

Abstract
Arabidopsis thaliana is an excellent model organism for studying epigenetic mechanisms. One of the reasons is the loss-of-function null mutant of DNA methyltransferases is viable, thus providing a system to study how loss of DNA methylation in a genome affects growth and development. Imprinting refers to differential expression of maternal and paternal alleles and plays an important role in reproduction development in both mammal and plants. DNA methylation is critical for determining whether the maternal or paternal alleles of an imprinted gene is expressed or silenced. In flowering plants, there is a double fertilization event in reproduction: one sperm cell fertilizes the egg cell to form embryo and a second sperm fuses with the central cell to give rise to endosperm. Endosperm is the tissue where imprinting occurs in plants. MEDEA, a SET domain Polycomb group gene, and FWA, a transcription factor regulating flowering, are the first two genes shown to be imprinted in endosperm and their expression is controlled by DNA methylation and demethylation in plants. In order to determine imprinting status of a gene and methylation pattern in endosperm, we need to be able to isolate endosperm first. Since seed is tiny in Arabidopsis, it remains challenging to isolate Arabidopsis endosperm and examine its methylation. In this video protocol, we report how to conduct a genetic cross, to isolate endosperm tissue from seeds, and to determine the methylation status by bisulfite sequencing.
Protocol
I. Genetic Crossing
1. Emasculating the female parent
In order to distinguish the maternal and paternal alleles by using DNA sequence polymorphism, two different ecotypes, e.g. Columbia-0 (Col-0) and Ler, will be chosen as female and male parents. The plants should be young and healthy. One can emasculate the female parent by using a dissecting microscope, magnifying visor, or naked eye. Locate stage-12 flowers (Smyth et al., 1990) and remove any flowers or siliques above and below them by clipping the base of the pedicel with the scissors. Sterilize forceps by dipping the base of the tip gently into a beaker of 95% ethanol, which will remove any pollen grains on the forceps and kill the pollen as well. Gently pry apart the flower buds using forceps, and gently remove 4 sepals, 4 petals, and 6 stamens, leaving the pistil bare and intact. Try to avoid damage to the carpels during this process.
2. Picking the pollen donor and carrying out pollination
The best pollen donors are anthesed open flowers at the stage 14 with the petals extending at a 90 ° angle to the pistil (Smyth et al., 1990), in which a lot of pollen is shedding. Grab the flower at the base and just above the pedicel, which will cause the flower to spread open. Dust the stigma of the prepared pistil with the anther. After the pollination, the stigma will be covered with the yellow pollen, which can be easily observed under a microscope.
3. Labeling the cross
After pollinating all the emasculated pistils on a plant, we label the cross with a jewelry tag and information of female and male parents and date. Place a stake in the soil close to the plant, use a string to tie the stem of the inflorescence to the stake, and cover the pollinated pistils with a plastic bag.
II. Isolation of Endosperm Tissues in Arabidopsis
1. Preparing materials
We usually get necessary materials ready before harvesting the endosperm and embryo tissues: a liquid nitrogen tank, liquid nitrogen, a dissecting microscope, two new pairs of fine-tip forceps (5 INOX. FST by Dumont biology, Switzerland), microscope glass slides (3" X 1" X 1.0 mm), a pH 5.7 solution of 0.3 M sorbitol and 5 mM MES.
2. Collecting siliques
At 7- or 8-day-after-pollination (DAP), the seeds are ready to be harvested at the mid- to late torpedo stage of embryogenesis and dissected for endosperm and embryo. Sometimes, it might take 9-10 DAP if the emasculated flowers are too young.
3. Isolating endosperm and embryo
Since Arabidopsis seeds are tiny, we use a dissecting microscope to isolate endosperm and embryo. Put an 8-DAP silique under a microscope, use a pair of forceps to hold the silique pedicel and use the tip of another pair of forceps to slide open the silique in the margin where two carpels fuse. Use a pair of forceps to pick up one seed onto pH 5.7 solutions of 0.3 M sorbitol and 5 mM MES, make a small cut at the micropyle end to slide out embryo, squeeze the uncut end to push out endosperm and separate endosperm from seed coat (Kinoshita et al., 2004). Put the embryo and endosperm into separate microtubes in liquid nitrogen. Continue this until we accumulate embryo or endosperm from 10-15 siliques and then store the tube in a -80°C freezer. In some cases, endosperm does not have to be separated from seed coat, i.e., one can isolate total RNA from the endosperm and seed coat mixture for gene imprinting analysis.
III. Bisulfite Sequencing
1. Required reagents
Cetyltrimethyl ammonium bromide (CTAB) for preparing genomic DNA, restriction enzymes, 3 M NaOH (Freshly made), 6.42 M urea/4 M sodium bisulfite (2 M sodium metabisulfite, Sigma-Aldrich, S9000, Na2S2O5, Molecular Weight: 190), 10 mM hydroquinone, a DNA purification kit (Promega Wizard DNA Clean-up System, Cat. # A7280), TE buffer, 6.3 M NaOH (freshly made), 10 M NH4OAc, 20 μg/μL tRNA, and 100% ethanol.
2. Details of the bisulfite treatment protocol
Isolate genomic endosperm or embryo DNA using a CTAB procedure (Rogers and Bendich, 1988).
Digest 100 ng - 2 μg of genomic DNA in 20 μL to 100 μL total volume with restriction enzymes that cut outside the region to be analyzed. For the MEA promoter, we use XhoI, NdeI, and PstI or HindIII.
Denature the restriction enzymes by boiling the DNA for five minutes and then quench on ice.
Add 1/9 volume (2.2 μL for 20 μL digested DNA) of 3 M NaOH and incubate at 37 °C for 15 minutes.
Transfer the solution to a 250 μL PCR tube.
Dissolve 7.5 g of urea in 10 mL of sterile distilled water; Slowly add 7.6 g of sodium metabisulfite over 1-2 hours and heating usually helps dissolving; Adjust the pH to 5 with freshly made 10 M NaOH; Add sterile distilled water to a final volume to 20 mL. This is 6.24 M urea/4 M sodium bisulfite solution.
Add 6.24 M urea/4 M sodium bisulfite solution to a final concentration of 5.36 M and 3.44 M, respectively (Paulin et al., 1998). For example, add 208 μL of 6.42 M urea/4 M sodium bisulfite solution to the above 22.2 μL of denatured genomic DNA (Xiao et al., 2003).
Add 10 mM hydroquinone to the DNA at a final concentration of 0.5 mM (12 μL for 20 μL digestion).
Conduct bisulfite treatment in a PCR machine: 30 cycles of 55 °C for 15 minutes and 95 °C for 30 seconds.
Desalt the bisulfite treated DNA using the Wizard DNA Clean-Up System from Promega and follow up the protocol (Jacobsen et al., 2000).
Measure the exact volume of TE recovered from the column after desalting and add 6.3 M NaOH to a final concentration of 0.3 M. Incubate at 37 °C for 15 minutes.
Add 10 M NH4OAc (pH 7.0) to a final concentration of 3 M, 2 μL of 20 μg/μL tRNA, and 3 volumes of 100% ethanol and then mix. Centrifuge for 15 minutes at 14,000 rpm.
Wash the pellet once with 70% ethanol, do a short centrifuge, and remove extra ethanol.
Dry pellet in a speedvac for 5-10 minutes and resuspend in 25 - 100 μL TE buffer depending on starting amount of DNA. The sodium bisulfite-treated DNA is now ready for PCR analysis.
3. PCR amplification
Since unmethylated cytosines are converted to uracil, it is difficult to amplify a large fragment using the bisulfite-treated DNA as a template. Thus, we usually design primers to amplify a product no longer than 500 bp. To sequence the 4-kb MEA promoter, we designed many sets of primers and amplified 14 overlapping fragments to cover the entire region (Xiao et al., 2003).
To sequence the top-strand, in designing a forward primer, I) choose a G (guanine)-rich region in order to have a higher annealing temperature without extra long nucleotides in the primers; II) change C (cytosine ) to Y (pyrimidine) at CG and CNG contexts and change the remaining C to T (thymine). In designing a reverse primer, I) choose a C-rich region; II) change G to R (purine) at CG and CNG contexts and change the remaining G to A (adenine).
To sequence the bottom-strand, in designing a forward primer, I) choose a C-rich region; II) change G to R at CG and CNG contexts and change the remaining G to A. In designing a reverse primer, I) choose a G-rich region; II) change C to Y (pyrimidine) at CG and CNG contexts and change the remaining C to T.
We usually use 1-2 μL of the sodium bisulfite-treated DNA as a template for each PCR amplification (Xiao et al., 2003). The PCR product needs to be analyzed by gel electrophoresis to confirm the correct size of the fragment, then to be gel purified and cloned into the TOPO TA cloning vector pCR2.1 (Invitrogen) as an insert. A single colony is picked up and cultured; plasmid DNA extracted and sent for sequencing.
4. Sequence analysis
The principle of bisulfite sequencing is that unmethylated cytosine will be converted to uracil due to hydrolytic deamination by high concentration of sodium bisulfite at pH5.0 which will be amplified as thymine in PCR product; while 5-methyl cytosine will not be modified by sodium bisulfite and remain to be cytosine after PCR amplification (Clark et al., 1994; Frommer et al., 1992). After obtaining the sequencing result, we compare it with the strand-specific template that is used for PCR amplification. If a cytosine residue in the template reads as a thymine in the sequencing result, it indicates that the cytosine is not methylated. If a cytosine residue in the template remains a cytosine in the sequencing, it means that the cytosine is methylated.
IV. Representative Results
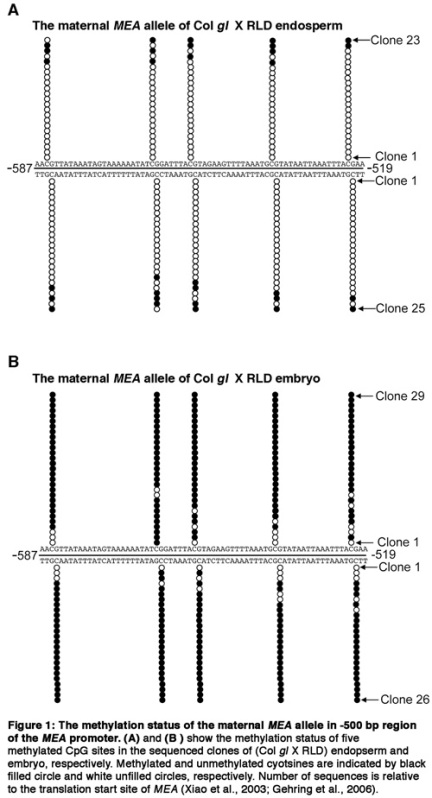 Figure 1.
Figure 1.
Discussion
It is relatively easy to separate embryo from endosperm and seed coat, but it is tedious to separate endosperm from seed coat, especially for seed at early or mid-torpedo stage of embryogenesis. Since seed coat only contributes a very small amount of tissue, for some genes, e.g., MEA and FWA, we do not have to separate endosperm from seed coat. That means that we can isolate RNAs from a mixture of endosperm and seed coat tissues, check expression of the maternal and paternal alleles of a gene, compare with expression in embryo, and determine the imprinting status of the gene.
For bisulfite sequencing, the designed primers need to be strand-specific. Sometimes, it is difficult to amplify a fragment from the bisulfite-treated genomic DNA due to damaged DNA template by sodium bisulfite treatment, and we usually try different sets of PCR primers in different regions and shorter fragments. After restriction enzyme digestion, the genomic DNA needs to be completely denatured into single-stranded since the bisulfite deamination almost does not work on the cytosine residues in double-stranded DNA structures. Another key is that bisulfite treatment needs to be complete. If a known unmethylated cytosine is not converted to thymine in the bisulfite sequencing, it indicates that the bisulfite treatment is not conducted properly. Alternatively, if many cytosine residues in an unknown region are not changed to thymine in the bisulfite sequencing, it is most likely due to amplification of unconverted DNA rather than indication of all methylated cytosine residues in the region.
Sodium bisulfite sequencing can precisely reveal whether a particular cytosine residue is methylated or not in a genome if the experiment is conducted correctly (Henderson et al., 2010). Combining the sodium bisulfite sequencing with recent high-throughput sequencing techniques, such as the Illumina Genome Analyzer high-throughput sequencing platform, one can map the epigenome at single-base resolution in plants and mammals (Cokus et al., 2008; Hsieh et al., 2009; Lister et al., 2008; Lister et al., 2009).
Disclosures
No conflicts of interest declared.
Acknowledgments
The authors thank Ms. Jennifer M. Lommel and Tara N. Rognan for maintenance of Arabidopsis plants. This work was supported by start-up funds from Saint Louis University and National Institutes of Health grants 1R15GM086846-01 and 3R15GM086846-01S1 to W. Xiao.
References
- Clark SJ, Harrison J, Paul CL, Frommer M. High sensitivity mapping of methylated cytosines. Nucleic Acids Res. 1994;22:2990–2997. doi: 10.1093/nar/22.15.2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cokus SJ, Feng S, Zhang X, Chen Z, Merriman B, Haudenschild CD, Sriharsa Pradhan S, Nelson SF, Pellegrini M, Jacobsen SE. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature. 2008;452:215–219. doi: 10.1038/nature06745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frommer M, McDonald LE, Millar DS, Collis CM, Watt F, Grigg GW. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc Natl Acad Sci USA. 1992;89:1827–1831. doi: 10.1073/pnas.89.5.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehring M, Huh JH, Hsieh TF, Penterman J, Choi Y, Harada JJ, Goldberg RB, Fischer RLDEMETER. DNA glycosylase establishes MEDEA polycomb gene self-imprinting by allele-specific demethylation. Cell. 2006;124:495–506. doi: 10.1016/j.cell.2005.12.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson IR, Chan SR, Cao X, Johnson L, Jacobsen SE. Accurate sodium bisulfite sequencing in plants. Epigenetics. 2010;5:47–49. doi: 10.4161/epi.5.1.10560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh TF, Ibarra CA, Silva P, Zemach A, Eshed-Williams L, Fischer RL, Zilberman D. Genome-wide demethylation of Arabidopsis endosperm. Science. 2009;324:1451–1454. doi: 10.1126/science.1172417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen SE, Sakai H, Finnegan EJ, Cao X, Meyerowitz EM. Ectopic hypermethylation of flower-specific genes in Arabidopsis. Curr. Biol. 2000;10:179–186. doi: 10.1016/s0960-9822(00)00324-9. [DOI] [PubMed] [Google Scholar]
- Kinoshita T, Miura A, Choi Y, Kinoshita Y, Cao X, Jacobsen SE, Fischer RL, Kakutani T. One-way control of FWA imprinting in Arabidopsis endosperm by DNA methylation. Science. 2004;303:521–523. doi: 10.1126/science.1089835. [DOI] [PubMed] [Google Scholar]
- Lister R, O'Malley RC, Tonti-Filippini J, Gregory BD, Berry CC, Millar AH, Ecker JR. Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell. 2008;133:523–536. doi: 10.1016/j.cell.2008.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo Q-M. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315–322. doi: 10.1038/nature08514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulin R, Grigg GW, Davey MW, Piper AA. Urea improves efficiency of bisulfite-mediated sequencing of 5'- methylcytosine in genomic DNA. Nucl. Acids Res. 1998;26:5009–5010. doi: 10.1093/nar/26.21.5009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers SO, & Bendich, J A. Extraction of DNA from plant tissues. Plant Molecular Biology Manual. 1988;A6:1–10. [Google Scholar]
- Smyth DR, Bowman JL, Elliot M, Meyerowitz EM. Early Flower Development in Arabídopsis. Plant Cell. 1990;2:755–767. doi: 10.1105/tpc.2.8.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao W, Gehring M, Choi Y, Margossian L, Pu H, Harada JJ, Goldberg RB, Pennell RI, Fischer RL. Imprinting of the MEA Polycomb gene is controlled by antagonism between MET1 methyltransferase and DME glycosylase. Dev. Cell. 2003;5:891–901. doi: 10.1016/s1534-5807(03)00361-7. [DOI] [PubMed] [Google Scholar]