

Abstract
The molecular chaperone αB-crystallin is a small heat-shock protein that is upregulated in response to a multitude of stress stimuli, and is found colocalized with Aβ amyloid fibrils in the extracellular plaques that are characteristic of Alzheimer's disease. We investigated whether this archetypical small heat-shock protein has the ability to interact with Aβ fibrils in vitro. We find that αB-crystallin binds to wild-type Aβ42 fibrils with micromolar affinity, and also binds to fibrils formed from the E22G Arctic mutation of Aβ42. Immunoelectron microscopy confirms that binding occurs along the entire length and ends of the fibrils. Investigations into the effect of αB-crystallin on the seeded growth of Aβ fibrils, both in solution and on the surface of a quartz crystal microbalance biosensor, reveal that the binding of αB-crystallin to seed fibrils strongly inhibits their elongation. Because the lag phase in sigmoidal fibril assembly kinetics is dominated by elongation and fragmentation rates, the chaperone mechanism identified here represents a highly effective means to inhibit fibril proliferation. Together with previous observations of αB-crystallin interaction with α-synuclein and insulin fibrils, the results suggest that this mechanism is a generic means of providing molecular chaperone protection against amyloid fibril formation.
Introduction
Alzheimer's disease (AD) is the most common cause of senile dementia, affecting >26 million people worldwide (1). This neurodegenerative condition is characterized by fibrillar protein deposition in both extracellular plaques and intracellular neurofibrillary tangles within the brain. The major constituents of the extracellular plaques are amyloid-β (Aβ) peptides, which are 39–43 amino acid residue cleavage products of the amyloid precursor protein (APP), an integral membrane protein. The aggregation of these peptides is thought to be critical to the pathogenesis of the disease (2). However, other proteins, including small heat-shock proteins (sHsps), a ubiquitous class of molecular chaperones, are also found colocalized with Aβ peptides in extracellular plaques (3–7). For example, the most extensively studied sHsp, αB-crystallin, (referred to as HSPB5 in the recently revised systematic nomenclature of the sHsps) is present at increased levels in the temporal and frontal lobes of postmortem AD brains (3,4), with the majority being localized within astrocytes and oligodendrocytes (4,8), and in the neurons surrounding senile plaques (8,9).
αB-Crystallin is found not only in the brain (10) but also in many other regions of the human body, including the retina, heart, skeletal muscle, skin, spinal cord, kidneys, lungs (10–12), cochleae of the mammalian ear (13), and the lacrimal gland duct and tears (14). It is also a crucial component of the eye lens, where it is found in particularly high concentrations (up to ∼200 mg/ml) together with its fellow sHsp αA-crystallin (15), with which it shares 57% sequence similarity (16). Of interest, αB-crystallin is associated with Aβ deposition in supranuclear cataracts in lenses from patients with AD (17).
In its native state, αB-crystallin assembles to form a heterogeneous mixture of oligomers ranging in size from 10 to 40 subunits, with the dominant populations containing 24–33 subunits (18). Each monomeric subunit is a 175 amino acid protein of ∼20 kDa in mass and is composed of an ∼90-residue central β-sandwich domain (19), which is termed the α-crystallin domain and is conserved between all sHsps (16). The α-crystallin domain is flanked by variable N-terminal and C-terminal regions, with the latter containing an unstructured and highly flexible 12 amino acid hydrophilic C-terminal extension at its extremity (20,21). The C-terminal region is important for maintaining the solubility of the protein (21,22) and regulating intersubunit interactions via the palindromic sequence centered about the conserved I-X-I motif within the native oligomer (23). As a result of the latter role, the C-terminal region may have an important function in chaperone action as regulated by subunit exchange (24,25). Transmission electron microscopy (TEM) shows native αB-crystallin oligomers to be roughly spherical (26), and cryo-EM reconstructions reveal a spherical protein shell averaging 13.5 nm in diameter with an 8.5 nm hollow interior (27,28).
αB-Crystallin is upregulated in response to a multitude of stress stimuli in vivo and has impressive chaperone abilities in vitro (21). Interactions of αB-crystallin with many amorphously aggregating proteins in vitro have been extensively documented (29). As an sHsp, it binds to partially folded proteins in an ATP-independent manner, sequestering them from aggregation until ATP-dependent chaperones, such as Hsp-70 and Hsp-90, have an opportunity to restore their native state (21). αB-Crystallin may also be involved in targeting substrates for degradation, as has been reported for other members of the sHsp chaperone family (30). Furthermore, αB-crystallin inhibits the growth of amyloid fibrils of the type found in disease-associated plaques, such as in AD (reviewed by Ecroyd and Carver (31)).
Amyloid fibrils are highly ordered linear protein aggregates with a cross-β core, typically having widths and lengths of a few nanometers and micrometers, respectively (32). Their assembly (from monomer) is a complex process, and a variety of different species of varying toxicity have been identified that may be intermediates in the pathway (33), particularly during the earlier stages of aggregation, including dimers and small oligomers (34–37), amyloid-β-derived diffusible ligands (soluble nonfibrillar oligomers) (38–40), and protofibrils (41,42) (soluble fibril-like oligomers). However, a highly effective model of fibril assembly can be described by just three kinetic processes: 1), nucleation of fibril seeds from monomeric protein (by unspecified pathways potentially involving oligomers); 2), elongation of these structures by incorporation of additional monomers; and 3), fragmentation of fibrils, which increases the number of growth sites at the fibril ends and results in exponential growth kinetics (43,44). Analysis of aggregation kinetics in terms of these fundamental microscopic processes is becoming increasingly important for elucidating the molecular basis of aggregate formation and the factors that can influence its rate, and a variety of methodologies have been developed to measure the individual processes in isolation (45,46). The lag phase in the growth kinetics can be bypassed if sufficient concentrations of preformed fibrils (or seeds) are provided as templates for elongation and subsequent fragmentation, resulting in immediate fibril growth (43). Amyloid fibrils are associated with many degenerative diseases; however, such fibrils can also be formed from nondisease-related proteins. The ability to form fibrils is believed to be a generic property of the polypeptide backbone, although the propensity for such formation varies significantly depending on factors such as the amino acid sequence and solution conditions (47).
The Aβ peptides have a favorable propensity for forming amyloid fibrils (48). The sequential cleavage of APP by β- and γ-secretases often yields Aβ peptides that are 40 or 42 residues long, with the latter isoform having greater aggregation potential (2). Genetic mutations in the APP-processing machinery, which increases the level of Aβ42 or the ratio of Aβ42/Aβ40, result in a dramatically earlier onset of AD (49,50), suggesting an important role for Aβ assembly in the pathogenic cascade. Genetic mutations within the Aβ sequence itself can also reduce the age of disease onset by increasing the aggregation propensity of the peptide. One such particularly aggregation-prone Aβ peptide is Aβ42 with the so-called Arctic mutation (E22G). This mutation, which was found in a single family in northern Sweden, gives rise to clinical features of early-onset AD, reportedly because of a propensity to form protofibrillar species over fibrillar ones (42).
The observation that Aβ and αB-crystallin are colocalized in vivo (3,4) has led to concerted investigations into the effect of αB-crystallin on Aβ aggregation in vitro. A number of apparently conflicting results have been reported (31), potentially because of differences in the experimental methods used, including the method of purification and handling of the Aβ peptide, and the incubation conditions. Most studies indicated that αB-crystallin is capable of inhibiting Aβ fibril formation (51–55), and recent work also showed a protective effect on Aβ-induced cytotoxicity (56). It is important to consider the mechanism by which αB-crystallin exerts this protective effect. As previously mentioned, several species of Aβ are present during the aggregation process. Differences in structure between the various Aβ species are likely to lead to different biophysical properties, such as exposed hydrophobicity, and as such the interaction between αB-crystallin and Aβ peptides is best examined for each Aβ species in isolation if possible. Investigators have examined interactions between αB-crystallin and the Aβ monomer using analytical ultracentrifugation, which did not reveal any complex formation (53). However, NMR studies identified a weak and transient interaction involving the hydrophobic core residues (17–21) of the Aβ40 peptide (57).
We recently demonstrated that αB-crystallin binds to amyloid fibrils formed by α-synuclein, the protein whose aggregation is linked to Parkinson's disease, and inhibits their elongation (58). Given that an interaction between αB-crystallin and Aβ40 fibrils was previously identified (53), with αB-crystallin preventing the seeded growth of Aβ40 and the nonseeded growth of the Aβ42 variant (53), we decided to study the interaction of these species to investigate whether fibril binding serves as a generic chaperone mechanism. We identified the existence of a similar chaperone mechanism for preventing aggregation of Aβ42 and its early-onset disease-associated Arctic variant, Aβ42arc. Through the use of fluorescence assays and immunoelectron microscopy, we found that αB-crystallin binds strongly to fibrils composed of both Aβ42 and Aβ42arc peptides. Furthermore, we investigated the mechanistic implications of this binding interaction directly by studying the effect of αB-crystallin on the Aβ42 amyloid fibril elongation phase examined in isolation, both in solution using thioflavin T (ThT) fluorescence, and on a surface using a quartz crystal microbalance (QCM). We found that αB-crystallin binding to Aβ fibrils strongly inhibits their elongation. Because the most effective means of extending the lag phase for amyloid fibril growth is to reduce the elongation (or fragmentation) rate (43), this is likely to represent a highly successful (and generic) chaperone mechanism for suppressing fibril proliferation.
Materials and Methods
The materials and methods used in this work are described in the Supporting Material.
Results
Quantification of αB-crystallin binding to Aβ amyloid fibrils
Initially, we investigated whether αB-crystallin was capable of binding to Aβ42 fibrils using a sedimentation assay. αB-Crystallin was incubated in the presence of Aβ42 fibrils for 1 h at room temperature, and then the mixture was centrifuged under conditions that cause sedimentation of Aβ fibrils but not free αB-crystallin oligomers (Fig. S1). Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis (see Fig. 1 A) showed that there was significantly less αB-crystallin in the supernatant in the presence of Aβ42 fibrils than in its absence, indicating that αB-crystallin copellets with Aβ42 fibrils. That this effect is not simply nonspecific protein binding was indicated by the fact that two control proteins of similar mass, i.e., green fluorescent protein (GFP) and ubiquitin carboxyl-terminal hydrolase isozyme L3 (UCH-L3), did not copellet under these conditions (Fig. 1 A). A purely electrostatic effect was also ruled out, because only a small change in sedimentation behavior was observed when the ionic strength of the solution was increased (Fig. S2).
Figure 1.
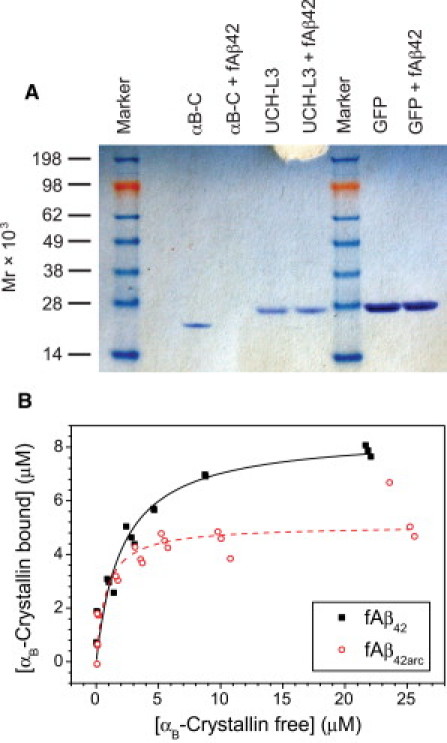
(A) Specificity of binding of αB-crystallin to Aβ42 fibrils. Solutions of 12 μM UCH-L3, GFP, and αB-crystallin in buffer A were incubated for 1 h at room temperature in the presence and absence of 12 μM Aβ42 fibrils, and then centrifuged for 30 min at 16,000 × g. The supernatants were separated by SDS-PAGE, and proteins were visualized by staining with Coomassie-Blue. (B) αB-Crystallin binding to Aβ42 fibrils (closed squares) and Aβ42arc fibrils (open circles). αB-Crystallin solutions of various concentrations were incubated with 15 μM Aβ fibrils at room temperature for 1 h and then centrifuged. The concentration of αB-crystallin in the supernatant was then determined by tryptophan fluorescence as described in Materials and Methods. The concentration of bound αB-crystallin was calculated as the difference between the estimated total concentration (from the tryptophan fluorescence of an identical solution prepared without Aβ fibrils and centrifugation) and the concentration determined for the supernatant concentration. The lines represent the line of best fit to the Scatchard equation for Aβ42 (solid line, Vmax = 8.48 ± 0.49 μM corresponding to MMBR = 0.57 ± 0.08, K = 2.09 ± 0.43 μM) and Aβ42arc (dotted line, Vmax = 5.08 ± 0.28 μM corresponding to MMBR = 0.34 ± 0.03, K = 0.77 ± 0.24 μM).
To quantify the stoichiometry and affinity of αB-crystallin binding to Aβ fibrils, we performed further sedimentation experiments over a range of different αB-crystallin concentrations (with Aβ fibril concentration held constant), and determined the concentration of αB-crystallin remaining in the supernatant by measuring its intrinsic tryptophan fluorescence (Aβ has no tryptophan residues). Consistent with the SDS-PAGE analysis (Fig. 1 A), a highly reproducible reduction in supernatant fluorescence was observed for αB-crystallin incubated with Aβ42 fibrils (Fig. 1 B, black squares). The concentration of bound αB-crystallin initially increased with total αB-crystallin concentration, and then reached a plateau indicating saturation of the available binding sites (Fig. 1 B, black squares). We analyzed these data using a single binding-site model with no cooperativity. The saturation isotherm was fitted to Eq. 1 directly (Fig. 1 B, solid black line, and Supporting Material) to determine both the dissociation constant for the interaction of 2.1 ± 0.4 μM and the maximum molar binding ratio (MMBR) of 0.57 ± 0.08 αB-crystallin monomers per Aβ42 monomer. A similar analysis was carried out for the binding of αB-crystallin to fibrils composed of the Aβ42arc peptide (Fig. 1 B, red circles). Again αB-crystallin was observed to copellet with the fibrils. Upon application of the single binding-site model (Fig. 1 B, red dotted line), both the dissociation constant and the MMBR were found to be slightly lower for these fibrils (0.77 ± 0.24 μM and 0.34 ± 0.03, respectively).
Imaging of the Aβ fibril complex with αB-crystallin
The high binding ratios reported above suggest that αB-crystallin binds along the entire length of the fibrils (rather than being restricted to, e.g., the fibril ends). To investigate this further, we visualized the fibril-chaperone complex using immuno-EM. αB-Crystallin was incubated with fibrils composed of Aβ42 and Aβ42arc peptides for 1 h at room temperature. The fibrils were readily observed by negative staining with uranyl acetate, and showed a morphology similar to that typically observed for Aβ fibrils (59). By contrast, the αB-crystallin oligomer was less readily resolved (due to its smaller relative size), and therefore samples were immunolabeled with an antibody directed against αB-crystallin, which was then stained with a secondary antibody conjugated to 10 nm gold nanoparticles. Immunoelectron micrographs of Aβ42 and Aβ42arc fibrils incubated with αB-crystallin (Fig. 2, A and C) all show αB-crystallin to be associated with the fibrils, with gold nanoparticles present along the entire length of the fibrils, without any apparent periodicity, and with occasional binding to the ends. Equivalent control samples incubated without αB-crystallin are shown in Fig. 2, B and D. No nonspecific immunolabeling was observed. Similar experiments with fibrils composed of Aβ40 peptide showed the same behavior (Fig. S3).
Figure 2.
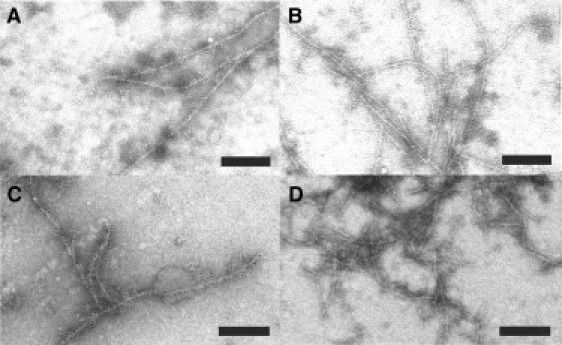
Immuno-EM of Aβ42 and Aβ42arc fibrils in the presence (A and C) and absence (B and D) of αB-crystallin, respectively. All scale bars represent 200 nm. Aβ fibrils were prepared and incubated in the presence or absence of an equal molar concentration of αB-crystallin for 1 h at room temperature. The mixtures were centrifuged at 16,000 × g for 30 min, and the pellets were resuspended and treated as described in Materials and Methods. Similar images were observed for Aβ40 fibrils (Fig. S2).
αB-Crystallin-mediated inhibition of Aβ fibril elongation
The binding of αB-crystallin inhibits the aggregation of α-synuclein in solution (60,61), and it was recently shown that binding to α-synuclein fibrils specifically and dramatically reduces the rate of fibril elongation (58). Given previous reports that αB-crystallin inhibits Aβ aggregation in solution (31), we investigated whether αB-crystallin binding to Aβ fibrils also directly inhibits fibril growth, using in situ ThT measurements of seeded growth (Fig. 3). In the absence of αB-crystallin, efficient seeding of fibril growth from both Aβ42 and Aβ42arc fibrils was observed (Fig. 3, red circles).
Figure 3.
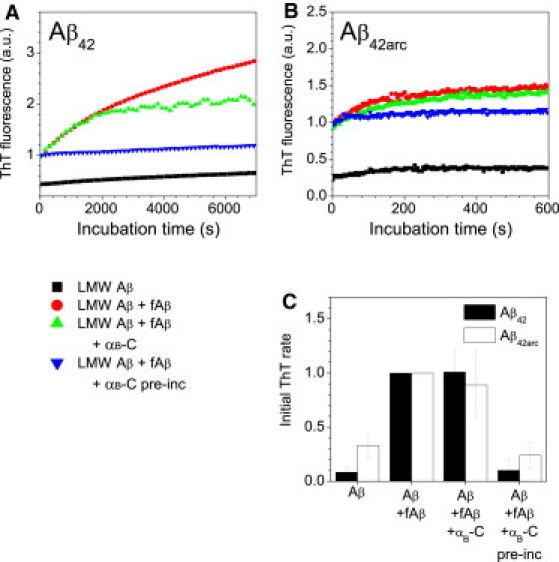
Inhibition of Aβ fibril elongation by αB-crystallin binding. Aβ fibril elongation kinetics for Aβ42 (A) and Aβ42arc (B) fibrils (fAβ) at room temperature in buffer A in the presence and absence of αB-crystallin. The solutions examined were LMW Aβ alone (black squares), LMW Aβ with fibril seed (red circles), LMW Aβ with fibril seed and a low concentration of αB-crystallin (green triangles), and LMW Aβ with αB-crystallin bound to fibril seeds (blue triangles). The fluorescence traces plotted are the average of three replicates and are reported relative to the starting fluorescence of solutions containing Aβ fibrils but no αB-crystallin. Control solutions of LMW Aβ with αB-crystallin displayed behavior similar to that observed for LMW Aβ alone. Solutions containing αB-crystallin and ThT alone showed no change in fluorescence over these timescales. (C) The initial elongation rates for Aβ42 (black bars) and Aβ42arc (white bars) were determined from the gradient of the linear fit to the first portion of data (1000 s for Aβ42, 200 s for Aβ42arc) for three repeats. Initial rates have been normalized with respect to Aβ elongation in the presence of Aβ seeds and the absence of αB-crystallin. Reported rates are the mean and standard deviation of the three repeats. The fibril seeds were preincubated with αB-crystallin for 1 h at room temperature. Concentrations of Aβ fibrils and αB-crystallin (when present) were ∼0.8 μM and 0.4 μM, respectively. Concentrations of LMW Aβ were 9.7 μM and 1.8 μM for Aβ42 and Aβ42arc, respectively.
Aβ fibrils were incubated in the presence of αB-crystallin (at a molar ratio of 2:1) for 1 h at room temperature to allow saturation of αB-crystallin binding to the fibrils. We then tested these fibrils for their seeding ability by introducing them into a solution of low-molecular-weight (LMW) Aβ (see Supporting Material) and monitoring fibril growth via ThT fluorescence. Fibril seeds that were preincubated with an equivalent concentration of αB-crystallin were completely unable to seed further Aβ fibril elongation. Moreover, the ThT fluorescence of the seed fibrils was unchanged in the presence of αB-crystallin, as judged by the fluorescence intensity at time zero (Fig. 3, A and B), indicating that the chaperone did not disaggregate the seeds. Upon addition of αB-crystallin (to a final concentration of 0.4 μM) to seeded Aβ42, a significant decrease in the elongation rate was only observed after ∼30 min (Fig. 3 A). Together, these findings implicate a slow binding of the chaperone to the fibril seeds as the origin of this inhibition. It is interesting that without preincubation of Aβ42 fibril seeds with the chaperone, inhibition of seeded growth occurred after ∼30 min after the addition of αB-crystallin (Fig. 3 A), which may correspond to the time required for αB-crystallin to bind to the fibrils in its active form. By contrast, for Aβ42arc, which aggregates much faster than Aβ42 (over ∼10 min, compared with ∼2 h), strong inhibition was observed only when the fibril seeds were preincubated with the chaperone (Fig. 3 B).
We also examined the inhibition of Aβ fibril growth using a QCM. In previous applications of this technique, we showed that when preformed seed fibrils are attached to the surface of the quartz crystal, this kinetic assay is highly specific for the elongation step of the overall fibrillization pathway (45,58,62,63). Aβ42 fibrils were covalently attached to the surface of the sensor crystal as previously described (63). Their rate of elongation was then observed as a change in the resonant frequency of the quartz sensor, which reflects an increase in the surface-bound mass (see Materials and Methods for details). A QCM sensor crystal covered with Aβ42 fibril seeds was exposed to solutions of LMW Aβ42 and αB-crystallin. Exposure of QCM sensors with no attached seeds to LMW Aβ does not result in a significant change in the resonant frequency of the system (63). In contrast, exposure of QCM sensors covered with Aβ42 fibril seeds to LMW Aβ42 yielded a linear decrease in resonant frequency over the course of several minutes, corresponding to a steady rate of fibril elongation (see Fig. 3 A). The introduction of 2.5 μM αB-crystallin caused a decrease in frequency that showed a different kinetic profile, namely, a saturable attachment of the chaperone to the surface (Fig. 4 A). Nonspecific adsorption of αB-crystallin was also observed for QCM sensors without attached fibril seeds; however, the observed increase in mass was less extensive than that found in the presence of bound seeds (Fig. S4 B). Once the fibrils had been exposed to αB-crystallin, they lost the ability to support further Aβ monomer deposition, when probed by renewed incubation of the sensor surface with Aβ42 solution (Fig. 4, A and B). Control experiments demonstrated that renewed incubation of the surface-bound seeds with LMW Aβ resulted in additional fibril elongation if there was no intermittent contact with αB-crystallin (Fig. S4 A). Because the chaperone and Aβ solutions were added sequentially with an intervening buffer wash of the liquid cell, the deactivation of fibril growth sites by chaperone binding, rather than the sequestration of amyloidogenic species in solution, must be responsible for the observed inhibition of increasing mass. After the QCM experiments, we acquired atomic force microscopy images of the functionalized sensors using tapping mode in air, and observed that the fibrils had lengthened (Fig. 4, C and D). Finally, we investigated the reversibility of the inhibition by allowing the αB-crystallin to dissociate from the fibril seeds by washing with a 3 M solution of the denaturant guanidinium chloride (this concentration was shown not to dissolve Aβ fibrils; data not shown). Subsequently, these seeds were able to elongate again when exposed to LMW Aβ42 (Fig S4 C).
Figure 4.
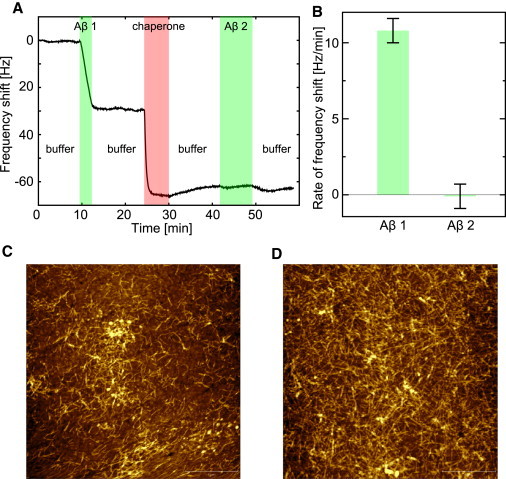
(A) Inhibition of Aβ42 fibril elongation by αB-crystallin probed by QCM. A decrease in resonant frequency (overtone with N = 3 shown) indicates attachment of protein. At 38°C, Aβ fibrils were incubated in buffer C (100 mM phosphate, pH 7.4) before injection of 1.7 μM LMW Aβ42. Buffer C then replaced the Aβ42 solution, quenching amyloid fibril elongation. After incubations with 2.5 μM αB-crystallin, the Aβ42 fibril chip was again exposed to a solution of 1.7 μM LMW Aβ42, which did not lead to a frequency shift comparable to that observed during the first incubation with LMW Aβ42, demonstrating the inhibitory effect of αB-crystallin. (B) The rate of frequency drop for the two periods of Aβ42 exposure determined from straight line fitting. The error bars represent the largest variation between the overall rate and local rates during the second exposure. AFM images of typical QCM sensor surfaces before (C) and after (D) exposure to Aβ solutions demonstrate fibril elongation.
Discussion
Amyloid fibrils are not simply inert products of protein aggregation; rather, they are dynamic species that have the ability not only to grow but also to fragment and dissociate (43,64). Given the association of αB-crystallin (7,24) and other sHsps with Aβ plaques in AD, we investigated whether this archetypical sHsp chaperone has the ability to influence one of the crucial stages of fibril formation, the elongation phase of amyloid growth. We demonstrated in this study that αB-crystallin selectively binds to Aβ42 and Aβ42arc fibrils with micromolar affinity, with high MMBRs for αB-crystallin (Aβ = 0.57 ± 0.08 and 0.34 ± 0.03, respectively). These values are highly consistent with immuno-EM data showing that the chaperone molecules bind along the length and at the ends of the fibrils. A previously described simple geometric model (58) indicated values of 0.2–0.7 for maximum coverage of the fibril. To elucidate the consequences of this interaction, we performed investigations into the effects of αB-crystallin on seeded Aβ fibril growth, both in solution and on the surface of a QCM biosensor. We observed that while bound, αB-crystallin strongly retards the rate of fibril elongation (Figs. 3 and 4), and that once it is removed, fibril elongation can be resumed (Fig. S4 C). The significant difference in seeding efficiency between fibrils with bound αB-crystallin and fibrils with unbound αB-crystallin (Fig. 3) demonstrates that αB-crystallin binding to fibrils can directly inhibit fibril growth. This result is inconsistent with inhibition due to chaperone binding to Aβ monomer or early oligomers, because a larger concentration of αB-crystallin is available for binding these species in the latter case, where no inhibition is observed.
The process of protein aggregation and amyloid formation is characterized by sigmoidal assembly kinetics, with a lag phase before rapid growth. The duration of the lag phase is usually shown to be heavily dominated by the rates of elongation and fragmentation while varying only logarithmically with the nucleation rate (43). With such a scenario, the most effective way to extend the lag phase would be to reduce the elongation or fragmentation rate, not the nucleation rate. Hence, the binding of a chaperone to fibrils to inhibit their further elongation, as observed here, is very likely to be critical and highly influential mechanistically, in terms of an effective inhibitory activity of the chaperone. Binding along the fibril length (l) may appear to be a fairly inefficient mechanism for inhibiting fibril elongation, because for a cylinder of radius r, only a proportion r/(r+l) of the area is at the ends. In the initial stages of aggregation, the fibrils/fibril precursors are much shorter, making it more efficient than in the case of mature fibrils. For example, for fibrils of radius 2–5 nm (as suggested by AFM images) and short fibrils of ∼20 nm, these calculations suggest ratios of 11:1 to 5:1. In addition, binding of the sHsp to the fibril could also serve additional roles, such as coverage of potentially toxic fibril surfaces (65), affecting fibril fragmentation rates, or targeting the fibrils for degradation (30).
We recently showed, using immunogold labeling TEM, that αB-crystallin can also bind to amyloid fibrils formed from an alternative target protein, α-synuclein (58). The binding interaction suppressed α-synuclein fibril elongation in a manner analogous to that found here for αB-crystallin with Aβ amyloid fibrils. This anti-aggregation activity of αB-crystallin toward the elongation phase of amyloid assembly could be a general mechanistic phenomenon involving sHsps and many or all amyloid fibrils. This conclusion is further supported by the observation from QCM studies that αB-crystallin inhibits the elongation of insulin amyloid fibrils (45).
It is interesting that fibrils formed from two different Aβ peptides showed similar MMBR values for αB-crystallin binding, despite significant differences in their capacity to bind ThT (data not shown). They are also comparable to the approximate MBR determined from α-synuclein fibrils (58), which may represent further evidence for a common mechanism of binding of αB-crystallin to amyloid fibrils. The fact that the measured ThT fluorescence of the fibril seeds is not diminished by the addition of αB-crystallin (Fig. 3) suggests that the mechanism and/or region of chaperone binding is distinct from that of the ThT dye. αB-Crystallin binds target proteins in part via exposed regions of hydrophobicity (21,31,66); hence, it is possible that αB-crystallin binds to amyloid fibrils through association with hydrophobic regions of the Aβ peptides or α-synuclein that protrude from the main cross-β amyloid fibril core.
Assuming that the chaperone activity of αB-crystallin is in part mediated by its ability to bind to patches of exposed hydrophobicity on aggregation-prone target proteins (21,31,66), it is likely that this sHsp associates with additional species on the amyloid formation pathway that also have exposed hydrophobic regions. Indeed, a weak and transient interaction was previously proposed for αB-crystallin with Aβ monomers (57). Consistent with this, the interaction of αB-crystallin with species early along the α-synuclein fibril-forming pathway has been well characterized (60,61).
Typically, sHsps (including αB-crystallin (31)) are localized inside cells as cytoplasmic proteins, although there have been reports of extracellular sHsps (67). The association of αB-crystallin with Aβ peptides in AD plaques, which are extracellular, may therefore be a result of their release from cells after damage associated with Aβ oligomers, which are proposed to be the primary neurotoxic species. Of interest, Friedrich et al. (68) recently showed that in cell culture, Aβ plaque formation occurs intracellularly and leads to cell death before the plaques are released into the extracellular space. Thus, because of the significant intracellular phase of Aβ aggregation, there may be ample opportunity for Aβ to interact with αB-crystallin and other sHsps. A number of studies have suggested that αB-crystallin can influence amyloid fibril assembly via interactions with early-stage fibrillar oligomers. For example, it has been shown that αB-crystallin can redirect the amyloidogenic α-synuclein protein toward an amorphous aggregation pathway with more easily degradable end products (60). One study indicated that αB-crystallin not only inhibits amyloid fibril formation by the Aβ peptides but also promotes the formation of alternative stable structures that are more toxic to cells (52). However, a more recent study showed that αB-crystallin inhibits amyloid fibril formation and its associated cell toxicity for both κ-casein and Aβ40 peptides (56). αB-Crystallin is much more efficient at inhibiting slowly aggregating target proteins along the amorphous (69) and fibril-forming (61,70–72) pathways. Consistent with this finding, we observed that because of its slower aggregation rate, Aβ42 enabled αB-crystallin to interact with its fibrils to partially inhibit fibril growth (Fig. 3 A), whereas LMW Aβ42arc and fAβ42arc did not (Fig. 3 B). Further investigations into the specific mechanistic processes involved in the interaction between αB-crystallin and isolated oligomeric Aβ amyloid intermediates will therefore be of interest.
It is interesting to speculate about the physiological implications of the interactions between αB-crystallin and elongating fibrils described here, and the influence these processes may have on their links with disease. As described earlier, the elongation phase is critical for determining the overall kinetics of the process of amyloid assembly (43), and hence a chaperone that is capable of inhibiting this stage of amyloid growth is likely to be a powerful suppressor of fibril proliferation. Further, the ability of αB-crystallin to bind fibrils with micromolar affinity is interesting in view of recent reports that sHsps are able not only to inhibit aggregation but also to target substrates for degradation (30). It is therefore possible that this binding interaction could be valuable for promoting the disposal of amyloidogenic species in vivo.
Conclusions
In summary, the data presented here reveal that αB-crystallin binds to Aβ amyloid fibrils, and through this interaction suppresses the elongation phase of Aβ42 fibril growth in a highly effective manner. Given the increasing number of amyloid fibril systems in which this behavior has now been observed (e.g., Aβ40, Aβ42, Aβ42arc, α-synuclein, and insulin), this activity appears to be a generic function of this archetypical sHsp, whereby it displays chaperone behavior toward all amyloid fibrils regardless of their constituent target proteins. Overall, therefore, it is likely that all species along the amyloid fibril-forming pathway, including oligomers and fibrils, will interact with molecular chaperones, such as sHsps, because all are misfolded and potentially expose hydrophobic regions. This process may offer a more comprehensive protection against the toxicity associated with amyloid fibrils than that obtained from interaction with the monomeric or oligomeric species only.
Acknowledgments
We thank Drs. Glyn Devlin, Andy Baldwin, Anthony Fitzpatrick, Jeremy Skepper, and Emma Evergreen for useful discussions, and Sharon Hook for providing Aβ42 prepared by the hexafluoroisopropanol method.
This study was supported by the Engineering and Physical Sciences Research Council, UK (S.S. and A.K.B.); Unilever and the Biotechnology and Biological Sciences Research Council (C.A.W.); the Wellcome and Leverhulme Trusts (C.M.D.); the Australian Research Council (J.A.C.); the Australian National Health and Medical Research Council; a Peter Doherty Fellowship (H.E.); a Herchel Smith Harvard Postgraduate Scholarship (S.W.); a Royal Society Dorothy Hodgkin Fellowship (S.M.); and a Bye Fellowship, Magdalene College, Cambridge (A.K.B.).
Supporting Material
References
- 1.Brookmeyer R., Johnson E., Arrighi H.M. Forecasting the global burden of Alzheimer's disease. Alzheimers Dement. 2007;3:186–191. doi: 10.1016/j.jalz.2007.04.381. [DOI] [PubMed] [Google Scholar]
- 2.Selkoe D.J. Alzheimer's disease: genes, proteins, and therapy. Physiol. Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 3.Yoo B.C., Kim S.H., Lubec G. Deranged expression of molecular chaperones in brains of patients with Alzheimer's disease. Biochem. Biophys. Res. Commun. 2001;280:249–258. doi: 10.1006/bbrc.2000.4109. [DOI] [PubMed] [Google Scholar]
- 4.Shinohara H., Inaguma Y., Kato K. αB crystallin and HSP28 are enhanced in the cerebral cortex of patients with Alzheimer's disease. J. Neurol. Sci. 1993;119:203–208. doi: 10.1016/0022-510x(93)90135-l. [DOI] [PubMed] [Google Scholar]
- 5.Renkawek K., Bosman G.J., de Jong W.W. Expression of small heat-shock protein hsp 27 in reactive gliosis in Alzheimer disease and other types of dementia. Acta Neuropathol. 1994;87:511–519. doi: 10.1007/BF00294178. [DOI] [PubMed] [Google Scholar]
- 6.Renkawek K., Bosman G.J., Gaestel M. Increased expression of heat-shock protein 27 kDa in Alzheimer disease: a preliminary study. Neuroreport. 1993;5:14–16. doi: 10.1097/00001756-199310000-00003. [DOI] [PubMed] [Google Scholar]
- 7.Muchowski P.J., Wacker J.L. Modulation of neurodegeneration by molecular chaperones. Nat. Rev. Neurosci. 2005;6:11–22. doi: 10.1038/nrn1587. [DOI] [PubMed] [Google Scholar]
- 8.Wilhelmus M.M.M., Otte-Höller I., Verbeek M.M. Specific association of small heat shock proteins with the pathological hallmarks of Alzheimer's disease brains. Neuropathol. Appl. Neurobiol. 2006;32:119–130. doi: 10.1111/j.1365-2990.2006.00689.x. [DOI] [PubMed] [Google Scholar]
- 9.Renkawek K., Voorter C.E., de Jong W.W. Expression of αB-crystallin in Alzheimer's disease. Acta Neuropathol. 1994;87:155–160. doi: 10.1007/BF00296185. [DOI] [PubMed] [Google Scholar]
- 10.Bhat S.P., Nagineni C.N. αB subunit of lens-specific protein α-crystallin is present in other ocular and non-ocular tissues. Biochem. Biophys. Res. Commun. 1989;158:319–325. doi: 10.1016/s0006-291x(89)80215-3. [DOI] [PubMed] [Google Scholar]
- 11.Bhat S.P., Horwitz J., Ding L. αB-crystallin exists as an independent protein in the heart and in the lens. Eur. J. Biochem. 1991;202:775–781. doi: 10.1111/j.1432-1033.1991.tb16432.x. [DOI] [PubMed] [Google Scholar]
- 12.Nagineni C.N., Bhat S.P. αB-crystallin is expressed in kidney epithelial cell lines and not in fibroblasts. FEBS Lett. 1989;249:89–94. doi: 10.1016/0014-5793(89)80022-5. [DOI] [PubMed] [Google Scholar]
- 13.May C.A., Arnold B., Lütjen-Drecoll E. αB-crystallin in the mammalian inner ear. ORL J. Otorhinolaryngol. Relat. Spec. 1998;60:121–125. doi: 10.1159/000027579. [DOI] [PubMed] [Google Scholar]
- 14.May C.A., Welge-Lüssen U., Lütjen-Drecoll E. αB-crystallin in lacrimal gland duct and tears. Curr. Eye Res. 2000;21:588–594. [PubMed] [Google Scholar]
- 15.Augusteyn R.C. On the growth and internal structure of the human lens. Exp. Eye Res. 2010;90:643–654. doi: 10.1016/j.exer.2010.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Jong W.W., Leunissen J.A., Voorter C.E. Evolution of the α-crystallin/small heat-shock protein family. Mol. Biol. Evol. 1993;10:103–126. doi: 10.1093/oxfordjournals.molbev.a039992. [DOI] [PubMed] [Google Scholar]
- 17.Goldstein L.E., Muffat J.A., Bush A.I. Cytosolic β-amyloid deposition and supranuclear cataracts in lenses from people with Alzheimer's disease. Lancet. 2003;361:1258–1265. doi: 10.1016/S0140-6736(03)12981-9. [DOI] [PubMed] [Google Scholar]
- 18.Aquilina J.A., Benesch J.L.P., Robinson C.V. Polydispersity of a mammalian chaperone: mass spectrometry reveals the population of oligomers in αB-crystallin. Proc. Natl. Acad. Sci. USA. 2003;100:10611–10616. doi: 10.1073/pnas.1932958100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jehle S., van Rossum B., Rajagopal P. αB-crystallin: a hybrid solid-state/solution-state NMR investigation reveals structural aspects of the heterogeneous oligomer. J. Mol. Biol. 2009;385:1481–1497. doi: 10.1016/j.jmb.2008.10.097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.MacRae T.H. Structure and function of small heat shock/α-crystallin proteins: established concepts and emerging ideas. Cell. Mol. Life Sci. 2000;57:899–913. doi: 10.1007/PL00000733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Treweek T.M., Morris A.M., Carver J.A. Intracellular protein unfolding and aggregation: the role of small heat-shock chaperone proteins. Aust. J. Chem. 2003;56:357–367. [Google Scholar]
- 22.Carver J.A., Aquilina J.A., Ralston G.B. Identification by 1H NMR spectroscopy of flexible C-terminal extensions in bovine lens α-crystallin. FEBS Lett. 1992;311:143–149. doi: 10.1016/0014-5793(92)81386-z. [DOI] [PubMed] [Google Scholar]
- 23.Laganowsky A., Benesch J.L.P., Eisenberg D. Crystal structures of truncated αA and αB crystallins reveal structural mechanisms of polydispersity important for eye lens function. Protein Sci. 2010;19:1031–1043. doi: 10.1002/pro.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun Y., MacRae T.H. Small heat shock proteins: molecular structure and chaperone function. Cell. Mol. Life Sci. 2005;62:2460–2476. doi: 10.1007/s00018-005-5190-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bova M.P., McHaourab H.S., Fung B.K. Subunit exchange of small heat shock proteins. Analysis of oligomer formation of αA-crystallin and Hsp27 by fluorescence resonance energy transfer and site-directed truncations. J. Biol. Chem. 2000;275:1035–1042. doi: 10.1074/jbc.275.2.1035. [DOI] [PubMed] [Google Scholar]
- 26.Burgio M.R., Bennett P.M., Koretz J.F. Heat-induced quaternary transitions in hetero- and homo-polymers of α-crystallin. Mol. Vis. 2001;7:228–233. [PubMed] [Google Scholar]
- 27.Haley D.A., Horwitz J., Stewart P.L. The small heat-shock protein, αB-crystallin, has a variable quaternary structure. J. Mol. Biol. 1998;277:27–35. doi: 10.1006/jmbi.1997.1611. [DOI] [PubMed] [Google Scholar]
- 28.Peschek J., Braun N., Buchner J. The eye lens chaperone α-crystallin forms defined globular assemblies. Proc. Natl. Acad. Sci. USA. 2009;106:13272–13277. doi: 10.1073/pnas.0902651106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Horwitz J. α-crystallin can function as a molecular chaperone. Proc. Natl. Acad. Sci. USA. 1992;89:10449–10453. doi: 10.1073/pnas.89.21.10449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Carra S., Brunsting J.F., Kampinga H.H. HspB8 participates in protein quality control by a non-chaperone-like mechanism that requires eIF2α phosphorylation. J. Biol. Chem. 2009;284:5523–5532. doi: 10.1074/jbc.M807440200. [DOI] [PubMed] [Google Scholar]
- 31.Ecroyd H., Carver J.A. Crystallin proteins and amyloid fibrils. Cell. Mol. Life Sci. 2009;66:62–81. doi: 10.1007/s00018-008-8327-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Knowles T.P., Fitzpatrick A.W., Welland M.E. Role of intermolecular forces in defining material properties of protein nanofibrils. Science. 2007;318:1900–1903. doi: 10.1126/science.1150057. [DOI] [PubMed] [Google Scholar]
- 33.Chiti F., Dobson C.M. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 2006;75:333–366. doi: 10.1146/annurev.biochem.75.101304.123901. [DOI] [PubMed] [Google Scholar]
- 34.Hung L.W., Ciccotosto G.D., Barnham K.J. Amyloid-β peptide (Aβ) neurotoxicity is modulated by the rate of peptide aggregation: Aβ dimers and trimers correlate with neurotoxicity. J. Neurosci. 2008;28:11950–11958. doi: 10.1523/JNEUROSCI.3916-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cleary J.P., Walsh D.M., Ashe K.H. Natural oligomers of the amyloid-β protein specifically disrupt cognitive function. Nat. Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- 36.Kawarabayashi T., Shoji M., Younkin S.G. Dimeric amyloid β protein rapidly accumulates in lipid rafts followed by apolipoprotein E and phosphorylated tau accumulation in the Tg2576 mouse model of Alzheimer's disease. J. Neurosci. 2004;24:3801–3809. doi: 10.1523/JNEUROSCI.5543-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sondag C.M., Dhawan G., Combs C.K. β amyloid oligomers and fibrils stimulate differential activation of primary microglia. J. Neuroinflammation. 2009;6:1. doi: 10.1186/1742-2094-6-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Catalano S.M., Dodson E.C., Kinney G.G. The role of amyloid-β derived diffusible ligands (ADDLs) in Alzheimer's disease. Curr. Top. Med. Chem. 2006;6:597–608. doi: 10.2174/156802606776743066. [DOI] [PubMed] [Google Scholar]
- 39.Hepler R.W., Grimm K.M., Joyce J.G. Solution state characterization of amyloid β-derived diffusible ligands. Biochemistry. 2006;45:15157–15167. doi: 10.1021/bi061850f. [DOI] [PubMed] [Google Scholar]
- 40.Wang H.-W., Pasternak J.F., Trommer B.L. Soluble oligomers of β amyloid (1-42) inhibit long-term potentiation but not long-term depression in rat dentate gyrus. Brain Res. 2002;924:133–140. doi: 10.1016/s0006-8993(01)03058-x. [DOI] [PubMed] [Google Scholar]
- 41.Lord A., Englund H., Nilsson L.N. Amyloid-β protofibril levels correlate with spatial learning in Arctic Alzheimer's disease transgenic mice. FEBS J. 2009;276:995–1006. doi: 10.1111/j.1742-4658.2008.06836.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nilsberth C., Westlind-Danielsson A., Lannfelt L. The ‘Arctic’ APP mutation (E693G) causes Alzheimer's disease by enhanced Aβ protofibril formation. Nat. Neurosci. 2001;4:887–893. doi: 10.1038/nn0901-887. [DOI] [PubMed] [Google Scholar]
- 43.Knowles T.P.J., Waudby C.A., Dobson C.M. An analytical solution to the kinetics of breakable filament assembly. Science. 2009;326:1533–1537. doi: 10.1126/science.1178250. [DOI] [PubMed] [Google Scholar]
- 44.Tanaka M., Collins S.R., Weissman J.S. The physical basis of how prion conformations determine strain phenotypes. Nature. 2006;442:585–589. doi: 10.1038/nature04922. [DOI] [PubMed] [Google Scholar]
- 45.Knowles T.P.J., Shu W., Welland M.E. Kinetics and thermodynamics of amyloid formation from direct measurements of fluctuations in fibril mass. Proc. Natl. Acad. Sci. USA. 2007;104:10016–10021. doi: 10.1073/pnas.0610659104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Orte A., Birkett N.R., Klenerman D. Direct characterization of amyloidogenic oligomers by single-molecule fluorescence. Proc. Natl. Acad. Sci. USA. 2008;105:14424–14429. doi: 10.1073/pnas.0803086105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dobson C.M. Protein folding and misfolding. Nature. 2003;426:884–890. doi: 10.1038/nature02261. [DOI] [PubMed] [Google Scholar]
- 48.Pawar A.P., Dubay K.F., Dobson C.M. Prediction of “aggregation-prone” and “aggregation-susceptible” regions in proteins associated with neurodegenerative diseases. J. Mol. Biol. 2005;350:379–392. doi: 10.1016/j.jmb.2005.04.016. [DOI] [PubMed] [Google Scholar]
- 49.Borchelt D.R., Thinakaran G., Sisodia S.S. Familial Alzheimer's disease-linked presenilin 1 variants elevate Aβ1-42/1-40 ratio in vitro and in vivo. Neuron. 1996;17:1005–1013. doi: 10.1016/s0896-6273(00)80230-5. [DOI] [PubMed] [Google Scholar]
- 50.Scheuner D., Eckman C., Younkin S. Secreted amyloid β-protein similar to that in the senile plaques of Alzheimer's disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer's disease. Nat. Med. 1996;2:864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- 51.Sgarbossa A., Buselli D., Lenci F. In vitro perturbation of aggregation processes in β-amyloid peptides: a spectroscopic study. FEBS Lett. 2008;582:3288–3292. doi: 10.1016/j.febslet.2008.08.039. [DOI] [PubMed] [Google Scholar]
- 52.Stege G.J., Renkawek K., de Jong W.W. The molecular chaperone αB-crystallin enhances amyloid β neurotoxicity. Biochem. Biophys. Res. Commun. 1999;262:152–156. doi: 10.1006/bbrc.1999.1167. [DOI] [PubMed] [Google Scholar]
- 53.Raman B., Ban T., Rao ChM. αB-crystallin, a small heat-shock protein, prevents the amyloid fibril growth of an amyloid β-peptide and β2-microglobulin. Biochem. J. 2005;392:573–581. doi: 10.1042/BJ20050339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wilhelmus M.M.M., Boelens W.C., Verbeek M.M. Small heat shock proteins inhibit amyloid-β protein aggregation and cerebrovascular amyloid-β protein toxicity. Brain Res. 2006;1089:67–78. doi: 10.1016/j.brainres.2006.03.058. [DOI] [PubMed] [Google Scholar]
- 55.Kudva Y.C., Hiddinga H.J., Eberhardt N.L. Small heat shock proteins inhibit in vitro A β(1-42) amyloidogenesis. FEBS Lett. 1997;416:117–121. doi: 10.1016/s0014-5793(97)01180-0. [DOI] [PubMed] [Google Scholar]
- 56.Dehle F.C., Ecroyd H., Carver J.A. αB-Crystallin inhibits the cell toxicity associated with amyloid fibril formation by κ-casein and the amyloid-β peptide. Cell Stress Chaperones. 2010;15:1013–1026. doi: 10.1007/s12192-010-0212-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Narayanan S., Kamps B., Reif B. αB-crystallin competes with Alzheimer's disease β-amyloid peptide for peptide-peptide interactions and induces oxidation of Aβ-Met35. FEBS Lett. 2006;580:5941–5946. doi: 10.1016/j.febslet.2006.09.063. [DOI] [PubMed] [Google Scholar]
- 58.Waudby C.A., Knowles T.P.J., Meehan S. The interaction of αB-crystallin with mature α-synuclein amyloid fibrils inhibits their elongation. Biophys. J. 2010;98:843–851. doi: 10.1016/j.bpj.2009.10.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fändrich M., Meinhardt J., Grigorieff N. Structural polymorphism of Alzheimer Aβ and other amyloid fibrils. Prion. 2009;3:89–93. doi: 10.4161/pri.3.2.8859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rekas A., Adda C.G., Carver J.A. Interaction of the molecular chaperone αB-crystallin with α-synuclein: effects on amyloid fibril formation and chaperone activity. J. Mol. Biol. 2004;340:1167–1183. doi: 10.1016/j.jmb.2004.05.054. [DOI] [PubMed] [Google Scholar]
- 61.Rekas A., Jankova L., Carver J.A. Monitoring the prevention of amyloid fibril formation by α-crystallin. Temperature dependence and the nature of the aggregating species. FEBS J. 2007;274:6290–6304. doi: 10.1111/j.1742-4658.2007.06144.x. [DOI] [PubMed] [Google Scholar]
- 62.Kotarek J.A., Johnson K.C., Moss M.A. Quartz crystal microbalance analysis of growth kinetics for aggregation intermediates of the amyloid-β protein. Anal. Biochem. 2008;378:15–24. doi: 10.1016/j.ab.2008.03.022. [DOI] [PubMed] [Google Scholar]
- 63.Buell A.K., Dobson C.M., Welland M.E. Interactions between amyloidophilic dyes and their relevance to studies of amyloid inhibitors. Biophys. J. 2010;99:3492–3497. doi: 10.1016/j.bpj.2010.08.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ban T., Goto Y. Direct observation of amyloid growth monitored by total internal reflection fluorescence microscopy. Methods Enzymol. 2006;413:91–102. doi: 10.1016/S0076-6879(06)13005-0. [DOI] [PubMed] [Google Scholar]
- 65.Matsuzaki K. Formation of toxic amyloid fibrils by amyloid β-protein on ganglioside clusters. Int. J. Alzheimers Dis. 2011;2011:956104. doi: 10.4061/2011/956104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Reddy G.B., Kumar P.A., Kumar M.S. Chaperone-like activity and hydrophobicity of α-crystallin. IUBMB Life. 2006;58:632–641. doi: 10.1080/15216540601010096. [DOI] [PubMed] [Google Scholar]
- 67.Fanelli M.A., Cuello Carrión F.D., Ciocca D.R. Serological detection of heat shock protein hsp27 in normal and breast cancer patients. Cancer Epidemiol. Biomarkers Prev. 1998;7:791–795. [PubMed] [Google Scholar]
- 68.Friedrich R.P., Tepper K., Fändrich M. Mechanism of amyloid plaque formation suggests an intracellular basis of Aβ pathogenicity. Proc. Natl. Acad. Sci. USA. 2010;107:1942–1947. doi: 10.1073/pnas.0904532106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Carver J.A., Lindner R.A., Redfield C. The interaction of the molecular chaperone α-crystallin with unfolding α-lactalbumin: a structural and kinetic spectroscopic study. J. Mol. Biol. 2002;318:815–827. doi: 10.1016/S0022-2836(02)00144-4. [DOI] [PubMed] [Google Scholar]
- 70.Ecroyd H., Carver J.A. The effect of small molecules in modulating the chaperone activity of αB-crystallin against ordered and disordered protein aggregation. FEBS J. 2008;275:935–947. doi: 10.1111/j.1742-4658.2008.06257.x. [DOI] [PubMed] [Google Scholar]
- 71.Ecroyd H., Meehan S., Carver J.A. Mimicking phosphorylation of αB-crystallin affects its chaperone activity. Biochem. J. 2007;401:129–141. doi: 10.1042/BJ20060981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Treweek T.M., Ecroyd H., Walker M.J. Site-directed mutations in the C-terminal extension of human αB-crystallin affect chaperone function and block amyloid fibril formation. PLoS ONE. 2007;2:e1046. doi: 10.1371/journal.pone.0001046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Meehan S., Knowles T.P.J., Carver J.A. Characterisation of amyloid fibril formation by small heat-shock chaperone proteins human αA-, αB-, and R120G αB-crystallins. J. Mol. Biol. 2007;372:470–484. doi: 10.1016/j.jmb.2007.06.060. [DOI] [PubMed] [Google Scholar]
- 74.Huang J., Craggs T.D., Jackson S.E. Stable intermediate states and high energy barriers in the unfolding of GFP. J. Mol. Biol. 2007;370:356–371. doi: 10.1016/j.jmb.2007.04.039. [DOI] [PubMed] [Google Scholar]
- 75.Buell A.K., Dobson C.M., Welland M.E. Interactions between amyloidophilic dyes and their relevance to studies of amyloid inhibitors. Biophys. J. 2010;99:3492–3497. doi: 10.1016/j.bpj.2010.08.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.