

Abstract
Cross-linking of the high-affinity IgE receptor (FcɛRI) on mast cells with IgE and multivalent antigen triggers mitogen-activated protein (MAP) kinase activation and cytokine gene expression. We report here that MAP kinase kinase 4 (MKK4) gene disruption does not affect either MAP kinase activation or cytokine gene expression in response to cross-linking of FcɛRI in embryonic stem cell-derived mast cells. MKK7 is activated in response to cross-linking of FcɛRI, and this activation is inhibited by MAP/ERK kinase (MEK) kinase 2 (MEKK2) gene disruption. In addition, expression of kinase-inactive MKK7 in the murine mast cell line MC/9 inhibits c-Jun NH2-terminal kinase (JNK) activation in response to cross-linking of FcɛRI, whereas expression of kinase-inactive MKK4 does not affect JNK activation by this stimulus. However, FcɛRI-induced activation of the tumor necrosis factor-α (TNF-α) gene promoter is not affected by expression of kinase-inactive MKK7. We describe an alternative pathway by which MEKK2 activates MEK5 and big MAP kinase1/extracellular signal-regulated kinase 5 in addition to MKK7 and JNK, and interruption of this pathway inhibits TNF-α promoter activation. These findings suggest that JNK activation by antigen cross-linking is dependent on the MEKK2-MKK7 pathway, and cytokine production in mast cells is regulated in part by the signaling complex MEKK2-MEK5-ERK5.
Mast cells express the high-affinity IgE receptor FcɛRI on their surface, and cross-linking of FcɛRI with multivalent antigen triggers degranulation and release of granules that contain histamine, serotonin, neutral proteases, and proteoglycans. This degranulation response contributes to immediate allergic responses. In addition, mast cells produce cytokines such as tumor necrosis factor-α (TNF-α), IL-4, IL-13, and granulocyte/macrophage colony-stimulating factor (GM-CSF) several hours after activation, contributing to chronic inflammation (1). Mitogen-activated protein kinase (MAPK) cascades play essential roles in the transduction of extracellular signals to cytoplasmic and nuclear effectors. There are numerous MAPK pathways in eukaryotic cells that lead to the activation of extracellular signal-regulated kinases (ERK1, ERK2), c-Jun-N-terminal kinases (JNK1, 2, 3), p38, or big MAPK (BMK1/ERK5) (2). In response to different external stimuli, these MAPKs are activated via a module consisting of MAPKKK-MAPKK-MAPK [MAP/ERK kinase (MEK) kinase (MEKK)-MEK-MAPK].
The JNK family members are activated by a large number of stimuli, including UV light, γ-irradiation, osmotic stress, or specific cytokines. Activation of JNKs is thought to be important in TNF-α gene activation, which results in phosphorylation of transcription factors c-Jun and activating transcription factor 2 (3). A number of MAPKKKs can activate JNK in different cell types. MEKK2 appears to be essential in mast cells. Studies of MEKK2−/− embryonic stem (ES) cell-derived mast cells (ESMCs) revealed that both JNK activation and TNF-α gene expression were markedly deficient after FcɛRI aggregation or c-Kit stimulation compared with wild-type (WT) ESMCs (4). These studies indicated that MEKK2 activation was critical for cytokine production.
Two specific JNK-activating MAPKKs, MAP kinase kinase 4 (MKK4) (JNKK1) and MKK7 (JNKK2), are known to directly activate JNK (5, 6). In different cell types, MKK4 and MKK7 may be differentially activated by external stimuli such as TNF-α or cellular stresses, and distinct upstream activators have been implicated in the activation of these responses (6, 7). For example, MKK4 was found to be a preferred substrate for the MAPKKK MEKK1 (8), whereas MKK7 could be activated by several of the MAPKKKs, including MEKK1, MEKK2, and MEKK3 (7). A recent study showed that the dual leucine zipper MAPKKK, MLK3/DLK can activate MKK7 but not MKK4 (9).
Recently, a MAPK pathway has emerged that is distinct from the traditional ERK pathways, BMK (also called ERK5) (10, 11). Unique structural features such as a large C terminus and a loop-12 sequence distinguish it from other ERKs. BMK1/ERK5 is activated by growth factors such as epidermal growth factor, oxidative stress, and hyperosmolarity, leading to the activation of myocyte enhancer factor-2C (MEF2C), a transcription factor that induces c-Jun expression (12, 13). These studies also determined that MEK5 is the upstream kinase of BMK1/ERK5 and is required for epidermal growth factor-induced signaling but is independent of Ras/Raf-induced activation of ERK1 and -2. Subsequently, one upstream kinase of MEK5 has been shown to be MEKK3 (14). MEKK3 physically interacts with and regulates MEK5, and its activity is required for the activation of BMK1/ERK5 and MEF2C. Recently, MEKK2 has been identified as a binding partner with MEK5 by yeast two-hybrid screening, and MEKK2 was shown to activate BMK1/ERK5 to a greater extent than does MEKK3 (15).
We investigated the role of MKK4, MKK7, MEK5, BMK1/ERK5, and MEKK2 in mast cell signaling of JNK activation and cytokine gene expression. In mast cells, the MEKK2-MKK7-JNK1 and MEKK2-MEK5-ERK5 complexes were activated in response to cross-linking of FcɛRI. Gene transcription after FcɛRI cross-linking in mast cells was regulated by the activation of the MEKK2-MEK5-ERK5 pathway.
Materials and Methods
Cells and Reagents.
Growth of MKK4−/− and MEKK2−/− ES cells was described previously (4, 16), and they were not discernibly different from WT cells (4). IL-3 was obtained from medium conditioned by X63 AG8-653 myeloma cells transfected with a vector encoding the IL-3 gene (17). c-Kit ligand (KL) was obtained from medium conditioned by Chinese hamster ovary cells transfected with a KL expression vector (kindly provided by the Genetics Institute, Cambridge, MA). Ovalbumin (OVA) (grade V) and anti-ERK5 antibody were purchased from Sigma. Recombinant murine KL and IL-3 were purchased from R & D Systems. Recombinant murine GM-CSF was purchased from PharMingen. Recombinant protein A-Sepharose was purchased from Zymed. Recombinant JNK1, anti-ERK2 antibody (C-14), and MKK4 antibody were purchased from Santa Cruz Biotechnology. Bovine myelin basic protein was purchased from Upstate Biotechnology (Lake Placid, NY). Anti- hemagglutinin (HA) antibody (12CA5) was purchased from Roche Molecular Biochemicals. Anti-ERK5 antibody was purchased from Sigma. Rabbit anti-MKK7 antibody was kindly provided by J. W. Schrader (University of British Columbia, Vancouver; ref. 5).
Plasmids and Transfection.
Kinase-inactive MKK7 (MKK7K63R) in a pCMV5 expression vector was kindly provided by H. Itoh (Tokyo Institute of Technology, Yokohama, Japan; ref. 8). The dominant negative MKK4 mutant (K116R) in LNCx expression vector was a kind gift from Lynn Heasley (University of Colorado School of Medicine, Denver) (18). The full-length promoter of the mouse TNF-α gene inserted upstream of a luciferase gene (pGL3TNF) was described (19). The dominant negative mutants of MEKK2 and MEKK3 were created by substitution of methionine for lysine in the ATP binding sites as described (20). Dominant negative MEK5 was created by replacing the two activating phosphorylation sites (Ser-311 and Thr-315) with alanine (15). Dominant negative BMK1/ERK5 (ERK5-NH2 terminus) was kindly provided by Silvio Gutkind (National Institutes of Health, Bethesda). Transient transfection of MC/9 was performed by electroporation at 320 V and 800 μF with Life Technologies (Rockville, MD) Cell-Porator.
In Vitro Differentiation of ES Cells into Mast Cells.
The conditions for mast cell differentiation from ES cells have been described (4).
Passive Sensitization and Stimulation of ESMCs and MC/9.
ESMCs or MC/9 were cultured with 500 ng/ml anti-OVA IgE (21) for 16 h. The cells were washed with medium three times and cultured with fresh medium for 2 h. OVA (10 μg/ml) dissolved in PBS was added to the passively sensitized cells, and PBS was used as a control.
Assay of JNK Activity.
Glutathione S-transferase–c-Jun fusion protein was prepared as described (22), and JNK activity was assayed as described (23).
Assay of p38 MAPK and ERK2 Activity.
p38 MAPK and ERK2 activities were measured as described previously, with the use of activating transcription factor 2 and myelin basic protein, respectively, as substrates (23).
Assay of MKK7 Activity.
Cells (3 × 106 cells) were lysed in 400 μl of lysis buffer as described for the JNK assay above. Cleared lysates were incubated with anti-MKK7 at 4°C for 2 h and then incubated with protein A-Sepharose beads for an additional 1 h (8). The mixtures were washed twice in lysis buffer and once in kinase buffer (15 mM Hepes, pH 7.5/12.5 mM β-glycerophosphate/12.5 mM p-nitrophenyl phosphate/12.5 mM MgCl2/1 mM DTT/100 μM sodium orthovanadate). Beads were suspended in 30 μl of kinase buffer containing 10 mM unlabeled ATP and 0.3 μg recombinant JNK1 and incubated at 30°C for 20 min. After the first reaction, glutathione S-transferase–c-Jun protein and 10 μCi of [γ-32P]ATP were added to the reaction mixtures and incubated at 30°C for 20 min. Reaction mixtures were added to Laemmli sample buffer, boiled, and resolved by SDS-PAGE.
Assay of BMK1/ERK5 Phosphorylation.
Cells (5–10 × 106) were lysed in 100 μl of RIPA buffer (25 mM Tris⋅HCl, pH 7.4/50 mM NaCl/0.5% Na deoxycholate/2% Nonidet P-40/0.2% SDS/1 μM phenylmethylsulfonyl fluoride/50 μg/ml aprotinin/50 μM leupeptin) at 4°C for 15 min. Samples were boiled and analyzed by 7% SDS-PAGE. The two different phosphorylation states were made visible by two distinct bands at 90 kDa, with the use of the anti-ERK5 antibody (Sigma).
Western Blot Analysis.
The cell lysates or immunoprecipitates were resolved by SDS-PAGE, and proteins were transferred to nitrocellulose membranes. Membranes were incubated overnight in blocking buffer containing 1% BSA at 4°C. The anti-MKK4 (1:200), anti-MKK7 (1:1,000), or anti-HA (1:1,000) antibody was added to the blocking buffer, and blots were incubated for an additional hour at room temperature. The blots were washed in TBST (25 mM Tris, pH 8.0/125 mM NaCl/0.025% Tween 20), and specific reactive proteins were detected by an enhanced chemiluminescence method, with the use of a sheep anti-rabbit or anti-mouse Ig antibody linked to horseradish peroxidase (Amersham Pharmacia).
RNA Isolation and RNase Protection Assay.
A S.N.A.P. Total RNA Isolation Kit (Invitrogen) was used to isolate total RNA. For the RNase protection assay, a RiboQuant MultiProbe RNase Protection Assay System (PharMingen) was used according to the manufacturer's directions.
Luciferase Assay.
Cell pellets were lysed according to the manufacturer's directions (Promega). The lysate was mixed with the Luciferase Assay Substrate containing beetle luciferin, and chemiluminescence was measured for 10 s as relative light units, with a luminometer (Monolight 2010; Analytical Luminescence Laboratory, San Diego). Relative light units were correlated with sample protein.
Results
MKK4 Gene Disruption Does Not Affect JNK Activation Through FcɛRI, c-Kit, or the IL-3 Receptor.
To determine the role that MKK4 plays in JNK activation, we studied the effects of MKK4 gene disruption on receptor-stimulated MAPK activation. Kinase activities of the endogenous MAPKs (JNK, p38, and ERK2) were examined in MKK4+/+ and MKK4−/− ESMCs after IgE/OVA stimulation in an in vitro kinase assay. To establish that the responses were not restricted to signaling via FcɛRI, KL stimulation and IL-3 stimulation were monitored in parallel. We did not observe any significant difference in the activities of JNK, p38, or ERK2 between MKK4+/+ and MKK4−/− ESMCs (Fig. 1). JNK activation by anisomycin was obviously decreased in MKK4−/− ESMCs (data not shown). The MKK4 dependency of anisomycin-induced JNK activation has previously been observed in ES cells (24, 25). Western blot analysis with anti-MKK4 antibody confirmed that MKK4 expression was abolished in MKK4−/− cells, and IgE/OVA stimulation did not affect MKK4 expression (data not shown). These data suggest that MKK4 is not required for JNK activation through FcɛRI, c-Kit, and the IL-3R in mast cells or that another JNK activator may compensate for MKK4 when its expression is abolished.
Figure 1.
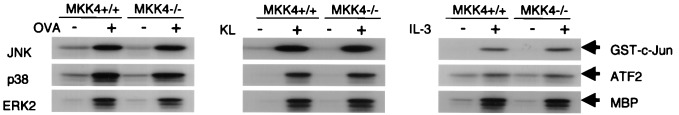
MKK4 gene disruption does not affect MAPK activation in response to cross-linking of FcɛRI or to KL and IL-3 in ESMCs. MKK4+/+ and MKK4−/− ESMC (3 × 106 cells) sensitized with anti-OVA IgE were stimulated by the addition of 10 μg/ml OVA. JNK activity was measured 15 min after the addition of OVA. Activities of p38 and ERK2 were measured 5 min after the addition of OVA. ESMCs were deprived of KL and IL-3 from culture medium for 3 h and stimulated with 100 ng/ml KL or 50 ng/ml IL-3. JNK activities were measured 15 min after the addition of KL or IL-3. p38 and ERK2 activities were measured 5 min after the addition of KL or IL-3. Representative autoradiographs from three independent experiments are shown.
MKK7 Is Activated in Response to Cross-Linking of FcɛRI or c-Kit and IL-3R Stimulation.
In addition to MKK4, MKK7 has also been demonstrated to serve as a potent activator of JNK in cell types other than mast cells (5, 26). To assess the role of MKK7 in JNK activation in MKK4−/− ESMC mast cells, in vitro kinase assays with anti-MKK7 immunoprecipitates were performed. Endogenous MKK7 was activated in both MKK4+/+ and MKK4−/− ESMCs after the addition of IgE/OVA, KL, and IL-3, and no obvious differences in MKK7-induced JNK activation were observed between MKK4+/+ and MKK4−/− ESMCs (Fig. 2). These data imply that MKK7 mediates JNK activation independently of MKK4 in response to these different receptors in mast cells.
Figure 2.

Activation of MKK7 in response to cross-linking of FcɛRI or to KL and IL-3 in MKK4+/+ and MKK4−/− ESMCs. Passively sensitized MKK4+/+ and MKK4−/− ESMC were stimulated with OVA for 10 min, and MKK7 activity was measured as described in Materials and Methods. Otherwise, MKK4+/+ and MKK4−/− ESMCs were stimulated with 100 ng/ml KL or 50 ng/ml IL-3 for 10 min and MKK7 activity was measured. Representative autoradiographs from three independent experiments are shown.
MEKK2 Gene Disruption Inhibits MKK7 Activation in Response to IgE/OVA, KL, or IL-3.
Targeted disruption of MEKK2 inhibits JNK activation in response to IgE/OVA and KL (4). We therefore examined endogenous MKK7 kinase activity in MEKK2−/− ESMCs. MKK7 activation in response to IgE/OVA, KL, and IL-3 was significantly inhibited in MEKK2−/− ESMCs compared with MEKK2+/+ ESMCs (Fig. 3). These data indicate that MEKK2 is an essential upstream regulator of MKK7 in mast cells stimulated by IgE/OVA, KL, or IL-3.
Figure 3.

The effect of MEKK2 gene disruption on MKK7 activation in response to cross-linking of FcɛRI to KL and IL-3 in ESMCs. Passively sensitized MEKK2+/+ and MEKK2−/− ESMCs were stimulated with OVA for 10 min, and MKK7 activity was measured as described in Materials and Methods. Otherwise, MEKK2+/+ and MEKK2−/− ESMCs were stimulated with 100 ng/ml KL or 50 ng/ml IL-3 for 10 min, and MKK7 activity was measured. MKK7 protein levels were determined by Western blot analysis. Representative autoradiographs from three independent experiments are shown.
Expression of Kinase-Inactive MKK7 Inhibits JNK Activation.
To directly address the importance of MKK7 for JNK activation, we examined the effect of kinase-inactive MKK7 (MKK7 with a point mutation in the catalytic site, MKK7K63R) on JNK kinase activity. MC/9 cells transfected with HA-tagged JNK1, together with kinase-inactive MKK7 or empty vector (pCMV5), were stimulated with IgE/OVA, KL, IL-3, or GM-CSF. Levels of MKK7 protein expression in each sample were confirmed to be similar in Western blots with the use of anti-Flag antibody. JNK1 activation in response to IgE/OVA was inhibited by kinase-inactive MKK7 expression (81% inhibition). KL-, IL-3-, and GM-CSF-induced JNK activation was also inhibited (90%, 83%, and 80% inhibition, respectively) (data not shown). As a control, the JNK1 response to osmotic shock (sorbitol) was not inhibited by the expression of kinase-inactive MKK7 (data not shown).
Consistent with our findings in MKK4−/− mast cells, expression of kinase-inactive MKK4 (MKK4 with a point mutation in the catalytic site, MKK4K116R) did not significantly affect the JNK response to any of these stimuli in transfected MC/9 cells (Fig. 4B). These data suggest that JNK activation by FcɛRI cross-linking or by KL, IL-3, or GM-CSF is preferentially mediated by MKK7.
Figure 4.

Expression of kinase-inactive MKK7 inhibits JNK activation in response to cross-linking of FcɛRI to KL, IL-3, and GM-CSF in MC/9 cells. MC/9 cells (5 × 106 cells) were transfected with 7.5 μg kinase-inactive MKK7 (MKK7K63R) together with 2.5 μg HA-epitope-tagged JNK1 (HA-JNK1). (A) Transfected cells were passively sensitized with anti-OVA IgE for 16 h, washed with culture medium three times, and treated with 10 μg/ml OVA or PBS as a control for 15 min. HA-JNK1 protein levels in the transfected cells were determined by Western blot analysis. (B) MC/9 cells (5 × 106 cells) were transfected with 7.5 μg kinase-inactive MKK4 (MKK4K116R) together with 2.5 μg HA-epitope-tagged JNK1 (HA-JNK1). The transfected cells were treated as described in A. HA-JNK1 activities were measured as described in Materials and Methods. Representative autoradiographs from three independent experiments are shown.
MKK4 Gene Disruption Does Not Affect Cytokine Gene Expression.
To examine the role of MKK4 in cytokine production, RNase protection assays were performed in MKK4+/+ and MKK4−/− ESMCs after IgE/OVA stimulation (Fig. 5). The RNA probes for IL-4, IL-5, TNF-α, IL-13, IL-15, IL-9, IL-2, IL-6, and IFN-γ were used in the assays. An increase in the expression of IL-4, TNF-α, IL-13, IL-9, and IL-6 was observed in response to IgE/OVA stimulation. The amount of mRNA for IL-4, IL-6, IL-9, and IL-13 was maximal at 30–60 min after stimulation. TNF-α mRNA was maximal at 30 min and had significantly decreased by 60 min. No apparent differences in cytokine mRNA levels were observed between MKK4+/+ and MKK4−/− ESMCs. These data suggest that MKK4 is not required for induction of these cytokine genes in mast cells.
Figure 5.

MKK4 gene disruption does not affect cytokine gene expression in ESMCs. MKK4+/+ or MKK4−/− ESMCs were passively sensitized with anti-OVA IgE and stimulated with OVA for 0, 30, 60, 120, and 180 min. Cells were harvested, and total RNA was isolated. Fifteen micrograms of total RNA was used in the RNase protection assay. The RNA probes for IL-4, IL-5, TNF-α, IL-13, IL-15, IL-9, IL-2, IL-6, and IFN-γ were used, in addition to L32 and glyceraldehyde-3-phosphate dehydrogenase as controls. Representative autoradiographs from two independent experiments are shown.
Expression of Kinase-Inactive MKK7 Does Not Affect TNF-α Gene Promoter Induction.
To define the role of MKK7 and JNK activation on cytokine gene promoter activity, we cotransfected a construct encoding the TNF-α promoter linked to a luciferase reporter gene (pGL3TNF) into MC/9 cells together with kinase-inactive MKK7 or empty vector and stimulated with IgE/OVA. No significant inhibition of TNF-α gene promoter activity was observed with kinase-inactive MKK7 expression in the same experiments where JNK activation was inhibited by at least 80–90% (Fig. 6). These data suggest that although FcɛRI-stimulated JNK activity is dependent on MKK7, JNK activation may not be essential for TNF-α gene expression. Alternatively, low levels of residual JNK activity may have been sufficient to activate the TNF-α gene promoter. Expression of kinase-inactive MKK4 also did not significantly affect the TNF-α promoter activity of the TNF-α gene (Fig. 6). This result is consistent with the data given in Fig. 5.
Figure 6.

Expression of kinase-inactive MKK7 does not affect promoter activity of TNF-α. MC/9 cells (5 × 106 cells) were transfected with 2.5 μg pGL3TNF together with 7.5 μg MKK7K63R or 7.5 μg MKK4K116R or an equivalent amount of empty vector (pCMV5, pLNCx). The transfected cells were passively sensitized with anti-OVA IgE for 16 h and washed three times and incubated for an additional 8 h with 10 μg/ml OVA (filled bars) or PBS (empty bars). Luciferase activities are shown as relative light units (luciferase units) (mean ± SD; n = 6–8; NS, not significant, paired two-tailed Student's t test).
Expression of Kinase-Inactive MEK5 Inhibits the Phosphorylation of BMK1/ERK5 and Inhibits Induction of the TNF-α Gene Promoter.
As kinase-inactive mutants of MKK4 and MKK7 did not appear to significantly inhibit the activation of the TNF-α promoter in mast cells, we sought possible candidate proteins in other signaling pathways that are regulated by MEKK2 and lead to changes in transcription factor activation. Previous reports have identified MEK5 as a substrate of MEKK2 and BMK1/ERK5 as a downstream target of MEK5 (15). Figure 7A confirms that in mast cells the cross-linking of FcɛRI or signaling through Kit (data not shown) results in the activation of endogenous BMK1/ERK5, as indicated by the mobility shift to the slower migrating phosphorylated BMK1/ERK5. We next cotransfected the construct encoding the TNF-α/luciferase reporter gene (pGL3TNF) into MC/9 cells together with either kinase-inactive MEK5 or BMK1/ERK5, with and without the WT MEKK2 gene. In these sensitized (IgE) cells challenged with OVA, there was a more than 100-fold reduction in TNF-α gene promoter activity when kinase-inactive MEK5 was transfected in equal amounts with MEKK2 (Fig. 7B). Protein expression of MEK5 was the same in each sample (as detected in Western blots with anti-Flag antibody) but slightly less than levels of MEKK2 protein expression (detected with anti-HA antibody) (data not shown). Similarly, in separate experiments, there was more than a 3-fold reduction in TNF-α promoter activity when dominant negative BMK1/ERK5 was cotransfected with WT MEKK2 (Fig. 7B). The inhibition of TNF-α promoter induction by dominant negative MEK5 and BMK1/ERK5 indicates the importance of this pathway in cytokine production in MC/9 cells. To confirm that MEK5 functioned as the upstream kinase regulating the BMK1/ERK5 pathway in these transfected MC/9 cells, we assessed the effect of this MEK5 mutant on the phosphorylation of BMK1/ERK5. We observed that the kinase-inactive MEK5 inhibits the mobility shift of BMK1/ERK5 to the same extent as the kinase-inactive mutants of MEKK2 and MEKK3 (Fig. 7 C and D). These data confirm the pivotal role of MEKK2 in mast cell TNF-α promoter activity and identify the MEKK2-MEK5-ERK5 signaling pathway in this cellular process.
Figure 7.

Expression of kinase-inactive MEK5 inhibits phosphorylation of BMK1 and the promoter activity of TNF-α. (A) MC/9 cells (10 × 106 cells) were stimulated with 10 μg/ml OVA (IgE sensitized) for a time course of 0, 10, 20, 30, or 60 min. BMK1 phosphorylation was analyzed by the mobility shift of proteins on a Western blot and visualization by chemiluminescence. (B) MC/9 cells (10 × 106 cells) were transfected with 5 μg pGL3TNF together with 7.5 μg MEKK2 or 7.5 μg of MEKK2 K/R; or 7.5 μg MEK5 S311A, S315A, or dominant negative ERK 5; or an equivalent amount of empty vector (pCMV5). The transfected cells were passively sensitized with anti-OVA IgE for 16 h and washed three times and incubated for an additional 6 h with 10 μg/ml OVA or PBS. Luciferase activities are shown as relative light units (luciferase units) (representative of three separate experiments). (C and D) MC/9 cells were transfected with 15 μg of WT MEKK2 and WT MEKK3 or 15 μg kinase-inactive MEKK2 (K-M) and kinase-inactive MEKK3 (K-M) or DN MEK5 (D), along with 5 μg of BMK1α plasmid or an equivalent amount of empty vector (pCMV5). Phosphorylation of BMK1 was analyzed by a shift in its electrophoretic mobility detected by Western blotting.
Discussion
MAPKs play an essential role in the regulation of several critical cellular processes. Diverse extracellular stimuli initiate signals via cell surface receptors that activate intracellular protein kinase cascades. These cascades involve the activation of MAPK family members that regulate an array of biological functions by phosphorylating specific downstream target molecules. Transcription factors make up one group of MAPK substrates, which increase transcriptional activity after phosphorylation. Hence, nuclear transcription factors are often the final target of such kinase signal transduction cascades, converting receptor, membrane, and cytoplasmic events into changes in gene expression. Given the number of kinases and multiple MAPK cascades, specificity is imparted by the activation of a restricted MAPK in response to a given external stimulus and the downstream substrate specificity of a given MAPK. Different cell types may also contribute to this specificity.
Cross-linking of FcɛR1, or addition of the cytokines KL or IL-3, activates MAPK pathways in MC/9 mast cells. We have previously shown that ERK, p38, and JNK MAPK pathways are activated in MC/9 cells in response to these mast cell stimuli (27, 28). We have also shown that MEKK2 is required for OVA/IgE, KL, and IL3 activation of JNK. Studies in cells that lack MEKK2 by targeted gene disruption have demonstrated that MEKK2 is also required for OVA/IgE and KL-stimulated cytokine production in mast cells (4). Furthermore, MEKK2 appears to be preferentially coupled to MKK7 relative to MKK4 in the activation of JNK after receptor stimulation in COS cells (29).
Our studies show that MEKK2 regulates the MEK5/BMK1 pathway. We also show that OVA/IgE and KL stimulate BMK1/ERK5 in MC/9 mast cells. Thus, in addition to JNK, ERK, and p38, receptor activation of mast cells activates the MEK5/BMK1 pathway. We have previously shown that p38 and ERK are not critical for the activation of the TNF-α promoter in response to IgE/OVA and KL (28). Using inhibitory mutants of MKK7, we were able to inhibit JNK activation by 80–90%. This level of inhibition had little or no effect on TNF-α promoter regulation, although we cannot eliminate the possibility that the residual JNK activity observed with expression of the inhibitory MKK7 mutants contributes to the regulation of TNF-α activation. The findings argue that the JNK pathway is not a dominant pathway whose significant inhibition suppresses TNF-α promoter activation in response to IgE/OVA in mast cells. In contrast and rather strikingly, we have shown that inhibition of the BMK1/ERK5 pathway strongly inhibits the activation of the TNF-α promoter.
With the use of stimulated MC/9 cells and ES-derived mast cells, a role for the MEKK2-MEK5-ERK5 pathway was shown in this study by several experimental findings. Cross-linking of FcɛR1 or the addition of KL resulted in the phosphorylation of endogenous BMK1/ERK5. This effect was demonstrated by a gel shift assay that has been correlated with phosphorylation and activation of BMK1/ERK5 (13). Furthermore, we have shown that MEKK2 activates MEK5, leading to BMK1/ERK5 activation in addition to MEKK2 activation of MKK7 and JNK. Kinase-inactive MEK5 was very effective in inhibiting FcɛR1 and MEKK2 activation of the TNF-α promoter. In contrast, kinase-inactive mutants of MKK7 or MKK4 were not effective in blocking TNF-α promoter activation in response to FcɛR1 or KL in MC/9 mast cells. MKK7 has been shown to be involved in FcɛRI-induced JNK1 activation, which is regulated in part by Btk (30). MKK4 has been excluded from playing an essential role in several in vitro and in vivo aspects of Ig-dependent mast cell function (31). Similar findings have been observed in activated T cells; although MKK7 was indispensable for JNK activation, the MKK7-JNK pathway was not required for T cell cytokine (IL-2) production (32). These findings indicate that MEKK2 coordinately regulates two MAPK pathways, JNK and ERK5, in mast cells. Thus, MEKK2 activation integrates the activation of transcriptional events controlled by JNK and BMK1/ERK5. The findings demonstrate the critical role of MEKK2 in controlling several mast cell signaling pathways and cytokine production.
The role that BMK1/ERK5 plays in mast cell cytokine production is not yet understood. To date the best characterized protein substrate for BMK1/ERK5 is the MEF2C transcription factor (12, 33). MEF2C is phosphorylated by BMK1/ERK5 and results in its activation. The p38 MAPK also phosphorylates and activates MEF2C (34). MEF2C is known to regulate the expression of c-Jun, apparently in a JNK-independent manner (34, 35). It is possible that BMK1/ERK5 and JNK are integrated in the control of c-Jun expression and the activation and induction of TNF-α promoter activity. The inability to inhibit TNF-α promoter activity with kinase-inactive MKK7 (or MKK4) contradicts this hypothesis, but because JNK activation was not completely inhibited this issue remains open. There is also no apparent MEF2C regulatory site in the TNF-α promoter, indicating that the regulation of TNF-α promoter activity is probably the result of MEF2C interactions with other transcription factors. The MEK5/ERK5 proteins are also expressed at modest levels in most cell types. This may be, in part, why the kinase-inactive MEK5 protein is such a potent inhibitor of BMK1/ERK5 activation and signaling.
The role of JNK in the regulation of cytokine production is complex. The findings with knockouts of JNK1 and JNK2 suggest that they are negative regulators of cytokine production in T cells (32, 36). Our data argue that JNK activation may not be required for TNF-α promoter activation. However, our data also do not suggest that JNKs are negative regulators of the TNF-α promoter in mast cells because their inhibition does not measurably increase TNF-α promoter activity. Rather, our findings indicate that activation of the TNF-α promoter is significantly suppressed by expression of an inhibitory kinase-inactive MEK5 or BMK1/ERK5. Cumulatively, our studies show that MEKK2 is critical for mast cell receptor signaling controlling cytokine production and that MEKK2 regulates two MAPK pathways, JNK and BMK1/ERK5, in mast cells. The integrated response to MEKK2 activation is probably critical for the proper control of the transcriptional events required for activation of the TNF-α promoter. Importantly, our results define a role for the MEKK2-MEK5-ERK5 pathway in mast cell receptor signaling and in the control of the TNF-α promoter.
Acknowledgments
We thank Hideki Kawasome for the luciferase gene construct for the TNF-α promoter; Anthony Joetham for purification of anti-OVA IgE; and Drs. John W. Schrader for anti-MKK7 antibody, Hiroshi Itoh for kinase-inactive MKK7 plasmid, Lynn Heasley for kinase-inactive MKK4 plasmid, Richard Flavell for reporter plasmids, and Sylvio Gutkind for dominant negative BMK1/ERK5. This work was supported by National Institutes of Health Grants AI-42246 (to E.W.G.) and DK-37871 (to G.L.J.) and the Cancer League of Colorado (J.C.P.).
Abbreviations
- MAP
mitogen-activated protein
- MAPK
MAP kinase
- BMK
big MAPK
- ERK
extracellular signal-regulated kinase
- MEK
MAP/ERK kinase
- ES cell
embryonic stem cell
- ESMC
ES cell-derived mast cell
- GM-CSF
granulocyte/macrophage colony-stimulating factor
- HA
hemagglutinin
- JNK
c-Jun NH2-terminal kinase
- KL
c-Kit ligand
- MEF
myocyte enhancer factor
- MEKK
MEK kinase
- MKK
MAP kinase kinase
- OVA
ovalbumin
- TNF
tumor necrosis factor
- WT
wild type
References
- 1.Metcalfe D D, Baram B, Mekori Y A. Phys Rev. 1997;77:1033–1079. doi: 10.1152/physrev.1997.77.4.1033. [DOI] [PubMed] [Google Scholar]
- 2.Robinson M J, Cobb M H. Curr Opin Cell Biol. 1997;9:180–186. doi: 10.1016/s0955-0674(97)80061-0. [DOI] [PubMed] [Google Scholar]
- 3.Tsai E Y, Jain J, Pesavento P A, Rao A, Goldfeld A E. Mol Cell Biol. 1996;16:459–467. doi: 10.1128/mcb.16.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Garrington T P, Ishizuka T, Papst P J, Chayama K, Webb S, Yujiri T, Sun W, Sather S, Russell D M, Gibson S B, et al. EMBO J. 2000;19:5387–5395. doi: 10.1093/emboj/19.20.5387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Foltz I N, Gerl R E, Wieler J S, Luckach M, Salmon R A, Schrader J W. J Biol Chem. 1998;273:9344–9351. doi: 10.1074/jbc.273.15.9344. [DOI] [PubMed] [Google Scholar]
- 6.Hirai S, Noda K, Moriguchi T, Nishida E, Yamashita A, Deyama T, Fukuyama K, Ohno S. J Biol Chem. 1998;273:7406–7412. doi: 10.1074/jbc.273.13.7406. [DOI] [PubMed] [Google Scholar]
- 7.Moriguchi T, Toyoshima F, Masuyama N, Hanafusa H, Gotoh Y, Nishida E. EMBO J. 1997;16:7045–7053. doi: 10.1093/emboj/16.23.7045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yamauchi J, Kaziro Y, Itoh H. J Biol Chem. 1999;274:1957–1965. doi: 10.1074/jbc.274.4.1957. [DOI] [PubMed] [Google Scholar]
- 9.Merritt S E, Mata M, Nihalani D, Zhu C, Hu X, Holzman LB. J Biol Chem. 1999;274:10195–10202. doi: 10.1074/jbc.274.15.10195. [DOI] [PubMed] [Google Scholar]
- 10.Lee J-D, Ulevitch R J, Han J. Biochem Biophys Res Commun. 1995;213:715–724. doi: 10.1006/bbrc.1995.2189. [DOI] [PubMed] [Google Scholar]
- 11.Zhou G, Bao Z Q, Dixon J E. J Biol Chem. 1995;270:12665–12669. doi: 10.1074/jbc.270.21.12665. [DOI] [PubMed] [Google Scholar]
- 12.Kato Y, Kravchenko V V, Tapping R I, Han J, Ulevitch R J, Lee J. EMBO J. 1997;16:7054–7066. doi: 10.1093/emboj/16.23.7054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kato Y, Tapping R I, Huang S, Watson M H, Ulevitch R J, Lee J. Nature (London) 1998;395:713–716. doi: 10.1038/27234. [DOI] [PubMed] [Google Scholar]
- 14.Chao T-H, Hayashi M, Tapping R I, Kato Y, Lee J-D. J Biol Chem. 1999;274:36035–36038. doi: 10.1074/jbc.274.51.36035. [DOI] [PubMed] [Google Scholar]
- 15.Sun W, Kesavan K, Schaefer B, Garrington T P, Ware M, Lassignal Johnson N, Gelfand E W, Johnson G L. J Biol Chem. 2001;276:5093–5100. doi: 10.1074/jbc.M003719200. [DOI] [PubMed] [Google Scholar]
- 16.Ganiatsas S, Kwee L, Fujiwara Y, Perkins A, Ikeda T, Labow M A, Zon L I. Proc Natl Acad Sci USA. 1998;95:6881–6886. doi: 10.1073/pnas.95.12.6881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karasuyama H, Melchers F. Eur J Immunol. 1988;18:97–104. doi: 10.1002/eji.1830180115. [DOI] [PubMed] [Google Scholar]
- 18.Chan E D, Winston B W, Uh S T, Wynes M W, Rose D M, Riches D W. J Immunol. 1999;162:415–422. [PubMed] [Google Scholar]
- 19.Ishizuka T, Kawasome H, Terada N, Takeda K, Gerwins P, Keller G M, Johnson G L, Gelfand E W. J Immunol. 1998;161:3624–3630. [PubMed] [Google Scholar]
- 20.Fanger G R, Johnson N L, Johnson G L. EMBO J. 1997;16:4961–4972. doi: 10.1093/emboj/16.16.4961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Oshiba A, Hamelmann E, Takeda K, Bradley K L, Loader J E, Larsen G L, Gelfand E W. J Clin Invest. 1996;97:1398–1408. doi: 10.1172/JCI118560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Minden A, Lin A, McMahon M, Lange-Carter C, Dérijard B, Davis R J, Johnson G L, Karin M. Science. 1994;266:1719–1723. doi: 10.1126/science.7992057. [DOI] [PubMed] [Google Scholar]
- 23.Ishizuka T, Terada N, Gerwins P, Hamelmann E, Oshiba A, Fanger G R, Johnson G L, Gelfand E W. Proc Natl Acad Sci USA. 1997;94:6358–6363. doi: 10.1073/pnas.94.12.6358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nishina H, Fischer K D, Radvanyi L, Shahinian A, Hakem R, Rubie E A, Bernstein A, Mak T W, Woodgett J R, Penninger J M. Nature (London) 1997;385:350–353. doi: 10.1038/385350a0. [DOI] [PubMed] [Google Scholar]
- 25.Yang D, Tournier C, Wysk M, Lu H T, Xu J, Davis R J, Flavell R A. Proc Natl Acad Sci USA. 1997;94:3004–3009. doi: 10.1073/pnas.94.7.3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Deacon K, Blank J L. J Biol Chem. 1997;272:14489–14496. doi: 10.1074/jbc.272.22.14489. [DOI] [PubMed] [Google Scholar]
- 27.Ishizuka T, Oshiba A, Sakata N, Terada N, Johnson G L, Gelfand E W. J Biol Chem. 1996;271:12762–12766. doi: 10.1074/jbc.271.22.12762. [DOI] [PubMed] [Google Scholar]
- 28.Ishizuka T, Chayama K, Takeda K, Hamelmann E, Terada N, Keller G M, Johnson G L, Gelfand E W. J Immunol. 1999;162:2087–2094. [PubMed] [Google Scholar]
- 29.Cheng J, Yang J, Xia Y, Karin M, Su B. Mol Cell Biol. 2000;20:2334–2342. doi: 10.1128/mcb.20.7.2334-2342.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kawakami Y, Hartman S E, Holland P M, Copper J A, Kawakami T. J Immunol. 1998;161:1795–1802. [PubMed] [Google Scholar]
- 31.Tsai M, Wedemeyer J, Ganiatsas S, Tam S-Y, Zon L I, Galli S J. Proc Natl Acad Sci USA. 2000;97:9186–9190. doi: 10.1073/pnas.160254997. . (First Published July 25, 2000; 10.1073/pnas.160254997) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dong C, Yang D D, Tournier C, Whitmarsh A J, Xu J, Davis R J, Flavell R A. Nature (London) 2000;405:91–94. doi: 10.1038/35011091. [DOI] [PubMed] [Google Scholar]
- 33.Martin J F, Schwarz J J, Olson E N. Proc Natl Acad Sci USA. 1993;90:5282–5286. doi: 10.1073/pnas.90.11.5282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marinissen M J, Chiariello M, Pallante M, Gutkind J S. Mol Cell Biol. 1999;19:4289–4301. doi: 10.1128/mcb.19.6.4289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chiariello M, Marinissen M J, Gutkind J S. Mol Cell Biol. 2000;20:1747–1758. doi: 10.1128/mcb.20.5.1747-1758.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dong C, Yang D D, Wysk M, Whitmarsh A J, Davis R J, Flavell R A. Science. 1998;282:2092–2095. doi: 10.1126/science.282.5396.2092. [DOI] [PubMed] [Google Scholar]