. Patch pipettes are filled with an internal solution of 125mM CsCl, 10mM EGTA, 2mM CaCl2, 1mM MgCl2, 4mM MgATP, 0.3mM Tris-GTP, and 10mM HEPES with a pH of 7.2 (Senatore and Spafford, 2010). The invertebrate L-type voltage-gated calcium channel (LCav1) can be recorded using an external solution containing barium as the charge carrier (15mM BaCl2, 1mM MgCl2, 10mM HEPES, 40mM TEA-Cl, 72.5mM CsCl, 10mM Glucose, with the pH adjusted to 7.2 with TEA-OH) and an internal solution containing 108mM Cs-methanesulfonate, 4mM MgCl2, 9mM EGTA, 9mM HEPES, pH adjusted to 7.2 with CsOH (Senatore et al., 2010).
4.5 Whole Cell Recording
A coverslip containing transfected cells (expressing heterologous ion channels or ion channel complexes) is transferred to a 35mm plastic petri dish containing ~3mL of external recording solution. The volume of recording solution must be measured accurately when performing drug experiments where known amounts of drug are to be added to the dish with a micropipette. Electrophysiological recordings are typically done at room temperature, although the researcher may wish to change this temperature depending on the experiment.
Cells are visualized through an epifluorescence microscope and an isolated, positively-transfected, adherent green cell is centered in the middle of the field of view.
The patch pipette is lowered into the bath solution using a micromanipulator, which completes the circuit between the electrode in the patch pipette and the reference electrode. While in bath mode, the pClamp10.1 software causes the amplifier to generate small repeating voltage steps between the pipette and the reference electrode, and displays the resulting current as a square wave trace (Figure 8A). This square wave trace must be zeroed using the pipette offset on the amplifier before patching a cell. The pipette resistance is measured by the pClamp10.1 software, and should be within 2-5MΩ.
While in bath mode, the pipette is brought into close proximity of the cell, which can be visualized as rapid fluctuations in the square wave trace, then a tiny amount of suction is applied to the patch pipette to form a gigaohm seal between the pipette and the membrane (causes a complete flat-line of the square wave trace; Figure 8B).
The program is then switched to patch mode, and pipette capacitance (visible as tiny blips of current on the leading and trailing edges of the flat current trace; Figure 8B) is compensated with the pipette capacitance compensation on the amplifier.
A small but sharp amount of suction is applied to rupture the membrane in the patch and allow for access of the electrode to the inside of the cell. Successful membrane rupture is indicated by the appearance of large capacitive transients also on the leading and trailing edges of the current wave trace (Figure 8C). Once break through has been achieved, the program is switched to cell mode and whole cell capacity compensation is adjusted on the amplifier to remove the capacitive transients. There is a delay of a few minutes to allow for equilibration of the intracellular recording solution into the cell prior to recording.
Voltage commands are generated and data is acquired using a computer equipped with a Digidata 1440A analog-to-digital converter interface in conjunction with pClamp10.1 software. Unique voltage clamp protocols are written for each experiment, and these are used to record ion channel currents from transfected cells. Figure 9 illustrates calcium currents recorded from an HEK-293T cell expressing the invertebrate LCav3 voltage-gated calcium channel. The cell was held at a voltage of -110mV, and incremental depolarisations between -70mV and +20mV were taken to elicit inward calcium currents.
Recorded currents are filtered at 10 kHz using a low-pass Bessel filter and digitized at a sampling frequency of 2 kHz. Only recordings with minimal leak (<10%) are used for analysis, and offline leak subtraction is carried out using the Clampfit 10.1 software.
4.6 Drug studies of recorded cells
Drugs may be directly pipetted into the dish being recorded. Concentration of drugs may be calculated if the volume of drug added and volume of bath solution are known. Expensive or limited quantities of drug may also be added by microperfusion from a multi-barrel manifold polyimide perfusion pencil with a 250 micron removable tip, using either an eight channel Smartsquirt pressure micro perfusion system (AutoMate Scientific) or gravity flow system operated through Teflon valves and Valvelink8 valve controllers (AutoMate Scientific; see Figure 5 for perfusion system setups). Flow rates from microperfusion systems are set at 0.1mL per minute in a stream exiting the perfusion pencil 400 microns from the recorded cell. The distance of the perfusion pencil tip from the recording electrode tip should be consistent from patch to patch; Figure 10 illustrates a suggested distance visible at 40x magnification. Adequate flow rates provide saturating perfusion of the recorded cell. Before a drug experiment is conducted using microperfusion, cells are equilibrated with the microperfusion of control bath solution in a continuous stream. Minimal interruption of the stream is avoided during the experiment by rapid switching from control bath, drug or wash solution. Suction mounted at the edge of the recording dish can be used to remove overflow of solutions, by means of manual suction with a syringe or using a peristaltic pump driven device (e.g. MINISTAR Miniature DC Peristaltic Pump, World Precision Instruments). Care must be taken since extremely rapid perfusion induces shear forces that may alter the calcium current (Peng et al. 2005), may wash away the cell being recorded, and depletes the drug reservoir too quickly.
Directly pipeting into dish vs. perfusion systems
| Direct pipetting | Micro Perfusion system | Gravity flow perfusion system | |
| Volume drug needed | Moderate | Low | High |
| Technical skill | Low | Medium | Medium |
| Equipment cost | Low | Significant | Significant |
| Daily Time to setup and cleanup | None | 20+ minutes | 20+ minutes |
| Contamination of equipment | None | Yes | Yes |
| Troubleshooting | Easy | Complex | Complex |
| Guaranteed drug in dish | Yes | No | No |
| Lose cell with drug addition | Yes | Yes | Yes |
| Greatly reduce drug conc. by washing | Difficult | Straightforward | Straightforward |
| Ease of doing a multiple dose response curve | Less than optimal | Straightforward | Straightforward |
| Reproducibility of results | OK - dependent on location of pippeting | Good | Good |
Discussion
Being able to record whole-cell currents using the described protocols and solutions indicates that the transfection was successful and that the desired voltage-gated ion channels or ion channel complexes are present with significant quantities at the cell membrane. Should the cells be positively transfected (i.e. fluorescent green) but not have recordable currents, additional time at 28°C may be required to allow for the channel protein to be properly packaged and inserted into the cell membrane. Note that prolonged incubation at 28°C causes cells to detach from the coverslips, and membranes destabilize and accumulate debris from dead cells, making patch clamp recording more challenging. All of the times suggested in this protocol have been optimized for our particular channels. Other channels and their subunits may require longer or shorter incubation times, and our protocol can be further adjusted as needed. For certain channels poor transfection efficiency (i.e. less fluorescent cells and lower fluorescence intensity per cell) produces better results than efficiently transfected, highly fluorescent cells. Should cells be fluorescent but not have recordable currents, troubleshooting points to consider include: a lack of accessory protein(s); improper mixing of transfection reagents; sub-optimal recording solutions; and mutation/recombination of channel or channel subunit cDNA constructs producing improperly folded or non-expressing channels.
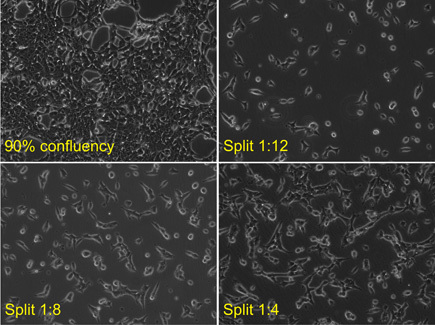 Figure 1: Human embryonic kidney 293T cells (HEK-293T) are grown in adherent monolayers at 37oC in CO2 incubators (5% CO2) in vented 25cm2 flasks to 90% confluency and split at 1:12, 1:8 and 1:4 dilutions. Cells are typically split bi-weekly, from a ~90% confluency. To ensure a similar ~90% confluency at splitting , cells are split in a 1:8 dilution on Mondays, and 1:12 on Thursdays. Prior to transfection, a fully confluent flask is split 1:4 into a new flask, and the cells are allowed to attach at 37oC in a CO2 incubator for 3-4 hrs.
Figure 1: Human embryonic kidney 293T cells (HEK-293T) are grown in adherent monolayers at 37oC in CO2 incubators (5% CO2) in vented 25cm2 flasks to 90% confluency and split at 1:12, 1:8 and 1:4 dilutions. Cells are typically split bi-weekly, from a ~90% confluency. To ensure a similar ~90% confluency at splitting , cells are split in a 1:8 dilution on Mondays, and 1:12 on Thursdays. Prior to transfection, a fully confluent flask is split 1:4 into a new flask, and the cells are allowed to attach at 37oC in a CO2 incubator for 3-4 hrs.
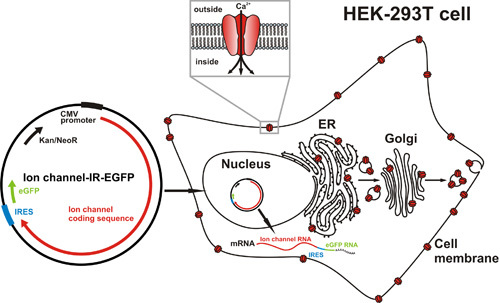 Figure 2: Strategy for heterologous expression of ion channels in HEK-293T. (Left) Ion channel cDNAs are cloned into the multiple cloning site of the pIRES2-EGFP plasmid. (Right) Transfection of these constructs into HEK cells results in the production of mRNA molecules containing the ion channel coding sequence (red) just upstream of an internal ribosome entry site (IRES; blue) and the eGFP coding sequence (green). Ion channels are translated and shuttled to the cell membrane via the ER and endomembrane system, while eGFP is translated and retained in the cytoplasm.
Figure 2: Strategy for heterologous expression of ion channels in HEK-293T. (Left) Ion channel cDNAs are cloned into the multiple cloning site of the pIRES2-EGFP plasmid. (Right) Transfection of these constructs into HEK cells results in the production of mRNA molecules containing the ion channel coding sequence (red) just upstream of an internal ribosome entry site (IRES; blue) and the eGFP coding sequence (green). Ion channels are translated and shuttled to the cell membrane via the ER and endomembrane system, while eGFP is translated and retained in the cytoplasm.
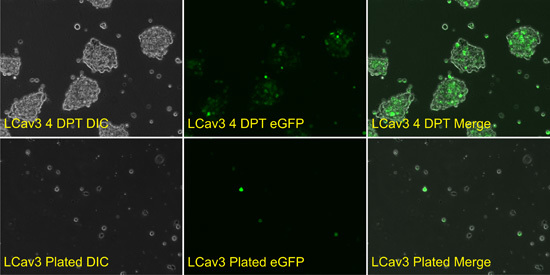 Figure 3: Transfection and expression of heterologous ion channel cDNAs in HEK-293T cells, and plating of cells onto coverslips for electrophysiological recording. (A) (Top panels) HEK-293T cells incubated at 28 C, four days post-transfection with the LCav3 calcium channels harbored in the pIRES2-EGFP mammalian CMV expression vector. (Bottom panels) The above cells trypsinized and plated on glass coverslips, isolated for electrophysiological recording. Differential Interference Contrast microscopy (DIC).
Figure 3: Transfection and expression of heterologous ion channel cDNAs in HEK-293T cells, and plating of cells onto coverslips for electrophysiological recording. (A) (Top panels) HEK-293T cells incubated at 28 C, four days post-transfection with the LCav3 calcium channels harbored in the pIRES2-EGFP mammalian CMV expression vector. (Bottom panels) The above cells trypsinized and plated on glass coverslips, isolated for electrophysiological recording. Differential Interference Contrast microscopy (DIC).
 Figure 4: A typical electrophysiological recording rig setup. Electrophysiological recording rig components include an amplifier (A), a personal computer (PC), Digidata 1440A analog-to-digital converter interface (C), motorized, dual pipette manipulators (PM), an epifluorescence microscope (M), perfusion systems (P), TMC 63-500 series vibration isolation table (VT), 40 tall Type II Faraday Cage (FC), and a free-standing 19 metal, electrical rack (R).
Figure 4: A typical electrophysiological recording rig setup. Electrophysiological recording rig components include an amplifier (A), a personal computer (PC), Digidata 1440A analog-to-digital converter interface (C), motorized, dual pipette manipulators (PM), an epifluorescence microscope (M), perfusion systems (P), TMC 63-500 series vibration isolation table (VT), 40 tall Type II Faraday Cage (FC), and a free-standing 19 metal, electrical rack (R).
 Figure 5: Perfusion systems. (A) A gravity flow system operated using Teflon valves and Valvelink8 controllers. (B) Eight channel Smartsquirt pressure micro perfusion system. Multi-Barrel Manifold Tip of either perfusion system is mounted on a motorized manipulator.
Figure 5: Perfusion systems. (A) A gravity flow system operated using Teflon valves and Valvelink8 controllers. (B) Eight channel Smartsquirt pressure micro perfusion system. Multi-Barrel Manifold Tip of either perfusion system is mounted on a motorized manipulator.
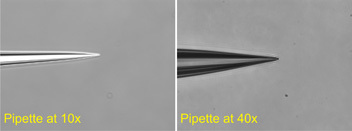 Figure 6: Polished, patch pipettes shown at 10x (left) and 40x (right) magnification. Patch pipettes were generated from borosilicate glass capillary tubes with filament and fire polished to smooth the pipette tip surfaces. A pipette-puller program was optimized to generate pipettes that gave a reading of 2-5MΩ in our electrophysiology set-up.
Figure 6: Polished, patch pipettes shown at 10x (left) and 40x (right) magnification. Patch pipettes were generated from borosilicate glass capillary tubes with filament and fire polished to smooth the pipette tip surfaces. A pipette-puller program was optimized to generate pipettes that gave a reading of 2-5MΩ in our electrophysiology set-up.
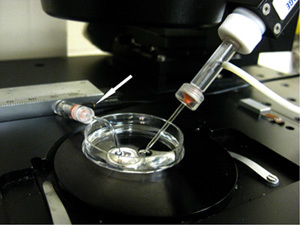 Figure 7: Ground electrode and patch pipettes in the recording dish. Patch pipette (right) and ground electrode (left; white arrow) in recording solution.
Figure 7: Ground electrode and patch pipettes in the recording dish. Patch pipette (right) and ground electrode (left; white arrow) in recording solution.
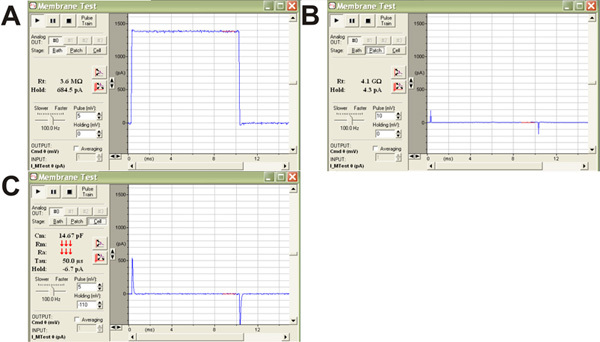 Figure 8: pClamp10.1 screen shots illustrating the steps of making a patch. (A) Square wave trace observed when in bath mode. (B) A Gigaohm seal in patch mode leads to a flattening of the square wave trace with pipette capacitance visible as tiny blips of current on the leading and trailing edges of the flat current trace. (C) After breakthrough and while in cell mode, large capacitive transients are seen.
Figure 8: pClamp10.1 screen shots illustrating the steps of making a patch. (A) Square wave trace observed when in bath mode. (B) A Gigaohm seal in patch mode leads to a flattening of the square wave trace with pipette capacitance visible as tiny blips of current on the leading and trailing edges of the flat current trace. (C) After breakthrough and while in cell mode, large capacitive transients are seen.
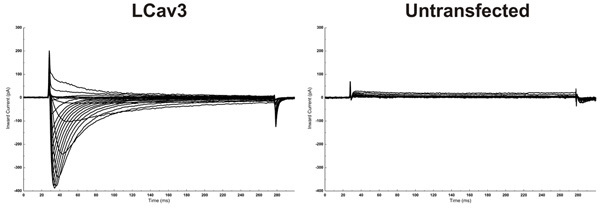 Figure 9: Electrophysiology results. (Left) Recordable inward voltage-gated calcium currents indicates successful transfection and expression of heterologous ion channels (e.g. LCav3). (Right) Untransfected cells have no recordable currents. Cells were subjected to an IV protocol that specifies that the software hold the cell at -110mV and then step to -70mV for 250msec then back down to -110mV holding potential. Each time the step increases by 10mV so that the steps are to -70mV, -60mV etc. until the last step is to +20mV. Results are for an invertebrate T-type (Cav3) voltage-gated calcium channel homologue from L. stagnalis termed LCav3.
Figure 9: Electrophysiology results. (Left) Recordable inward voltage-gated calcium currents indicates successful transfection and expression of heterologous ion channels (e.g. LCav3). (Right) Untransfected cells have no recordable currents. Cells were subjected to an IV protocol that specifies that the software hold the cell at -110mV and then step to -70mV for 250msec then back down to -110mV holding potential. Each time the step increases by 10mV so that the steps are to -70mV, -60mV etc. until the last step is to +20mV. Results are for an invertebrate T-type (Cav3) voltage-gated calcium channel homologue from L. stagnalis termed LCav3.
 Figure 10: Arrangement of electrodes and microperfusion pencil in recording solution. (A) Patch pipettes crafted from borosilicate glass (right) and polyimide multi-barrel manifold tip for drug perfusion (left). Ground electrode (back) of borosilicate glass filled with 3M CsCl and 1.5% agarose solution bent as hockey stick held in Microelectrode Holder Half-Cells. (B) 40x view through a microscope of the perfusion pencil tip and recording microelectrode.
Figure 10: Arrangement of electrodes and microperfusion pencil in recording solution. (A) Patch pipettes crafted from borosilicate glass (right) and polyimide multi-barrel manifold tip for drug perfusion (left). Ground electrode (back) of borosilicate glass filled with 3M CsCl and 1.5% agarose solution bent as hockey stick held in Microelectrode Holder Half-Cells. (B) 40x view through a microscope of the perfusion pencil tip and recording microelectrode.
Disclosures
No conflicts of interest declared.
Acknowledgments
This work was supported by a Discovery Operating Grant to JDS from the Natural Science and Engineering Research Council (NSERC) of Canada, an NSERC Alexander Graham Bell Canada Graduate Scholarships (doctoral) (to A. Senatore) and the Heart and Stroke Foundation, Grant-In-Aid NA#6284.
References
- Thomas P, Smart TG. HEK293 cell line: A vehicle for the expression of recombinant proteins. J. Pharmacological and Toxicological Methods. 2005;51:187–200. doi: 10.1016/j.vascn.2004.08.014. [DOI] [PubMed] [Google Scholar]
- Senatore A, Spafford JD. Transient and big are key features of an invertebrate T-type channel (LCav3) from the central nervous system of Lymnaea stagnalis. Journal of Biological Chemistry. 2010;285:7447–7458. doi: 10.1074/jbc.M109.090753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogden D, Stanfield P. Microelectrode techniques: the Plymouth workshop handbook. Cambridge: The Company of Biologists; 1987. Patch clamp techniques for single channel and whole-cell recording. [Google Scholar]
- Molleman A. England: John Wiley & Sons Ltd; 2003. Patch clamping: an introductory guide to patch clamp electrophysiology. [Google Scholar]
- Peng S-Q, Hajela RK, Atchison WD. Fluid flow-induced increase in inward Ba2+ current expressed in HEK293 cells transiently transfected with human neuronal L-type Ca2+channels. Brain Research. 2005;1045:116–123. doi: 10.1016/j.brainres.2005.03.039. [DOI] [PubMed] [Google Scholar]