

Abstract
The multidomain protein Thermus aquaticus MutS and its prokaryotic and eukaryotic homologs recognize DNA replication errors and initiate mismatch repair. MutS actions are fueled by ATP binding and hydrolysis, which modulate its interactions with DNA and other proteins in the mismatch-repair pathway. The DNA binding and ATPase activities are allosterically coupled over a distance of ∼70 Å, and the molecular mechanism of coupling has not been clarified. To address this problem, all-atom molecular dynamics simulations of ∼150 ns including explicit solvent were performed on two key complexes—ATP-bound and ATP-free MutS⋅DNA(+T bulge). We used principal component analysis in fluctuation space to assess ATP ligand-induced changes in MutS structure and dynamics. The molecular dynamics-calculated ensembles of thermally accessible structures showed markedly small differences between the two complexes. However, analysis of the covariance of dynamical fluctuations revealed a number of potentially significant interresidue and interdomain couplings. Moreover, principal component analysis revealed clusters of correlated atomic fluctuations linking the DNA and nucleotide binding sites, especially in the ATP-bound MutS⋅DNA(+T) complex. These results support the idea that allosterism between the nucleotide and DNA binding sites in MutS can occur via ligand-induced changes in motion, i.e., dynamical allosterism.
Introduction
DNA mismatch repair (MMR) is a highly conserved pathway in prokaryotes and eukaryotes that corrects replication errors and thereby suppresses deleterious mutations and genome instability. Defective MMR is associated with 10–30% spontaneous cancers in various tissues and hereditary nonpolyposis colorectal cancer (Lynch syndrome) (1–3). MMR is also involved in mediating cellular responses to DNA damage, including cell death, as well as somatic hypermutation in B cells (4–7).
The multidomain MutS protein is the first component of the MMR pathway and its function is to recognize replication errors such as basepair mismatches, insertion/deletion loops (IDLs), and damage lesions, such as O6-methylguanine and 6-thioguanine in DNA. After mismatch/IDL recognition, MutS initiates DNA repair, which includes activating a second protein component, MutL, followed by nicking of the error-containing strand, and strand excision and resynthesis by other DNA repair and replication proteins. In case of lesions, MutS binding to DNA can trigger cell-cycle checkpoints and apoptosis via mechanisms not yet fully resolved (4,5).
MutS actions in MMR are fueled by its ATP binding and hydrolysis activity, which modulates its interactions with DNA and other proteins in the pathway (8–11). Mismatch/IDL recognition at the DNA binding site in MutS is allosterically coupled to ATPase activity at a nucleotide-binding site separated by ∼70 Å. Mismatch/IDL binding by MutS proteins is followed by a marked suppression of ATP hydrolysis and a reduction in DNA binding affinity (12–16). It has been hypothesized that a MutS⋅DNA complex bound by two ATP molecules is a key species that signals initiation of DNA repair (17). X-ray crystallography (18–24), deuterium-exchange mass spectrometry (25), and computational studies (26,27) have been used to investigate various DNA- and nucleotide-bound/free MutS complexes, but the conformational dynamics have yet to be resolved in molecular detail, particularly for the MutS⋅DNA⋅ATP signaling complex. Since communication between the DNA- and nucleotide-binding sites in MutS is essential for triggering downstream MMR events, understanding the mechanism of allosteric signaling between the two sites is a matter of particular significance. However, it has proven difficult to experimentally discern the nature of allostery within the ∼180-kDa MutS protein and determine exactly how ATP binding/hydrolysis influences DNA binding and vice versa.
MutS is a multidomain dimeric protein, and several crystal structures have been solved for Thermus aquaticus and Escherichia coli MutS, as well as the human MutS homolog, MSH2-MSH6, in complex with different mismatches/IDLs (19–24). The crystal structure of T. aquaticus (Taq) MutS homodimer bound to a 23-basepair DNA containing a T bulge (+T), and ADP⋅beryllium fluoride in the nucleotide binding sites, forms the basis of our computational modeling (19). Each MutS subunit (Fig. 1, S1 and S2) contains ∼800 amino acids grouped historically into five domains (I–V). The structure of MutS resembles the Greek letter Θ, with enclosed upper and lower channels in the orientation shown in Fig. 1. The DNA(+T) binds in the lower channel and is observed to be kinked by ∼60° at the site of the inserted T. The N-terminal domain I (Fig. 1, Mismatch Binding) contains a highly conserved Phe-X-Glu motif that serves as a reading head and makes base-stacking and hydrogen-bonding contacts with the inserted T (28,29). This base-specific interaction occurs only between one subunit and DNA (S1 in MutS, MSH6 in MSH2-MSH6). Together with domain I, domain IV (Fig. 1, Clamp) in both subunits make up the complete DNA binding site, surrounding the duplex and making nonspecific contacts with the sugar-phosphate backbone. The C-terminal domain V (Fig. 1, ATPase) comprises the nucleotide binding site that belongs to the ABC transporter (ATP binding cassette) superfamily for ATP binding and hydrolysis; the site includes highly conserved motifs including the Walker A (phosphate-binding) and Walker B (Mg2+-binding) loops (20). The two nucleotide binding sites in the dimer are located adjacent to each other at the S1-S2 interface that closes the upper channel and, as noted earlier, are separated by ∼70 Å from the DNA binding site. Completing the domain decomposition of MutS as suggested by the crystal structure are domains II, IIIa, and IIIb (Fig. 1, Connector (between I and III), Lever, and Lever-Clamp, respectively), which link the DNA binding and ATPase sites.
Figure 1.
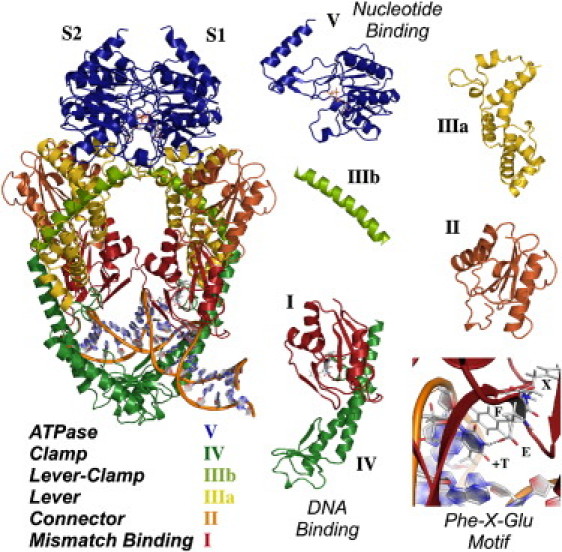
Crystal structure of T. aquaticus MutS shown with decomposition of domains (PDB code 1nne).
To further investigate the nature of allosterism in MutS, we carried out a series of all-atom molecular dynamics (MD) simulations with explicit consideration of solvent on a model of Taq MutS complexed with DNA containing a thymine insertion (+T) as well as ATP, and the corresponding complex without ATP. These forms of MutS were chosen for study because of experimental evidence that nucleotide-free MutS binds mismatched basepairs/IDLs with high affinity and selectivity, whereas ATP-bound MutS has lower affinity for DNA and is capable of interacting with other repair proteins (9,14,30). Moreover, results from this study would provide new information to complement recent MD analysis of DNA-free and G:T-mismatch-bound human MSH2-MSH6 (performed without nucleotide ligands) (26). Leading models of MMR postulate that ATP binding to MutS triggers allosteric communication from the nucleotide to the DNA binding site and is critical for MutS to transition from mismatch recognition to further actions on DNA that initiate repair (10,11,17). The MD ensembles were analyzed to predict solution structures and equilibrium dynamics, and to elucidate ATP-binding-induced changes in dynamical structure that are potentially important for the mechanism of allosterism in MutS (31,32).
Materials and Methods
System setup
The crystal structure of T. aquaticus MutS (PDB code 1nne) (19) serves as the reference structure for all simulations. The protein structure consists of a homodimer (765 amino acids/peptide chain) in complex with a 23-basepair +T bulge duplex-DNA of the sequence (dGCGACGCTAGCGTGCGGCTCGTC) and one ADP⋅BeF3− crystallographic ligand/nucleotide-binding site. Coordinates of each ADP⋅BeF3− ligand were removed and replaced with ATP for the model-built MutS⋅DNA(+T)⋅ATP complex (ATP-bound), and were removed entirely for the MutS⋅DNA(+T) complex (ATP-free). For the MutS⋅DNA(+T)⋅Mg-ATP complex, the coordinates for Mg-ATP were derived from an E. coli MutS crystal structure (PDB code 1w7a) (18). The crystallographic DNA was retained in all simulations. Coordinates of missing atoms and residues were added with the LEap module of AMBER 9.0 (33) and PYMOL (DeLano Scientific, 2006) (for additional details, see sections S6 and S7 in the Supporting Material.
MD simulation protocol
We carried out all-atom simulations on model-built ATP-bound and ATP-free MutS⋅DNA(+T) complexes with the PMEMD version of the AMBER 9.0 suite of programs (33) (150 ns for MutS⋅DNA(+T) and MutS⋅DNA(+T)⋅ATP; 40 ns for MutS⋅DNA(+T)⋅Mg-ATP). The ff99 force field (34,35) with ff99SB (36), ff99bsc0 (37), and ff99ions08 (38) corrections were used for proteins, DNA, and monovalent ions, respectively. Polyphosphate parameters were used for ATP (39). The complexes were solvated in a 12-Å-thick truncated octahedron box of ∼65,000 TIP3P water molecules (40) and treated under periodic boundary conditions. The complex was neutralized with 81 Na+ ions and an additional 182 NaCl ions were added to achieve an ionic strength of 150 mM (total box size, 129 Å × 82 Å × 108 Å). Long-range electrostatic interactions were treated with the particle mesh Ewald (PME) algorithm (41–43). The Lennard-Jones cutoff was set to 10 Å. Initial calculations involved two separate energy minimizations of first the solvent and then the solute-solvent, followed by slow heating to 300 K and equilibration. Production steps of the MD were carried out using the NPT ensemble conditions and Berendsen algorithm (44). We applied SHAKE (45) constraints to all bonds involving hydrogen atoms, permitting an integration time step of 2 fs. Trajectory snapshots were saved every 2 ps, and the simulations were run in successive 1 ns intervals. MD trajectories for MutS⋅DNA(+T)⋅ATP and MutS⋅DNA(+T) complexes were obtained for 150 ns and repeated for ∼100 ns starting from a different configuration. The duplicate simulations were initiated from snapshots at 120 ns and 35 ns of the equilibrated MutS⋅DNA(+T)⋅ATP (without Mg2+) and MutS⋅DNA(+T) simulations, respectively; solvent molecules and ions were stripped from each snapshot, solvent boxes rebuilt, and ions added to match the original simulation conditions. MD quality (46) for the simulations under study was assessed with root mean-square deviation (RMSD), radius of gyration (RG), and principal component analysis (PCA) of covariance matrices in Cartesian space (47,48) (see Fig. S3, Fig. S4, Fig. S5, Fig. S6, and Fig. S7 in the Supporting Material).
MD analysis
To elucidate ATP-binding-induced changes in the dynamical structure, we first compared the overall backbone RMSD of ATP-bound and ATP-free average structures, as well as the thermal dispersion of corresponding MD ensembles. As described fully in the Supporting Material, we calculated the dynamical cross-correlation map (DCCM) for each simulation to extract groups of correlated atoms (49). Matrices with pairwise correlations of atomic fluctuations averaged by residue were generated in the ptraj module of AmberTools 1.2; pairwise correlations of atomic fluctuations are computed using backbone atoms (N, Cα, and C in amino acids, and P, O5′, C5′, C4′, C3′, and O3′ in nucleosides) (50). We assessed the temporal convergence of these results by comparing the RMSD (Fig. S6 and Fig. S7) and correlation matrices (Fig. S8 and Fig. S9) calculated over several 10 ns average blocks of the equilibrated portion of each simulation. Next, we obtained the eigenvectors and eigenvalues of the correlation matrices by carrying out PCA on each matrix of correlated atomic fluctuation. PCA of correlated atomic fluctuations is similar to PCA in Cartesian coordinate space (48), except that the correlation-matrix elements are normalized covariances in the space of atomic fluctuations as opposed to covariances in Cartesian space. Each eigenvector represents a cluster of mutually correlated atomic motions and its eigenvalue represents the weight of that cluster (see Supporting Material for additional details).
Results
Effect of ATP binding on MutS structure and dynamics
We monitored the stability of the MutS⋅DNA(+T) and MutS⋅DNA(+T)⋅ATP simulations by following the time series for RMSDs and radius of gyration (RG). All trajectory snapshots were aligned to the first, and the RMSD(t) plots for the protein and backbone atoms of ATP-bound and ATP-free simulations are shown in Fig. 2 A. The RMSD exhibits good stability with thermal fluctuations ranging from ∼2.0 to 3.5 Å, and minor fluctuations (∼±0.5) in the last 100 ns of the simulations. The relatively long timescale for equilibration may indicate that MutS requires time to reorganize in response to removal of ADP⋅BeF3− (and replacement with ATP). The radius of gyration also shows stabilization (Fig. 2 B); a small expansion in RG(t) (ΔRG ∼ 2.0 Å) occurs initially, followed by minor fluctuations (ΔRG ± 0.5 Å) through the remaining trajectory. The overall structure of the MD-simulated ATP-bound (−/+ Mg2+) and ATP-free MutS⋅DNA(+T) complexes is quite similar to that of the MutS⋅DNA⋅ADP⋅BeF3− crystal structure (1nne) (Fig. 2 C; MutS⋅DNA(+T)⋅ATP shown superimposed with 1nne). The backbone RMSD between the structures is ∼3–4 Å, a reasonable difference, considering the different construction of the MD model system. The changes in MutS and DNA conformation during the simulations are also quite subtle; indeed, the thermal dispersions of structures in the MD ensembles of the ATP-bound and ATP-free forms overlap well (Fig. 2, D and E, respectively), and the RG values for the equilibrated portions for both simulations are almost identical (RG ∼ 41.0 Å) (Fig. 2 B). The differences between MutS⋅DNA(+T)⋅ATP and MutS⋅DNA(+T)⋅Mg-ATP structures are also small (Fig. S12). We compare mainly the MutS⋅DNA(+T) and MutS⋅DNA(+T)⋅ATP complexes, which are both 150 ns simulations, to elucidate the effects of ATP binding on MutS and DNA.
Figure 2.
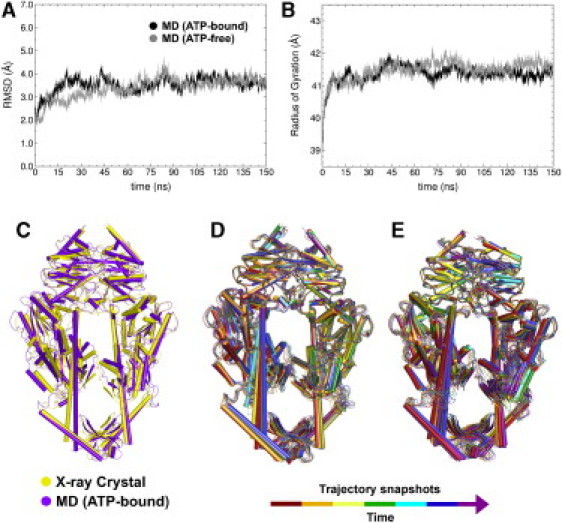
(A) RMSD of protein and DNA backbone atoms with respect to the first simulation snapshot for MutS⋅DNA(+T)⋅ATP (black) and MutS⋅DNA(+T) (gray) simulations. (B) Radius of gyration of protein backbone for the corresponding simulations. (C) Crystal structure of MutS⋅ADP⋅BeF3− (PDB code 1nne) (yellow) superimposed with the calculated average structure for MD on MutS⋅DNA(+T)⋅ATP (ATP-bound) (purple). (D and E) Simulation snapshots of ATP-bound (D) and ATP-free (E) complexes.
The ATP-bound and ATP-free MD average structures are shown superimposed in Fig. 3 A. The protein backbones in the nucleotide binding sites (domain V) are similar in these complexes, with an RMSD range of 2.0–2.7 Å for each subunit (Fig. 3 B). We do find small changes in the positions of loops, especially in subunit S1; for example, in the presence of ATP, the SDDLAGGKST loop (Fig. 3 B and Fig. S1 a) containing the highly conserved N-2 motif extends by 10 Å and makes contact with the Connector (domain IIIa) (20). This movement may enhance communication between the nucleotide-binding sites and other domains in MutS (19). The orientation of the active-site residues also remains quite similar, including the conserved Walker A (Lys589, Ser590) and Walker B (Asp662) residues (Fig. S1 B). One significant difference is that Phe587, which stacks against the adenine base in the ATP-bound complex, rotates ∼80° away from the binding pocket in the ATP-free complex. The structure of the DNA binding site is also quite similar in both complexes, except for a small but distinct change in the Clamp domain (domain IV) of S2, as well as a loop (Val98–Gly106) in the Mismatch binding domain (domain I) of S1 (Fig. 3 C). These small changes are consistent with subtle differences observed in the proteolysis pattern of DNA-bound MutS in the absence versus the presence of ATP (51). In general, the nonspecific interactions of domains I and IV (S1 and S2) with DNA, including the stacking and hydrogen-bonding interactions of the Phe-X-Glu motif (S1) with the +T base are similar in both structures. However, the kink angle at the +T site in DNA decreases from ∼74° in the ATP-free complex to ∼60° in the ATP-bound complex (Fig. 3 D), indicating slight straightening of DNA, which may underlie the reduction in MutS affinity for DNA upon ATP binding (1). Current models of DNA mismatch repair propose that ATP binding leads to conversion of MutS into a clamp form that can slide away from the mismatch on DNA (52). We speculate that movement of the glutamate-rich Val98–Gly106 loop ∼10 Å away from the major groove in ATP-bound MutS (Fig. 3 C), and the associated straightening of DNA may facilitate this conversion.
Figure 3.
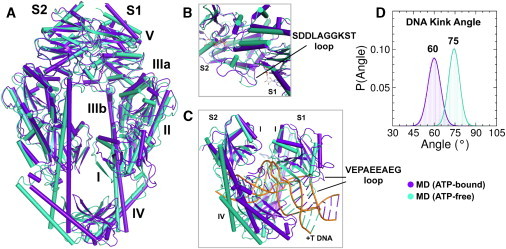
(A–C) Calculated average structures for MD on MutS⋅DNA(+T)⋅ATP (ATP-bound) (purple) and MutS⋅DNA(+T) (ATP-free) (cyan) complexes, with close-ups of the nucleotide- (B) and DNA-binding (C)sites. (D) DNA kink angles for ATP-bound (purple) and ATP-free (cyan) complexes.
Atomic fluctuations and MutS allostery
The lack of dramatic ATP-induced changes in the overall structure of MutS⋅DNA(+T) complexs pointed this study in the direction of allosterism without significant change in structure (53–55), in which communication between two sites on a macromolecule occurs via changes in dynamics, such as changes in thermal fluctuations. We first probed the ligand-induced changes in MutS dynamics by considering the relative change in B-factors upon ATP-binding (Fig. 4; MD-calculated B-factors are atomic positional fluctuations multiplied by (8/3)π2; see Supporting Material). Residues with B-factors >30 Å2 and exhibiting >30% change are mapped onto the MutS structure in brown (increase) and green (decrease) (corresponding B-factor plots are shown in Fig. S14). Our results show an ATP-induced increase in the Mismatch binding domain I B-factors (both S1 and S2 subunits) and, conversely, a decrease in the nucleotide-binding-site B-factors, especially in the S1 subunit. This result is broadly consistent with experimental data indicating increased organization of the nucleotide-binding site in the presence of ATP (18,25) and the ATP-induced loss of MutS DNA-binding affinity (1).
Figure 4.
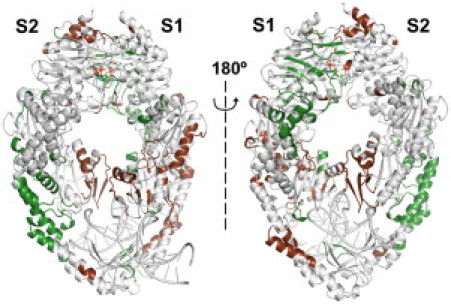
Relative changes in B-factors upon ATP binding, showing areas with a >30% increase (brown) and those with a >30% decrease (green). Only residues with a B-factor of >30 Å2 are shown.
Support for intramolecular communication via dynamics is provided by the covariance of fluctuations in the MD ensembles for the ATP-bound and ATP-free MutS⋅DNA(+T) complexes. Following previous analyses, we formed the normalized covariance matrix of atomic fluctuations averaged with respect to amino acids and nucleotides, and we present the results in a dynamical cross-correlation map (DCCM) (49,56). We also generated correlation matrices for 10 ns blocks of the equilibrated portion of the simulations to assess convergence (the maps are shown in Fig. S8 and Fig. S9). The MD-calculated DCCM for MutS⋅DNA(+T)⋅ATP complex is shown in Fig. 5 A. The map is subdivided into four quadrants—the two along the diagonal show motional correlations within subunits S1 and S2, and the off-diagonal blocks describe correlations between S1 and S2. The correlation matrix is symmetric, and following a previously defined convention (49,57–63), positive correlations are shown in the upper triangle and negative correlations in the lower triangle for clarity of interpretation. MD-calculated correlated atomic fluctuations within S1 and S2 subunits are shown in the lower left and upper right blocks, respectively. There is an abundance of correlations indicated on the map, some of which are highlighted here. Within the subunits, the blocks of correlations along the diagonal correspond well with the domain architecture derived by inspection of the MutS crystal structure (Fig. 1). The idea that blocks of motional correlations might be a useful method to define functionally relevant structural domains has been noted previously (58), and our results with MutS show that this idea has traction when extended to a complex multidomain protein. The dynamical correlations within each of the domains show evidence of well-defined interdomain structures. For example, within S1, there is a prominence of positive correlations between domains I and II, between II and IIIa and IIIb, between IIIa and IIIb (confirming the previous annotation of domain III as a single domain though not contiguous in sequence), and between domains IIIa and V (similar observations apply to S2 as well (Fig. 5 A)). These correlation values are well converged, as seen from the similar patterns across most of the 10 ns blocks of the equilibrated simulation (Fig. S8). With regard to the DNA substrate, MutS DNA-binding domain I shows correlations with fluctuations in the DNA(+T) per se, a likely consequence of their direct contacts. Finally, correlations between S1 and S2 are detected in domains IV and V across the subunit interface. There is also evidence, albeit weak, for coupling of the DNA-binding and ATPase domains (e.g., domain IV in S1 and domain V in S2), consistent with experimental evidence that these active sites are in allosteric communication with each other (1).
Figure 5.
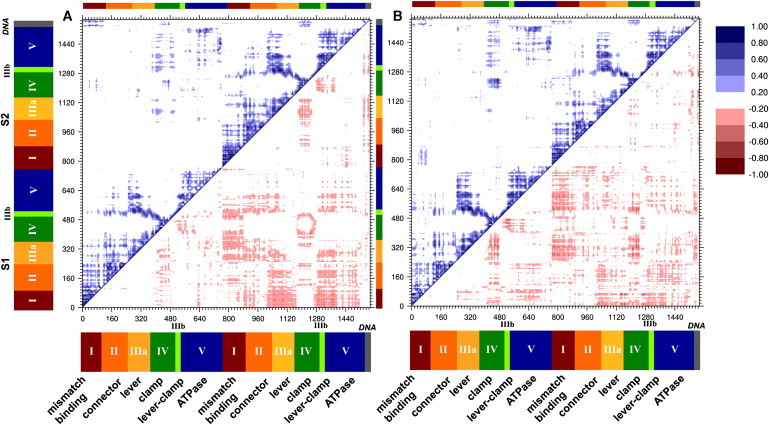
Calculated correlation of atomic fluctuations (by residue) for ATP-bound (A) and ATP-free (B) complexes. Magnitudes of calculated cross-correlations are indicated by (Cij = ±0.20–1.00).
Next, we compared the DCCMs for the ATP-bound and ATP-free complexes and found a number of differences between the two. Because the significance of these differences is difficult to assess visually (cf. Fig. 5, A and B), we performed PCA on the DCCMs from the MD runs of each complex to determine specific motional changes associated with ATP binding to MutS⋅DNA(+T). The eigenvectors of the DCCM matrices revealed clusters of mutually correlated fluctuations. Each of the first four eigenvectors with the largest eigenvalues (Fig. 6) from the correlation matrices of ATP-bound and ATP-free complexes was mapped onto its corresponding structure and color-coded according to the magnitude of the contribution from each residue motion to the eigenvector (Fig. S15). The intensity of the color represents the coefficient of residue motion to each eigenvector. Blue/green versus red/orange corresponds to combinations with opposite phase (sign is arbitrary) and the color intensity varies with degree of correlation. We calculated the overlap to match eigenvectors from the ATP-bound simulation with those from the ATP-free simulation (Table S2, A and B). From these four sets of structures, we focus on two that reveal clusters of mutually correlated residues connecting the ATP-binding sites with the DNA-binding site (Fig. 7, A–D). Although the other eigenvectors may also have functional relevance, they do not link the two active sites (Fig. S15). In the first set of structures (Fig. 7, A and B) (both eigenvector 1), a cluster of correlated fluctuations involving the DNA- and nucleotide-binding sites, along with links to other regions of the protein, is clearly evident in the ATP-bound complex (Fig. 7 A). This cluster encompasses the mismatch-binding site in S1 (including Phe-X-Glu motif), as well as the DNA-binding and nucleotide-binding sites in S2. The cluster also includes prominent contributions from domains II, V, and III in S2, supporting the idea that these domains play key roles in allosteric communication within MutS (23). The corresponding cluster from the ATP-free complex (Fig. 7 B) has a similar cluster of correlated motions through the protein, but the cluster is overall less extensive and does not involve the mismatch-binding site (including the Phe-X-Glu motif) nor the nucleotide-binding sites. This finding suggests that ATP binding could act as a switch to couple the motions of the nucleotide-binding site with this cluster of correlated motions through the protein and thus establish communication with the distant DNA-binding site. The existence of a robust dynamic framework for relaying signals between the two functional sites in MutS is consistent with experimental evidence that ATP binding alters MutS interaction with DNA, which is essential for initiating downstream events in the MMR pathway.
Figure 6.
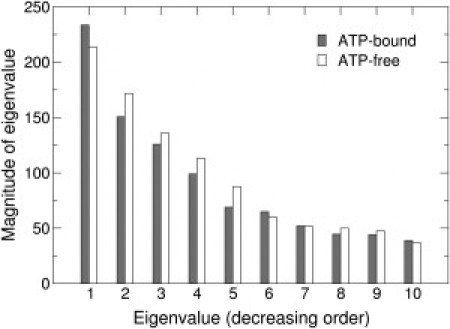
Graph of the 10 largest eigenvalues of each complex plotted in descending order. Gray and white bars represent ATP-bound and ATP-free complexes, respectively.
Figure 7.
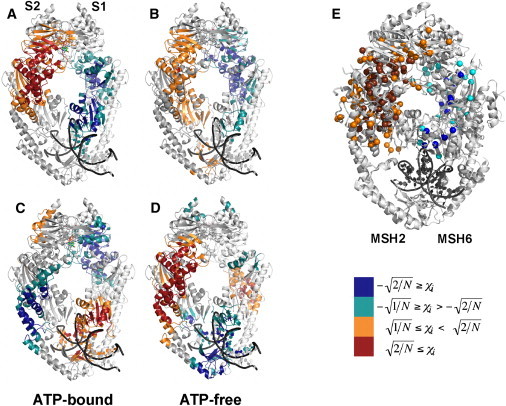
Eigenvectors with the first (A and B) and fourth (C and D) largest eigenvalues of the correlation matrices from ATP-bound and ATP-free complexes mapped onto the corresponding structures. The intensity of the color represents the coefficient (χi) of residue motion i to each eigenvector. Blue/green versus red/orange correspond to combinations with opposite phase (sign is arbitrary). (E) Structure of human MSH2-MSH6 showing colon-cancer-linked mutations that map by structural alignment (23) to Taq MutS residues in clusters from eigenvector 1 (shown in A and B).
The second set of structures of the ATP-bound (eigenvector 4) and ATP-free (eigenvector 3) complexes (Fig. 7, C and D) also reveals clusters of correlated residues with potential functional significance. These structures have been highlighted because of the predominance of the long α-helical levers of domains IIIb and IV within the clusters. The levers are highly conserved structural features of all MutS proteins, including the human homolog Msh2–Msh6, and have been previously proposed to play an important role in communication between the nucleotide- and DNA-binding sites in MutS (19,20). In this set, the clusters in both structures include the mismatch-binding site (including the Phe-X-Glu motif); however, the cluster in the ATP-bound complex is more extensive and includes the nucleotide-binding site, whereas the ATP-free complex structure clearly does not. Another striking difference is the absence of S2 domain IV residues specifically in the ATP-bound complex clusters. These results support the hypothesis that frameworks of mutually correlated motions specifically connect the two active sites in the MutS⋅DNA(+T)⋅ATP complex and could therefore mediate the allosteric signal generated by binding of ATP to MutS after mismatch recognition.
Discussion
MD simulations were carried out for the ATP-bound complex of MutS⋅DNA(+T), in which allosteric communication between the nucleotide-binding and DNA-binding sites is considered critical for initiation of MMR, and the corresponding MutS⋅DNA(+T) complex without ATP ligands. The MD averaged structures indicate slight differences in MutS conformation between the two complexes, for example, in the Clamp domain (IV) that cradles the DNA duplex (Fig. 3 C) and in the kink angle at the +T base in DNA (Fig. 3, C and D). We also found an increase in B-factors in the mismatch-binding domains of S1 and S2 subunits in the ATP-bound MutS⋅DNA(+T) complex (Fig. 4). It is tempting to speculate that these subtle structural changes are related to the decrease in affinity between MutS and DNA on ATP binding. In addition, we detected changes in B-factors in the nucleotide-binding sites (decrease in S1, increase in S2), which indicate local changes in dynamics on ATP binding that could in turn affect MutS ATPase activity and interaction with DNA. This finding is consistent with the subtle differences in the nucleotide-binding sites of ATP-bound and nucleotide-free E. coli MutS⋅G:T mismatch crystal structures (18). Overall, however, ATP binding causes little alteration in the structure of the MutS⋅DNA(+T) complex in the timescale of the MD simulations. These MD results do not preclude significant ATP-induced changes in MutS conformation, perhaps on a longer timescale. A recent protein cross-linking study of E. coli MutS indicates domain I rearrangement in the presence of ATP (64), and preliminary kinetic data with fluorescently labeled Taq MutS also indicates movement of domain I on the order of seconds (P. Sharma and A. Hingorani, unpublished results).
The MD data enable us to investigate whether the nucleotide- and DNA-binding sites in MutS can communicate via a mechanism involving ligand-induced changes in dynamics (53,55). Thus, we analyzed the MD trajectories for the MutS⋅DNA(+T) complexes in terms of the normalized covariance of dynamical fluctuations and displayed the results as DCCMs. Visual inspection of the DCCM revealed numerous interresidue and interdomain motional couplings, and PCA analysis in fluctuation space based on the DCCMs provided evidence of clusters of correlated atomic fluctuations linking the nucleotide and the DNA-binding sites in MutS. The motions in these clusters were found to be more extensive in the ATP-bound MutS⋅DNA(+T) complex relative to the ATP-free complex. Thus, MD simulations support the hypothesis that ATP binding to Taq MutS can affect its interaction with DNA at a distance of ∼70 Å by sending signals dynamically, as has been proposed recently for other, smaller proteins (53,65–67).
The clusters of correlated fluctuations discovered from the MD simulation data provide support for aspects of previous speculations about the allosteric mechanism of MutS (20,23), which were based on the idea that interfaces between relatively rigid domains may facilitate communication across the protein (68). The clusters described in Fig. 7, C and D, encompass the long α-helix spanning the protein from domain IV to domain V, which is considered to be a likely transmitter of allosteric signals. Also included in the clusters is the connector domain (II), whose position in a cleft surrounded by domains V and III can be considered as favorable for allosteric signaling. Another interesting structural element in the clusters is a conserved loop in domain III at the intersection of domains II and IV (Fig. 1); although it has low sequence conservation, it is speculated that this loop also plays a role in allosteric signaling between the nucleotide- and DNA-binding domains in human MutS (20,23).
This MD analysis also provides a framework to determine 1), which regions (and residues) of the protein are involved or not involved in clusters in any nucleotide occupancy state; and 2), which regions exhibit altered involvement depending on nucleotide occupancy. For example, the lower α-helix in the S1 clamp IV domain is not part of any cluster within any of the structures shown in Fig. 7; thus, one can predict that mutations in this structural element may have little impact on allosteric communication between the two active sites in MutS. In other examples, S2 domains IIIa, IIIb, and (partially) V exhibit higher correlation in clusters within the ATP-bound versus the ATP-free structure (compare Fig. 7, A and B), and the lower portion of S2 domain I is part of the clusters in the ATP-free, but not the ATP-bound, structure (compare Fig. 7, A and B, with Fig. 7, C and D, respectively); thus, one can predict that mutations in these structural elements will have differential effects on allosteric communication in ATP-bound versus ATP-free MutS. These findings can aid development of experimentally testable hypotheses about MutS residues that may play a key role in coupling the mismatch binding and ATPase activities. Moreover, using the structural alignment of human MSH2-MSH6 and Taq MutS reported by Beese et al. (23), we have found that a large number of mutations in msh2 and msh6 genes from the Lynch syndrome database (http://www.insight-group.org/home/) map onto the clusters shown in Fig. 7, A and B (Fig. 7 E). Many of these residues are in low sequence-homology regions and are located far from the nucleotide- and DNA-binding active sites, which makes it difficult to ascribe specific roles to them based on crystal structure alone. The MD analysis suggests that these mutations are likely to affect MutS allostery.
Our findings extend other recently published computational studies of MutS structure and dynamics (26,27,69), specifically normal-mode analysis of E. coli MutS and human MSH2-MSH6 (27), as well as all-atom MD simulations of MSH2-MSH6 (26) free and bound to a G:T-mismatch-containing DNA in the absence of any nucleotides. A noteworthy observation is that the overall structures of the Taq and E. coli MutS homodimers and human MSH2-MSH6 heterodimer are similar in all computational calculations thus far, and comparable to the crystal structures, despite significant differences in protein sequence, model construction, and mode of analysis. The reported normal-mode calculations and MD simulations focused on the effects of DNA binding to MutS and found the Clamp (IV) and Lever (III) domains to be open in the DNA-free structure, consistent with the x-ray structure reported for DNA-free Taq MutS, in which these domains are partially unresolved (20). Based on a comparison of the nucleotide-free MD structures and ADP-Mg2+-bound crystal structure, it was proposed that DNA binding to MSH2-MSH6 is coupled with subtle reorientation of amino acids in the nucleotide-binding site (26). This proposal is consistent with allosteric communication between the two active sites such that DNA binding affects MutS structure/dynamics and function. This report addresses the complementary question of how ATP binding affects MutS and presents evidence for subtle changes in the DNA-binding site that could alter MutS affinity for DNA; more significantly, it presents evidence for clusters of correlated amino acid residue motions that form a dynamic framework for transmission of allosteric signals from the nucleotide-binding to DNA-binding sites in MutS.
Conclusion
The protein dynamics-based model for allosteric signaling proposed here is plausible for MutS function after mismatch recognition and ATP binding, since radical changes in MutS structure at this point in the reaction could compromise its processive actions on DNA, which include interactions with other proteins and apparent movement on DNA to initiate repair. This view is consistent with experimental data indicating that ATP-bound MutS can release the mismatch but retain hold of the DNA and slide on it (14,17,70). By comparing nucleotide-free and ATP-bound forms of MutS⋅DNA(+T) complexes, our MD analysis has revealed significant ligand-induced changes in clusters of correlated residue fluctuations in the protein, and has also identified clusters specifically linking the DNA- and nucleotide-binding sites within the ATP-bound complex. These findings provide new ideas for experimental investigation of allostery in MutS. Beyond MutS, these findings support the broader emerging paradigm that allosterism between two sites on a large multidomain protein may occur via ligand-induced changes in motion, i.e., dynamical allosterism.
Acknowledgments
We thank Fernando R. Clemente for helpful discussions.
This work was supported by grants from the National Science Foundation (MCB 1022203 to M.M.H) and the National Institutes of Health (GM-076490 to D.L.B). S.N.P. was supported by a National Institutes of Health National Research Service Award postdoctoral fellowship (F32-GM-87101). We acknowledge partial support from the National Science Foundation through the TeraGrid resources provided by grant number CHE10004, utilizing the Ranger Cluster at the Texas Advanced Computing Center.
Supporting Material
References
- 1.Kunkel T.A., Erie D.A. DNA mismatch repair. Annu. Rev. Biochem. 2005;74:681–710. doi: 10.1146/annurev.biochem.74.082803.133243. [DOI] [PubMed] [Google Scholar]
- 2.Renkonen E., Zhang Y., Peltomaki P. Altered expression of MLH1, MSH2, and MSH6 in predisposition to hereditary nonpolyposis colorectal cancer. J. Clin. Oncol. 2003;21:3629–3637. doi: 10.1200/JCO.2003.03.181. [DOI] [PubMed] [Google Scholar]
- 3.Kolodner R.D. Mismatch repair: mechanisms and relationship to cancer susceptibility. Trends Biochem. Sci. 1995;20:397–401. doi: 10.1016/s0968-0004(00)89087-8. [DOI] [PubMed] [Google Scholar]
- 4.Jiricny J. MutLalpha: at the cutting edge of mismatch repair. Cell. 2006;126:239–241. doi: 10.1016/j.cell.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 5.Hsieh P., Yamane K. DNA mismatch repair: molecular mechanism, cancer, and ageing. Mech. Ageing Dev. 2008;129:391–407. doi: 10.1016/j.mad.2008.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Iyer R.R., Pluciennik A., Modrich P. MutLα and proliferating cell nuclear antigen share binding sites on MutSβ. J. Biol. Chem. 2010;285:11730–11739. doi: 10.1074/jbc.M110.104125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kunz C., Saito Y., Schär P. DNA repair in mammalian cells: mismatched repair: variations on a theme. Cell. Mol. Life Sci. 2009;66:1021–1038. doi: 10.1007/s00018-009-8739-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kolodner R.D., Mendillo M.L., Putnam C.D. Coupling distant sites in DNA during DNA mismatch repair. Proc. Natl. Acad. Sci. USA. 2007;104:12953–12954. doi: 10.1073/pnas.0705698104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mendillo M.L., Mazur D.J., Kolodner R.D. Analysis of the interaction between the Saccharomyces cerevisiae MSH2-MSH6 and MLH1-PMS1 complexes with DNA using a reversible DNA end-blocking system. J. Biol. Chem. 2005;280:22245–22257. doi: 10.1074/jbc.M407545200. [DOI] [PubMed] [Google Scholar]
- 10.Acharya S., Foster P.L., Fishel R. The coordinated functions of the E. coli MutS and MutL proteins in mismatch repair. Mol. Cell. 2003;12:233–246. doi: 10.1016/s1097-2765(03)00219-3. [DOI] [PubMed] [Google Scholar]
- 11.Selmane T., Schofield M.J., Hsieh P. Formation of a DNA mismatch repair complex mediated by ATP. J. Mol. Biol. 2003;334:949–965. doi: 10.1016/j.jmb.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 12.Antony E., Hingorani M.M. Mismatch recognition-coupled stabilization of Msh2-Msh6 in an ATP-bound state at the initiation of DNA repair. Biochemistry. 2003;42:7682–7693. doi: 10.1021/bi034602h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Antony E., Hingorani M.M. Asymmetric ATP binding and hydrolysis activity of the Thermus aquaticus MutS dimer is key to modulation of its interactions with mismatched DNA. Biochemistry. 2004;43:13115–13128. doi: 10.1021/bi049010t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gradia S., Subramanian D., Fishel R. hMSH2-hMSH6 forms a hydrolysis-independent sliding clamp on mismatched DNA. Mol. Cell. 1999;3:255–261. doi: 10.1016/s1097-2765(00)80316-0. [DOI] [PubMed] [Google Scholar]
- 15.Martik D., Baitinger C., Modrich P. Differential specificities and simultaneous occupancy of human MutSα nucleotide binding sites. J. Biol. Chem. 2004;279:28402–28410. doi: 10.1074/jbc.M312108200. [DOI] [PubMed] [Google Scholar]
- 16.Studamire B., Quach T., Alani E. Saccharomyces cerevisiae Msh2p and Msh6p ATPase activities are both required during mismatch repair. Mol. Cell. Biol. 1998;18:7590–7601. doi: 10.1128/mcb.18.12.7590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mazur D.J., Mendillo M.L., Kolodner R.D. Inhibition of Msh6 ATPase activity by mispaired DNA induces a Msh2(ATP)-Msh6(ATP) state capable of hydrolysis-independent movement along DNA. Mol. Cell. 2006;22:39–49. doi: 10.1016/j.molcel.2006.02.010. [DOI] [PubMed] [Google Scholar]
- 18.Lamers M.H., Georgijevic D., Sixma T.K. ATP increases the affinity between MutS ATPase domains. Implications for ATP hydrolysis and conformational changes. J. Biol. Chem. 2004;279:43879–43885. doi: 10.1074/jbc.M406380200. [DOI] [PubMed] [Google Scholar]
- 19.Alani E., Lee J.Y., Yang W. Crystal structure and biochemical analysis of the MutS.ADP.beryllium fluoride complex suggests a conserved mechanism for ATP interactions in mismatch repair. J. Biol. Chem. 2003;278:16088–16094. doi: 10.1074/jbc.M213193200. [DOI] [PubMed] [Google Scholar]
- 20.Obmolova G., Ban C., Yang W. Crystal structures of mismatch repair protein MutS and its complex with a substrate DNA. Nature. 2000;407:703–710. doi: 10.1038/35037509. [DOI] [PubMed] [Google Scholar]
- 21.Lamers M.H., Winterwerp H.H., Sixma T.K. The alternating ATPase domains of MutS control DNA mismatch repair. EMBO J. 2003;22:746–756. doi: 10.1093/emboj/cdg064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Natrajan G., Lamers M.H., Sixma T.K. Structures of Escherichia coli DNA mismatch repair enzyme MutS in complex with different mismatches: a common recognition mode for diverse substrates. Nucleic Acids Res. 2003;31:4814–4821. doi: 10.1093/nar/gkg677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Warren J.J., Pohlhaus T.J., Beese L.S. Structure of the human MutSalpha DNA lesion recognition complex. Mol. Cell. 2007;26:579–592. doi: 10.1016/j.molcel.2007.04.018. [DOI] [PubMed] [Google Scholar]
- 24.Junop M.S., Obmolova G., Yang W. Composite active site of an ABC ATPase: MutS uses ATP to verify mismatch recognition and authorize DNA repair. Mol. Cell. 2001;7:1–12. doi: 10.1016/s1097-2765(01)00149-6. [DOI] [PubMed] [Google Scholar]
- 25.Mendillo M.L., Putnam C.D., Kolodner R.D. Probing DNA- and ATP-mediated conformational changes in the MutS family of mispair recognition proteins using deuterium exchange mass spectrometry. J. Biol. Chem. 2010;285:13170–13182. doi: 10.1074/jbc.M110.108894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mukherjee S., Feig M. Conformational change in MSH2-MSH6 upon binding DNA coupled to ATPase activity. Biophys. J. 2009;96:L63–L65. doi: 10.1016/j.bpj.2009.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mukherjee S., Law S.M., Feig M. Deciphering the mismatch recognition cycle in MutS and MSH2-MSH6 using normal-mode analysis. Biophys. J. 2009;96:1707–1720. doi: 10.1016/j.bpj.2008.10.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Malkov V.A., Biswas I., Hsieh P. Photocross-linking of the NH2-terminal region of Taq MutS protein to the major groove of a heteroduplex DNA. J. Biol. Chem. 1997;272:23811–23817. doi: 10.1074/jbc.272.38.23811. [DOI] [PubMed] [Google Scholar]
- 29.Dufner P., Marra G., Jiricny J. Mismatch recognition and DNA-dependent stimulation of the ATPase activity of hMutSα is abolished by a single mutation in the hMSH6 subunit. J. Biol. Chem. 2000;275:36550–36555. doi: 10.1074/jbc.M005987200. [DOI] [PubMed] [Google Scholar]
- 30.Zhai J., Hingorani M.M. Saccharomyces cerevisiae Msh2-Msh6 DNA binding kinetics reveal a mechanism of targeting sites for DNA mismatch repair. Proc. Natl. Acad. Sci. USA. 2010;107:680–685. doi: 10.1073/pnas.0908302107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koshland D.E.J., Jr., Némethy G., Filmer D. Comparison of experimental binding data and theoretical models in proteins containing subunits. Biochemistry. 1966;5:365–385. doi: 10.1021/bi00865a047. [DOI] [PubMed] [Google Scholar]
- 32.Monod J., Wyman J., Changeux J.P. On the nature of allosteric transitions: a plausible model. J. Mol. Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 33.Case D.A., Darden T.A., Kollman P.A. University of California; San Francisco: 2006. AMBER 9. [Google Scholar]
- 34.Cornell W.D., Cieplak P., Kollman P.A. A second generation force field for the simulation of proteins, nucleic acids, and organic molecules. J. Am. Chem. Soc. 1995;117:5179–5197. [Google Scholar]
- 35.Wang J., Cieplak P., Kollman P.A. How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem. 2000;21:1049–1074. [Google Scholar]
- 36.Hornak V., Abel R., Simmerling C. Comparison of multiple Amber force fields and development of improved protein backbone parameters. Proteins. 2006;65:712–725. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pérez A., Marchán I., Orozco M. Refinement of the AMBER force field for nucleic acids: improving the description of α/γ conformers. Biophys. J. 2007;92:3817–3829. doi: 10.1529/biophysj.106.097782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Joung I.S., Cheatham T.E., 3rd Determination of alkali and halide monovalent ion parameters for use in explicitly solvated biomolecular simulations. J. Phys. Chem. B. 2008;112:9020–9041. doi: 10.1021/jp8001614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meagher K.L., Redman L.T., Carlson H.A. Development of polyphosphate parameters for use with the AMBER force field. J. Comput. Chem. 2003;24:1016–1025. doi: 10.1002/jcc.10262. [DOI] [PubMed] [Google Scholar]
- 40.Jorgensen W.L., Chandrasekhar J., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926–935. [Google Scholar]
- 41.Darden T.A., York D., Pedersen L. Particle mesh Ewald—an Nlog(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- 42.Essmann U., Perera L., Pedersen L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995;103:8577–8593. [Google Scholar]
- 43.Crowley M.F., Darden T.A., Deerfield D.W., II Adventures in improving the scaling and accuracy of a parallel molecular dynamics program. J. Supercomput. 1997;11:255–278. [Google Scholar]
- 44.Berendsen H.J.C., Postma J.P.M., Haak J.R. Molecular dynamics with coupling to an external heath bath. J. Chem. Phys. 1984;81:3684–3690. [Google Scholar]
- 45.Ryckaert J.-P., Ciccotti G., Berendsen H.J. Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 1977;23:327–341. [Google Scholar]
- 46.Grossfield A., Zuckerman D.M. Quantifying uncertainty and sampling quality in biomolecular simulations. Annu Rep Comput Chem. 2009;5:23–48. doi: 10.1016/S1574-1400(09)00502-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Levy R.M., Srinivasan A.R., McCammon J.A. Quasi-harmonic method for studying very low frequency modes in proteins. Biopolymers. 1984;23:1099–1112. doi: 10.1002/bip.360230610. [DOI] [PubMed] [Google Scholar]
- 48.Amadei A., Linssen A.B., Berendsen H.J. Essential dynamics of proteins. Proteins. 1993;17:412–425. doi: 10.1002/prot.340170408. [DOI] [PubMed] [Google Scholar]
- 49.Ichiye T., Karplus M. Collective motions in proteins: a covariance analysis of atomic fluctuations in molecular dynamics and normal mode simulations. Proteins. 1991;11:205–217. doi: 10.1002/prot.340110305. [DOI] [PubMed] [Google Scholar]
- 50.Cheatham III, T. E. 2008. Ptraj, AmberTools 1.2.
- 51.Biswas I., Vijayvargia R. Heteroduplex DNA and ATP induced conformational changes of a MutS mismatch repair protein from Thermus aquaticus. Biochem. J. 2000;347:881–886. [PMC free article] [PubMed] [Google Scholar]
- 52.Jeong C., Cho W.K., Lee J.B. MutS switches between two fundamentally distinct clamps during mismatch repair. Nat. Struct. Mol. Biol. 2011;18:379–385. doi: 10.1038/nsmb.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tsai C.J., del Sol A., Nussinov R. Allostery: absence of a change in shape does not imply that allostery is not at play. J. Mol. Biol. 2008;378:1–11. doi: 10.1016/j.jmb.2008.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gunasekaran K., Ma B.Y., Nussinov R. Is allostery an intrinsic property of all dynamic proteins? Proteins. 2004;57:433–443. doi: 10.1002/prot.20232. [DOI] [PubMed] [Google Scholar]
- 55.Cooper A., Dryden D.T. Allostery without conformational change. A plausible model. Eur. Biophys. J. 1984;11:103–109. doi: 10.1007/BF00276625. [DOI] [PubMed] [Google Scholar]
- 56.Karplus M., Ichiye T. Comment on a “fluctuation and cross correlation analysis of protein motions observed in nanosecond molecular dynamics simulations”. J. Mol. Biol. 1996;263:120–122. doi: 10.1006/jmbi.1996.0562. [DOI] [PubMed] [Google Scholar]
- 57.Harte W.E., Jr., Swaminathan S., Beveridge D.L. Domain communication in the dynamical structure of human immunodeficiency virus 1 protease. Proc. Natl. Acad. Sci. USA. 1990;87:8864–8868. doi: 10.1073/pnas.87.22.8864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Swaminathan S., Harte W.E., Jr., Beveridge D.L. Investigation of domain structure in proteins via molecular dynamics simulation: application to HIV-1 protease dimer. J. Am. Chem. Soc. 1991;113:2717–2721. [Google Scholar]
- 59.Kormos B.L., Baranger A.M., Beveridge D.L. Do collective atomic fluctuations account for cooperative effects? Molecular dynamics studies of the U1A-RNA complex. J. Am. Chem. Soc. 2006;128:8992–8993. doi: 10.1021/ja0606071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kormos B.L., Baranger A.M., Beveridge D.L. A study of collective atomic fluctuations and cooperativity in the U1A-RNA complex based on molecular dynamics simulations. J. Struct. Biol. 2007;157:500–513. doi: 10.1016/j.jsb.2006.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Knaggs M.H., Salsbury F.R., Jr., Fetrow J.S. Insights into correlated motions and long-range interactions in CheY derived from molecular dynamics simulations. Biophys. J. 2007;92:2062–2079. doi: 10.1529/biophysj.106.081950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Abseher R., Nilges M. Are there non-trivial dynamic cross-correlations in proteins? J. Mol. Biol. 1998;279:911–920. doi: 10.1006/jmbi.1998.1807. [DOI] [PubMed] [Google Scholar]
- 63.Hünenberger P.H., Mark A.E., van Gunsteren W.F. Fluctuation and cross-correlation analysis of protein motions observed in nanosecond molecular dynamics simulations. J. Mol. Biol. 1995;252:492–503. doi: 10.1006/jmbi.1995.0514. [DOI] [PubMed] [Google Scholar]
- 64.Winkler I., Marx A.D., Friedhoff P. Chemical trapping of the dynamic MutS-MutL complex formed in DNA mismatch repair in Escherichia coli. J. Biol. Chem. 2011;286:17326–17337. doi: 10.1074/jbc.M110.187641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Smock R.G., Gierasch L.M. Sending signals dynamically. Science. 2009;324:198–203. doi: 10.1126/science.1169377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Popovych N., Sun S.J., Kalodimos C.G. Dynamically driven protein allostery. Nat. Struct. Mol. Biol. 2006;13:831–838. doi: 10.1038/nsmb1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Changeux J.P., Edelstein S.J. Allosteric mechanisms of signal transduction. Science. 2005;308:1424–1428. doi: 10.1126/science.1108595. [DOI] [PubMed] [Google Scholar]
- 68.Schirmer T., Evans P.R. Structural basis of the allosteric behaviour of phosphofructokinase. Nature. 1990;343:140–145. doi: 10.1038/343140a0. [DOI] [PubMed] [Google Scholar]
- 69.Salsbury F.R., Jr., Clodfelter J.E., Scarpinato K.D. The molecular mechanism of DNA damage recognition by MutS homologs and its consequences for cell death response. Nucleic Acids Res. 2006;34:2173–2185. doi: 10.1093/nar/gkl238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gorman J., Chowdhury A., Greene E.C. Dynamic basis for one-dimensional DNA scanning by the mismatch repair complex Msh2-Msh6. Mol. Cell. 2007;28:359–370. doi: 10.1016/j.molcel.2007.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.