

Abstract
The secretory pathway maintains multiple quality control checkpoints. Initially, endoplasmic reticulum-associated degradation pathways monitor protein folding to retain and eliminate aberrant products. Despite its broad client range, some molecules escape detection and traffic to Golgi membranes. There, a poorly understood mechanism termed Golgi quality control routes aberrant proteins for lysosomal/vacuolar degradation. To better understand Golgi quality control, we examined the processing of the obligate substrate Wsc1p. Misfolded Wsc1p does not use routes of typical vacuolar membrane proteins. Instead, it partitions into intralumenal vesicles of the multivesicular body (MVB) pathway, mediated by the E3 ubiquitin ligase Rsp5p. Its subsequent transport to the vacuolar lumen is essential for complete molecule breakdown. Surprisingly, the transport mode plays a second crucial function in neutralizing potential substrate toxicity. Eliminating the MVB sorting signal diverted molecules to the vacuolar limiting membrane, resulting in the generation of toxic by-products. These data demonstrate a new role of the MVB pathway in protein quality control.
Keywords: Lysosomes, Membrane Trafficking, Protein Degradation, Protein Folding, Protein Turnover, Protein Misfolding, Protein Quality Control
Introduction
Protein quality control mechanisms ensure the fidelity of the proteome by partitioning polypeptides based on conformational states. Correctly folded proteins can proceed to and remain at their sites of function, whereas aberrant molecules are slated for elimination. In the early secretory pathway, endoplasmic reticulum-associated degradation (ERAD)2 and autophagic pathways specialize in seeking misfolded polypeptides and mediate their degradation (for a review, see Ref. 1). Although these mechanisms seemed sufficiently comprehensive, reports of aberrant molecules trafficking out of the ER undetected suggest that the “molecular sieve” is somewhat porous (2–5). Even a minor flaw could be disastrous because misfolded proteins are often toxic if allowed to accumulate. This scenario is averted because analyses of ERAD-independent substrates revealed post-ER quality control mechanisms that efficiently capture the wayward molecules. Some molecules are caught in the Golgi apparatus, whereas others continue on to the plasma membrane. At both sites, specialized mechanisms sort and transport aberrant proteins to the lysosomes and vacuoles (lysosome-like organelles in fungi) for degradation.
The surveillance mechanism at the Golgi apparatus is termed Golgi quality control (GQC) (for reviews, see Refs. 6–8). Like ERAD, it recognizes a variety of aberrant soluble and membrane proteins (2, 3, 9–12). To be effective, its stringency must be greater than ERAD. This is apparently the case because GQC efficiently ensnares escaped ERAD substrates (2, 13) and recognizes a form of bovine pancreatic trypsin inhibitor with increased structural flexibility but not grossly misfolded (12). The plasma membrane mechanism, called peripheral quality control, is dedicated to the surveillance of membrane proteins (4, 5). This mechanism is distinct from the clearance of folded receptor proteins in the early steps due to its reliance on the Hsp70/Hsp90 molecular chaperone systems (4, 5). This is notable because the same chaperone classes are employed for the quality control of ER and cytosolic proteins (for a review, see Ref. 14). Substrate ubiquitination is used to signal endocytosis and sorting into the multivesicular body (MVB) endosomal pathway for degradation in lysosomes (for a review, see Ref. 15).
The transport route for GQC substrates from the Golgi is not known. In budding yeast, proteins traffic to the vacuole using one of two general routes. The first involves trafficking proteins in Golgi-derived vesicles to an endosomal prevacuolar compartment. This pathway was defined genetically in part by the class E vps (vacuolar protein sorting) mutants (16–18). Class E mutants cause formation of exaggerated prevacuolar compartments containing soluble and membrane vacuolar hydrolases, unrecycled Golgi proteins, and endocytosed proteins (16, 17, 19–21). Thus, the prevacuolar compartment represents a major trafficking hub for vacuolar/lysosomal transport that also includes the MVB pathway. This pathway is termed the “CPY pathway” because carboxypeptidase Y is a well studied client. A distinct vacuolar transport pathway was defined by the membrane protein alkaline phosphatase (20, 22, 23). The alkaline phosphatase pathway is not as well understood as the CPY pathway, but there are clear differences. Whereas the alkaline phosphatase pathway is known only to deliver membrane-bound cargo to vacuolar limiting membrane, the CPY pathway can do that and also places membrane proteins entirely into the lumen using the MVB pathway. For membrane-integrated GQC substrates, there is no obvious restriction for the route used. The MVB pathway provides a good mechanism because molecules are transported entirely into the vacuolar lumen on intralumenal vesicles. By contrast, the alternative alkaline phosphatase pathway results in membrane protein insertion into vacuolar limiting membrane. Although this might present a topological problem for degradation, the invagination and hydrolysis of vacuolar membranes and their contents through microautophagy would overcome the limitation if coupled to protein quality control (24, 25).
We previously reported that a plasma membrane protein, Wsc1p, is an endogenous, obligate substrate of post-ER quality control (26). Wsc1p is a single-spanning integral membrane protein that functions as a sensor for cell wall integrity in budding yeast (27, 28). Mutations disrupting the luminal folding of Wsc1p are not detected by ERAD, in part due to the inability of the ERAD chaperone Kar2p/BiP to recognize the variants (26). Instead, the Wsc1p cytoplasmic domain contains a dominant ER export signal that directs its transport regardless of the luminal folding state. For Wsc1p quality control, sorting occurs in the Golgi apparatus. Folded Wsc1p progresses to the plasma membrane, whereas misfolded molecules partition to the vacuole, dependent on the Vps10p cargo-sorting factor (26). These characteristics make Wsc1p an ideal model to uncover the mechanisms underlying GQC. Although both soluble and membrane GQC substrates can utilize Vps10p, the subsequent steps of transport are unknown (10, 29). In this study, Wsc1p variants were used to uncover the sorting and transport mechanism from the Golgi apparatus. We show that GQC and peripheral quality control pathways converge at the MVB endosomal sorting compartment. Importantly, we demonstrate that the use of the MVB pathway ensures whole molecule degradation and prevents toxic degradation products that would otherwise form through other vacuolar transport routes.
EXPERIMENTAL PROCEDURES
Strains, Antibodies, and Reagents
Yeast strains used in this study are listed in supplemental Table S1. All strains are in the W303 background except for strains used in supplemental Figs. S2B and S4, which are in the BY4741 background. Anti-HA mAb (HA.11) and anti-GFP monoclonal antibodies were purchased from Covance Research Products and Roche Applied Science, respectively. Anti-V-ATPase 60-kDa subunit and anti-3-phosphoglyceric phosphokinase (PGK) monoclonal antibodies were from Molecular Probes. Anti-Kar2p and anti-Sec61p antibodies were provided by P. Walter (University of California, San Francisco).
Plasmids Used in This Study
Plasmids used in this study were constructed using standard protocols (30). Primer sequences are listed in supplemental Table S2. All plasmid inserts were confirmed by nucleotide sequencing. pES67, expressing the HA epitope-tagged CPY* driven by the GAL1 promoter, was described previously (2). pSW104 and pSW148 (encoding Wsc1* driven by the PRC1 and GAS1 promoters, respectively) were described previously (26). Wsc1* contains a point mutation (L63R) in the luminal/extracellular domain of Wsc1p. All substrate constructs were engineered with an HA epitope tag at their C termini except for pSW177, which is fused to GFP.
pSW177
A 1.7-kb fragment containing the PRC1 promoter and Wsc1* was amplified from pSW104 using primers SWN99/SWN5 and digested with NotI and ClaI. Next, a 0.7-kb fragment (GFPS65T,Q80R) was amplified from pDN330 (Ng plasmid collection (31)) with primers SWN38/SWN39 and digested with ClaI and XbaI. The two fragments were ligated into pSW148 digested with NotI and XbaI.
pSW252
pSW252 expresses Wsc1*-6R using the PRC1 promoter. Wsc1*-6R contains six point mutations (K293R, K301R, K308R, K315R, K338R, and K365R) in the cytoplasmic domain made by site-directed mutagenesis as described previously (32). Primers SWN103, SWN104, SWN105, SWN106, SWN107, and SWN108 were used to direct mutagenesis.
pSW257
pSW257 encodes Wsc1*-6R regulated by the GAS1 promoter. A 600-bp fragment containing the GAS1 promoter was amplified from genomic DNA using SWN87/SWN88 primers and digested with NotI and BamHI. Next, a 1.7-kb fragment digested with BamHI/SalI was purified from pSW252. It contains the Wsc1*-6R coding sequence and the ACT1 terminator. The two fragments were ligated into pRS315 digested with NotI and SalI.
pSW182 and pSW263
pSW182 and pSW263 encode Wsc1* and Wsc1*-6R regulated by the GAL1 promoter. A 450-bp fragment containing the GAL1 promoter was amplified from pYES2.1 (Invitrogen) using SWN100/SWN101 primers and digested with NotI and BamHI. A second 1.7-kb fragment containing the Wsc1*-HA or Wsc1*-6R-HA gene followed by the ACT1 terminator sequence was released from pSW148 or pSW252 digested with BamHI and SalI. The fragments were ligated to pRS315 digested with NotI and SalI.
Indirect Immunofluorescence
Indirect immunofluorescence was performed as described previously (26) with minor modifications. Primary antibody working concentrations were as follows: HA.11 mAb (1:200) or anti-V-ATPase (1:100) in TBS containing 1% BSA. Alexa Fluor 488 goat anti-mouse antibody was used at 1:500 (Molecular Probes) in TBS containing 1% BSA. Samples were visualized using an Axio Imager M1 microscope with a ×100, 1.4 numerical aperture oil DIC Plan-Apochromat objective (Carl Zeiss MicroImaging, Inc.). HA/V-ATPase and DAPI fluorescence was detected with a 488- and 405-nm laser line, respectively. Images were archived using ImageJ (National Institutes of Health) and Adobe Photoshop CS3 (Adobe Systems). In some cases, linear adjustments were applied to enhance the brightness of images using levels in the image adjustment function of Photoshop. z-Stack images were taken with 0.38-μm intervals, and only a 488-nm argon laser was used during acquisition of z-stack images to minimize signal bleaching. A montage of z-stack images was created using ImageJ (National Institutes of Health) with an increment of 1.
Live Cell Imaging
Cells expressing Wsc1* fused to GFP were harvested at an A600 of 0.4–0.6 and resuspended in the same selective medium for visualization. Vacuolar membrane staining was performed, including FM4-64 (Molecular Probes) in YPD medium (2% peptone, 1% yeast extract, 2% dextrose) at 30 °C for 20 min, and chased for 45 min (supplemental Fig. S3) or 90 min (Fig. 7) at 30 °C as described (33). Cells were visualized using an Axio Imager M1 microscope with a ×100, 1.4 numerical aperture oil DIC Plan-Apochromat objective (Carl Zeiss MicroImaging, Inc.). GFP and FM4–64 fluorescence was captured with a 488- and 543-nm laser line, respectively. Images were processed using Adobe Photoshop CS3 (Adobe Systems). In some instances, image brightness was enhanced using levels and curves in the image adjustment function of Photoshop.
FIGURE 7.

Wsc1*-6R degradation by-products disrupt vacuolar membrane morphology. Wild type cells with an empty vector (pRS315), (PGAS1)Wsc1*, or (PGAS1)Wsc1*-6R and Δpep4 cells expressing (PGAS1)Wsc1* or (PGAS1)Wsc1*-6R were grown to log phase and stained with FM4-64 at 30 °C. Cell imaging was performed using confocal and DIC microscopy. Scale bar, 5 μm.
LD540 Staining of Lipid Droplets
One A600 unit of log phase cells was stained by incubation in 0.5 μg/ml LD540 for 5 min at 30 °C (provided by Dr Marcus R. Wenk, National University of Singapore) (34). After washing, LD540 fluorescence was visualized using an Axio Imager M1 microscope with a ×100, 1.4 numerical aperture oil DIC Plan-Apochromat objective (Carl Zeiss MicroImaging, Inc.) and a 488-nm laser line.
Transmission Electron Microscopy of Yeast
Sample preparation for electron microscopy analysis was performed as described with minor modifications (35). One A600 unit of log phase cells grown at 30 °C was collected and fixed with glutaraldehyde overnight at 4 °C. Next, cells were treated with 2% potassium permanganate for 1 h at room temperature. After dehydration in ethanol, cells were infiltrated with Spurr's resin and incubated for 24 h at 60 °C to allow polymerization. Thin sections were prepared using an Ultracut UCT (Leica) microtome and stained with lead citrate. Micrographs were taken using a transmission electron microscope (Jeol JEM-1230) and archived using Adobe Photoshop CS3 (Adobe Systems).
Preparation of Yeast Extract and Western Blotting
Two A600 units of log phase cells were harvested by centrifugation and resuspended in 1.0 ml of 10% trichloroacetic acid (TCA; Sigma-Aldrich) followed by the addition of 0.5 cm3 of 0.45-mm zirconium beads. Cells were disrupted by two 30 s cycles in a mini-bead beater (BioSpec Products Inc.). The lysate was transferred to a new tube and combined with a 10% TCA bead wash. The precipitate was pelleted by centrifugation and vortexed in 80 μl of TCA resuspension buffer (100 mm Tris, pH 11.0, 3% SDS, 1 mm PMSF). The sample was heated to 100 °C for 10 min and given a 16,000 × g spin to separate insolubles. A portion of the extract was separated by SDS-PAGE using a 15% gel and transferred to nitrocellulose. The blots were probed with anti-HA antibody, stripped, and reprobed with anti-PGK antibody. Proteins were visualized by enhanced chemiluminescence and exposure to x-ray film (Pierce). For quantitative immunoblotting experiments, proteins were first separated on a 4–15% gradient gel and transferred to PVDF membranes. The blots were subsequently incubated with anti-HA and anti-Sec61p antibodies followed by anti-mouse IRDye 680 and anti-rabbit IRDye 800 secondary antibodies (LI-COR Biosciences). Direst infrared fluorescence was detected and quantified by using the Odyssey infrared imaging system (LI-COR Biosciences).
Metabolic Pulse-Chase Analysis
Metabolic pulse-chase experiments were performed as described previously (36) except for the following modifications. Proteins were immunoprecipitated using anti-HA or anti-GFP antibodies and separated by SDS-PAGE. Visualization and quantification was performed using a Typhoon 8600 scanner and ImageQuantTM TL software (GE Healthcare). Plotted data reflect three independent experiments with the mean ± S.D. indicated.
Cycloheximide Chase Analysis
Cycloheximide chase experiments were performed as described previously (37) with the following modifications. Briefly, cycloheximide was added to log phase cultures to initiate the chase (200 μg/ml final concentration; Sigma-Aldrich). 1.8 ml of cell culture was collected at indicated times, pelleted, and resuspended in 1.0 ml of 10% TCA. Detergent lysates were prepared as described above for Western analysis. Blots were probed with anti-HA, stripped, reprobed with anti-PGK antibody, and visualized using the ECL system.
Alkaline Carbonate Extraction
Five A600 units of wild type and Δpep4 cells expressing (PGAS1)Wsc1*-6R were harvested and pelleted by centrifugation. Cells were resuspended in 1.2 ml of 10 mm sodium phosphate, pH 7.0, containing 1 mm PMSF and protease inhibitor mixture (CompleteTM, Roche Applied Science). 0.5 cm3 of zirconium beads was added, and cells were disrupted by 4 × 15 s cycles in a mini-bead beater. The collected lysate was centrifuged at 800 × g for 5 min, and the supernatant was transferred to a fresh tube. An equal volume of 0.2 m sodium carbonate (pH 11.0) was added and incubated on ice for 30 min. One-fifth of the mixture was saved, and the remaining fraction was subject to ultracentrifugation (100,000 × g) for 30 min at 4 °C. The high speed supernatant was transferred to a fresh tube. Pelleted membranes were solubilized in 3% SDS, 100 mm Tris, pH 7.4, 3 mm DTT and heated to 100 °C for 10 min. Proteins in the reserved lysate and high speed supernatant were precipitated in 10% TCA for 30 min on ice. Proteins were collected by centrifugation, resuspended in TCA resuspension buffer, and heated to 100 °C for 10 min. Proteins were analyzed by immunoblotting after adjustments to normalize protein concentrations. Primary antibody concentrations were as follows: 1:5000 anti-HA; 1:10,000 anti-Kar2p; and 1:5000 anti-Sec61p.
RESULTS
Misfolded Wsc1 Proteins Are Transported en Bloc to the Vacuolar Lumen
The variant Wsc1-L63R (Wsc1* hereafter) contains a single point mutation in its luminal domain that prevents proper folding (Fig. 1A). Wsc1* traffics efficiently from the ER to Golgi via COPII vesicles and is degraded in the vacuole (26). To determine its transport route to the vacuole, indirect immunofluorescence was performed in wild type and Δpep4 cells. The rapid degradation of Wsc1* in wild type cells results in low steady state levels, making visualization difficult (26). For this reason, expression was enhanced using the moderate GAS1 promoter for imaging studies (38). Wsc1* intracellular trafficking patterns and degradation are indistinguishable from molecules expressed from the weaker PRC1 promoter (supplemental Fig. S1, A and B). In wild type cells, Wsc1p variants localized to small punctate structures reminiscent of Golgi and endosomal membranes (Fig. 1B, a) (39–42). Vacuoles, which are visualized as depressions in DIC images (Fig. 1D), are deficient in signal because of rapid substrate degradation there (26). In Δpep4 cells, where vacuolar proteases are inactivated, Wsc1* accumulated strongly in vacuoles (Fig. 1B, e–h). This pattern differs from V-ATPase (Fig. 1D), a resident of the vacuolar limiting membrane, suggesting luminal localization. To confirm this notion, sequential z-stack confocal scans of Wsc1* in Δpep4 cells were collected. As shown in Fig. 1C, staining can be seen across the organelle in all of the z-stacks with significant signals. Taken together, these data show that Wsc1* is delivered to the lumen of the vacuole.
FIGURE 1.

Wsc1* localizes to the vacuolar lumen. A, schematic representation of Wsc1*. The L63R mutation is indicated by the asterisk, and the position of the HA epitope tag is shown in black. S.S., signal sequence; TM, transmembrane domain; Cyt, cytoplasmic domain. B, Wsc1* in wild type and Δpep4 cells was localized by indirect immunofluorescence using anti-HA monoclonal antibody and Alexa Fluor 488 goat anti-mouse antibody (green channel). Nuclear DNA was stained by DAPI to indicate positions of nuclei (blue channel). Visualization was performed using confocal and DIC microscopy as indicated. Scale bar, 5 μm. C, Wsc1* localization in Δpep4 cells as described in B. A z-series was captured, with the middle plane, its corresponding DIC image, and merged images shown in a–c. d–k show individual z-stacks of the series from top to bottom. Scale bars, 1 μm. D, wild type and Δpep4 cells were processed as in B and bound with anti-V-ATPase (60-kDa subunit) antibody as a vacuolar membrane marker followed by Alexa Fluor 488 goat anti-mouse antibody. Cells were visualized by confocal and DIC microscopy. Scale bar, 5 μm.
Misfolded Wsc1p Is Sorted into the MVB Pathway for Complete Molecule Breakdown
The most direct route for luminal delivery of Wsc1* is through the MVB pathway. To test this possibility, a panel of class E vacuolar protein sorting (vps) mutants was analyzed using fluorescence-based imaging. The simple assay takes advantage of the fact that these mutants share the same terminal phenotype. Regardless of the step disrupted in MVB-dependent transport, each mutant forms an aberrant prevacuolar organelle devoid of vesicles called the class E compartment (16–18). Cargo that normally uses the MVB pathway accumulates there with a portion continuing on to the vacuolar limiting membrane (17, 19). A Wsc1*-GFP fusion protein was constructed for examination in live cells. Wsc1*-GFP behaves indistinguishably from Wsc1* in wild type cells except that the GFP moiety accumulates in the vacuole because it is resistant to proteolysis (supplemental Fig. S2, A and B, WT panel). Among four well characterized endosomal sorting complexes required for transport (ESCRT) involved in cargo clustering (ESCRT-0), membrane budding (ESCRT-I and ESCRT-II), and membrane scission (ESCRT-III) (for review, see Ref. 43), most mutants caused the accumulation of Wsc1*-GFP in class E compartments (seen prominently in the micrographs as a strongly staining structure juxtaposed to the vacuolar membrane) and the vacuolar limiting membrane (supplemental Fig. S2B). Interestingly, Δhse1 cells had no effect, indicating that the Vps27p partner is dispensable for Wsc1*-GFP transport (44, 45). Among the panel, Δdid2, Δvta1, Δvps60, Δist1, and Δmvb12 strains are described as not displaying prominent class E compartments, and portions of their MVB cargo distribute into the vacuolar lumen (46–51). Accordingly, the Wsc1*-GFP trafficking defect is weaker or unaffected, as was the case for the ESCRT-I component Mvb12p, which is important for the trafficking of CPS, Ste3p, and Sna3p (50). Transport proficiency in Δist1 cells was not unexpected because it was not known to cause defects unless combined with other MVB mutants (51). Taken together, the data show that misfolded Wsc1p is sorted into the MVB pathway for trafficking into the vacuolar lumen.
For more detailed analysis, Wsc1* was expressed in the well characterized Δvps27 ESCRT-0 mutant (Wsc1*-GFP was unsuitable because of rapid bleaching). Wsc1* accumulates unambiguously in the characteristic prevacuolar class E compartment and some in the vacuolar membrane (Fig. 2A (i–l) and supplemental Fig. S3). The absence of signal in the vacuolar lumen of Δpep4Δvps27 cells suggests that Wsc1* molecules use the MVB pathway for its primary vacuolar transport (Fig. 2, A (m–p) and B). However, these data do not rule out the possibility that some molecules insert directly into the vacuolar limiting membrane and degrade in the lumen following uptake through microautophagy. We tested this question by determining the fate of Wsc1* in strains lacking EGO1, EGO3, and GTR2, genes encoding positive effectors of microautophagy (25). Wsc1* degrades similarly to wild type in these strains and traffics normally to the vacuolar lumen. These data confirm the notion that vacuolar localization and degradation are exclusively via the MVB pathway (supplemental Fig. S4).
FIGURE 2.

ESCRT mutants alter trafficking and degradation patterns of Wsc1*. A, wild type and Δpep4, Δvps27, and Δpep4Δvps27 mutants expressing Wsc1* were processed for indirect immunofluorescence, as described in the legend to Fig. 1B. Cells were visualized by confocal and DIC microscopy. Scale bar, 5 μm. B, a–c display the middle plane of a z-series as described for Fig. 1B captured from Δpep4Δvps27 cells expressing Wsc1*. d–o display the z-stack series from top to bottom. Scale bars, 1 μm. C, Wsc1* expression in wild type and Δpep4, Δvps27, and Δpep4Δvps27 mutants was analyzed by immunoblotting. Membranes were probed with anti-HA antibody (top), stripped, and reprobed with anti-PGK antibody as the loading control (bottom).
We next sought to understand the physiological role of directing misfolded proteins through the MVB pathway. For this, it was necessary to analyze substrate processing in detail. Many plasma membrane proteins are regulated through lysosomal/vacuolar degradation via the MVB pathway. In turn, MVB mutants are known to stabilize a variety of MVB cargo by disrupting their vacuolar transport or by causing enhanced recycling to the plasma membrane (4, 52–57). To assess the fate of Wsc1* in wild type, Δpep4, Δvps27, and Δpep4Δvps27 cells, immunoblots were prepared from cell extracts and probed using anti-HA monoclonal antibody to detect substrates. Strains lacking PEP4 yield greater steady state levels, as expected (Fig. 2C, lanes 1 and 2). Surprisingly, the Δvps27 mutant accumulated fragmented Wsc1* molecules with sizes ranging from full-length to just above the 20 kDa marker (Fig. 2C, lane 3). The identical pattern was also observed in Δvps36 and Δvps37 ESCRT mutants (data not shown). We conclude that these represent in vivo vacuolar or prevacuolar proteolytic products because also deleting the PEP4 gene eliminated fragmentation (Fig. 2C, lane 4). Membrane localization of substrate, its absence from the vacuolar lumen, and the smallest fragment being slightly larger than the predicted Wsc1p cytosolic domain suggest degradation of just the luminal domain. We were intrigued because it suggests that substrate insertion into the vacuolar limiting membrane creates a topologic constraint that prevents degradation of the whole molecule. However, ESCRT mutants severely disrupt endosomal trafficking, so it is also plausible that the data reflect the compromised proteolytic capacity of the vacuole due to defective sorting of some vacuolar hydrolases (16).
MVB cargo proteins are often ubiquitinated to form a signal recognized by ESCRT-0 and ESCRT-I components (for reviews, see Refs. 43 and 58). We tested if this mode of sorting is also used for misfolded Wsc1p. If so, it might provide an experimental means to control Wsc1* transport without compromising general trafficking and vacuolar functions. In yeast, CPS is a vacuolar luminal resident whose biogenesis requires transiting through the MVB pathway (21, 59). The E3 ubiquitin ligases Rsp5p and Tul1p were reported to play roles in CPS ubiquitination and transport (60, 61). In addition, Rsp5p is required for the direct Golgi-vacuole transport of Gap1p and Pma1-7p (62–65). For Wsc1*, Tul1p is not required because transport and degradation in Δtul1 cells were indistinguishable from wild type (supplemental Fig. S2).3 In rsp5–1 cells, however, Wsc1* was detected at the vacuolar limiting membrane (Fig. 3A, i). A similar pattern with an accompanying weak luminal signal was observed in the Δpep4 rsp5-1 mutant (Fig. 3A, m) consistent with a leaky phenotype, as reported previously (61). These data suggest that ubiquitination mediated by Rsp5p is required for the trafficking of misfolded Wsc1p to the vacuolar lumen. We tested this idea further by analyzing Wsc1* in Δdoa4 cells. Doa4p is a ubiquitin hydrolase recruited by ESCRT components to deubiquitinate cargo (66–69). Because deubiquitination is a requirement for entry into intralumenal vesicles, some MVB cargo proteins missort into vacuolar membranes in Δdoa4 cells (68–70). Indeed, Wsc1* localized to the vacuolar membrane of Δdoa4 cells (Fig. 3B, i). In the Δpep4Δdoa4 double mutant, some substrate was also observed in the vacuolar lumen, indicating that a fraction can enter the MVB pathway (Fig. 3B, m). This is consistent with observations of folded Doa4p-dependent MVB cargo (66, 70, 71). Substrates do not accumulate in a class E compartment because rsp5-1 and Δdoa4 mutants leave the MVB pathway intact (61, 68, 70). Thus, the vacuole is expected to function normally. Indeed, Wsc1* is processed to fragments in a range more narrow than the ESCRT mutants, with the smallest remaining above the 20 kDa marker (Fig. 3, C and D, lanes 3). The processing is vacuolar because also deleting the PEP4 gene stabilizes full-length Wsc1* (Fig. 3, C and D, lanes 4). These data show that transporting misfolded Wsc1 proteins to the vacuolar lumen via the MVB pathway is required for complete substrate degradation.
FIGURE 3.

Wsc1* requires Rsp5p and Doa4p for entry into the MVB pathway. A, wild type, Δpep4, rsp5-1, and Δpep4 rsp5-1 cells expressing Wsc1* were grown to log phase and shifted to 37 °C for 1 h. Cell preparation and image acquisition were performed as described in the legend to Fig. 1B. Scale bar, 5 μm. B, indirect immunofluorescence of wild type, Δpep4, Δdoa4, and Δpep4Δdoa4 strains expressing Wsc1*. Scale bar, 5 μm. C, Western blot analysis of wild type, Δpep4, rsp5-1, and Δpep4 rsp5-1 cells expressing Wsc1* after a 1-h shift to 37 °C. D, Wsc1* expression in wild type, Δpep4, Δdoa4, and Δpep4Δdoa4 cells was analyzed by immunoblotting as described in the legend to Fig. 2C.
Substrate Missorting Causes the Generation of Toxic Degradation Products
Localization of Wsc1* to the vacuolar limiting membrane in rsp5-1 and Δdoa4 mutants indicates that ubiquitination is required for sorting into intralumenal vesicles but not for vacuolar transport. This suggested that misfolded Wsc1p substrate sorting can be bypassed by eliminating the ubiquitin signal. The Wsc1p cytoplasmic domain has six potential lysine sites of modification (57, 62, 63, 65, 72, 73). To determine whether any of these lysine residues are required for sorting, each was mutated individually to arginine in Wsc1* to prevent ubiquitination. All six variants were efficiently sorted into the vacuolar lumen, suggesting that multiple sites can be used for signaling.3 To eliminate modification, all six sites were mutated to arginine to generate the Wsc1*-6R variant. Because cytoplasmic domains of membrane proteins must properly fold to avoid ERAD and display export signals (if any), Wsc1*-6R localization was determined by indirect immunofluorescence. In wild type cells, Wsc1*-6R was found primarily in the vacuolar limiting membrane, indicating that the mutations did not affect ER export (Fig. 4A and supplemental Fig. S5a). However, we noted that vacuolar morphology appeared abnormal in many of the cells (see below). No luminal signal was observed in Δpep4 cells, indicating a complete bypass of the MVB pathway (Fig. 4B and supplemental Fig. S5e). Thus, the creation of Wsc1*-6R provided the means to query the consequences of trafficking a GQC substrate through routes normally taken by resident vacuolar membrane proteins (74). By immunoblotting, Wsc1*-6R in wild type cells accumulated C-terminal fragments migrating between the 20 and 25 kDa markers (Fig. 4C, lane 3). Processing Wsc1*-6R to these forms require vacuolar proteases because they do not form in Δpep4 cells (Fig. 4C, lane 4).
FIGURE 4.

A Wsc1* ubiquitination-deficient variant fails to enter the MVB pathway. Wsc1*-6R localization in wild type (A) and Δpep4 cells (B). Images in a–c are from the middle plane of a z-series displaying Wsc1* (HA), vacuoles (DIC), and their merged images. d–m (A) and d–k (B) show a series of z-stacks from top to bottom. Scale bars, 1 μm. C, Wsc1* and Wsc1*-6R expression in wild type and Δpep4 cells analyzed by immunoblotting using the anti-HA antibody.
Next, we examined the nature of the degradation products. For this, we performed cycloheximide chase experiments to measure stability. Wsc1* in wild type cells is rapidly degraded as previously observed by pulse-chase analysis (compare supplemental Fig. S1A and Fig. 5A, lanes 1–3). In Δvps27 cells, where Wsc1* accumulates mostly in the class E compartment, the broad range of degradation products is mostly stable except for continued processing to smaller species over the chase period (Fig. 5A, lanes 4–6). Wsc1*-6R in wild type cells shows a similar high molecular weight form as Wsc1* before cycloheximide addition (Fig. 5, A and B, lanes 1). In addition, the low molecular weight fragments are in much greater abundance, suggesting accumulation. This is confirmed in the chase, where the high molecular weight form is converted rapidly to degradation fragments that are remarkably stable (Fig. 5B, lanes 1–3). In the Δpep4 strain, only the full-length Wsc1*-6R is detected even as both forms are localized to the vacuolar membrane (Figs. 4B and 5B, lanes 4–6).
FIGURE 5.
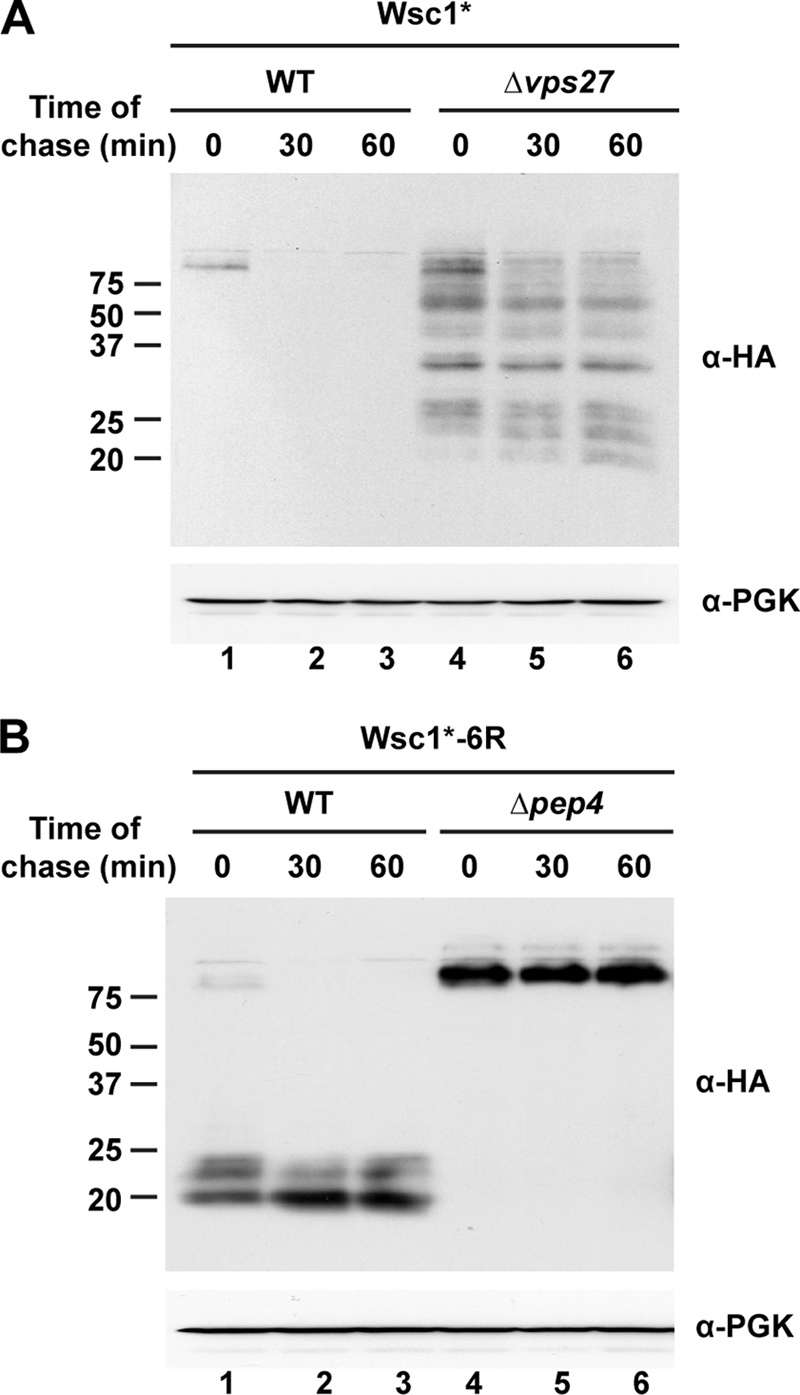
Wsc1* degradation intermediates are stable in Δvps27 cells. A, cycloheximide chase analysis of wild type and Δvps27 cells expressing Wsc1*. Chase times following the cycloheximide addition are shown above the blots. Wsc1* was detected using anti-HA antibodies (top). Anti-PGK antibody was used to probe the stripped blot as a loading control (bottom). B, cycloheximide chase analysis was performed on wild type and Δpep4 cells expressing Wsc1*-6R as described in A.
In our experiments, we observed that MVB mutants expressing Wsc1* grew more slowly than in its absence. This suggested that the MVB pathway could be required to alleviate toxicity of the misfolded protein. To test this notion directly, Wsc1* was placed under the tightly regulated GAL1 promoter and transformed into wild type, Δpep4, and Δvps27 cells. When spotted onto glucose media to repress expression, the strains grew indistinguishably (Fig. 6A, upper Glucose panel). When Wsc1* expression was induced with galactose, Δvps27 cells were killed. Surprisingly, Δpep4 cells, which strongly stabilize Wsc1* in the vacuole, grew as well as control (Fig. 6A, upper Galactose panel). The effect is specific to Wsc1* because expression of the misfolded soluble protein CPY*, which traffics the vacuole under galactose induction (2), had no adverse effect on Δvps27 cells (Fig. 6A, lower panels). Although these data are consistent with the MVB pathway playing an important role in detoxifying GQC substrates, the lack of toxicity after accumulation in the Δpep4 strain seemed paradoxical. This can be explained because Wsc1* stabilization in the two strains is dramatically different. In Δpep4 cells, Wsc1* is stabilized in the vacuolar lumen after trafficking through a functioning MVB pathway. By contrast, Wsc1* accumulates as aberrant degradation fragments in the membranes of the class E compartments and vacuoles of Δvps27 cells. Wsc1* toxicity in MVB mutants is probably caused by the accumulation of degradation products because moderate expression of Wsc1*-6R, which bypasses the MVB pathway, strongly compromises the growth of even wild type cells (Fig. 6, B and C). Interestingly, toxicity is less severe in the Δpep4 mutant, which allows trafficking of Wsc1*-6R to the vacuolar limiting membrane (Fig. 4B) but prevents its processing into fragments (Fig. 4C).
FIGURE 6.

Accumulation of Wsc1* fragments causes toxicity. A, wild type, Δpep4, and Δvps27 cells containing (PGAL1)Wsc1* or (PGAL1)CPY* grown in raffinose media were spotted as 10-fold serial dilutions onto glucose (promoter-repressed) and galactose (promoter-activated) medium plates and incubated at 30 °C for 2 and 3 days, respectively. B, wild type and Δpep4 cells containing the control vector, (PGAS1)Wsc1*, or (PGAS1)Wsc1*-6R were spotted on selective synthetic plates and incubated at 30 °C for 2 days. C, growth analysis of wild type and Δpep4 cells containing the empty vector (pRS315), (PGAS1)Wsc1*, or (PGAS1)Wsc1*-6R. Cells were grown in selective synthetic media to log phase and diluted to 0.1 A/ml. Growth was measured as 0.1 A600/ml readings from cultures at regular intervals over 10 h. The data plotted reflect three independent experiments with the mean ± S.D. (error bars) indicated. *, p < 0.01, Student's t test. D, membranes prepared from wild type and Δpep4 cells expressing (PGAS1)Wsc1*-6R were treated with 0.1 m sodium carbonate, pH 11.0, for 30 min on ice. A portion was reserved as total (T), and the remaining was subjected to centrifugation at 100,000 × g. Supernatant (S) and membrane pellet (P) fractions were collected and analyzed by immunoblotting. Wsc1*-6R was detected using anti-HA antibody. Kar2p and Sec61p serve as soluble and integral membrane protein controls, respectively.
To determine the effect of protein concentration on toxicity, Wsc1* and Wsc1*-6R were expressed using the PRC1, GAS1, and GAL1 promoters corresponding to weak, moderate, and strong expression. Quantitative immunoblotting of substrates confirmed the relative strength of these promoters for Wsc1* and Wsc1*-6R (supplemental Fig. S6, C–E). In liquid culture, weak expression of either protein had no effect on cell growth compared with control (supplemental Fig. S6A). Moderately expressed Wsc1*-6R (PGAS1) accumulated to the same extent in wild type and Δpep4 cells, demonstrating that the difference in toxicity is not due to unequal protein levels. At high expression from the GAL1 promoter, Wsc1*-6R is strongly toxic to both strains compared with controls (supplemental Fig. S6, B and F). In this case, the difference in toxicity between wild type and Δpep4 strains persists but is diminished. These data demonstrate a dose-dependent relationship to the toxicity of aberrant proteins inserted into the vacuolar membrane.
We next wished to understand how Wsc1* fragments trigger toxicity. Immunolocalization indicates that degradation products are at the vacuolar membrane, but it was unclear if they remained integrated or became peripherally associated following proteolysis (Fig. 4A). To answer this question, cell extracts were prepared from wild type and Δpep4 strains expressing Wsc1*-6R and treated with alkali to strip soluble and peripherally associated proteins from membranes. Membranes and alkali-extracted proteins were separated by high speed centrifugation and detected on immunoblots. Full-length Wsc1*-6R fractionated with the membrane pellet, as expected (Fig. 6D, α-HA panel, lane 6). The stable Wsc1*-6R degradation products were detected exclusively in the membrane fraction, demonstrating that they remain integrated in the vacuolar membrane even after extensive luminal proteolysis (Fig. 6D, α-HA panel, lane 3).
To analyze the integrity of vacuolar membranes, we incubated cells expressing Wsc1* or Wsc1*-6R with the lipophilic dye FM4–64 to visualize vacuolar membranes (33). Wild type and Δpep4 cells expressing Wsc1* display vacuolar morphologies similar to control cells (Fig. 7, compare d and g with a). By contrast, wild type cells expressing Wsc1*-6R contain distorted or fragmented vacuoles (Fig. 7j). Δpep4 cells, which accumulate full-length Wsc1*-6R in the vacuolar membrane, display relatively normal vacuoles (Fig. 7m).
Ultrastructural analysis of Wsc1*-6R expressing wild type cells shows dramatic disruption of vacuolar morphologies and the accumulation of aberrant membrane structures (supplemental Fig. S7, compare A and D). In addition, these cells strongly accumulate lipid droplets, an indicator of lipid disequilibrium (supplemental Fig. S8) (75, 76). These data show that the by-products of a missorted GQC substrate exert toxicity by physically disrupting the membranes to which they are inserted.
DISCUSSION
The range of protein quality control mechanisms is far wider than previously thought. The breadth is not surprising, given their importance in maintaining cellular protein homeostasis. Although early studies focused on those stationed at sites of de novo protein synthesis, there is now great interest in less understood pathways that can be found throughout the cell (3, 5, 9, 10, 38, 77–80). Instead of just monitoring the folding of polypeptides as they are made, these mechanisms also police populations of functional proteins for those going bad. Thus, strategies suited only for actively folding proteins, like the mannose timer hypothesis in ERAD (81–83), would not apply there.
The peripheral quality control system appears to be charged with both roles. For example, the multispanning protein Pma1p is subject to ERAD if it is grossly misfolded by mutation. One allele, pma1-7, is caught by GQC and diverted to the vacuole for turnover (9). Other mutants, undetected by either pathway, traffic to the plasma membrane, where they rapidly endocytose and traffic to the vacuole (84, 85). The reasons for these sorting patterns are unclear because specific features, recognized by one system and not others, are not known. The plasma membrane system can also detect post-maturation conformational changes. In mammalian cells, the most common disease allele of the cystic fibrosis transconductance regulator is ΔF508 (86). At 37 °C, ΔF508 is retained in the ER and degraded by ERAD. However, shifting cells to 26 °C allows the maturation and trafficking of the mutant to the plasma membrane, where it is at least partially functional (87–90). Because of this effect, getting mutant forms of cystic fibrosis transconductance regulator to bypass the ERAD checkpoint has been a major research goal as a therapeutic strategy. However, the existence of a plasma membrane quality control mechanism complicates the strategy. Returning cells to 37 °C, and presumably the corresponding conformation, triggers ΔF508 recognition, endocytosis, and transport to lysosomes by the MVB pathway (5, 55, 91). Thus, the importance of understanding these mechanisms cannot be overstated. Lukacs and co-workers (4, 5) recently reported the role of cytosolic chaperones in recognizing misfolded proteins at the plasma membrane. Interestingly, some of these are known components of other quality control systems suggesting mechanistic overlaps. These studies demonstrate the necessity to account for peripheral quality control before the therapeutic strategy can be viable.
In this study, we report that the GQC substrate Wsc1* sorts to the MVB pathway for turnover (Fig. 8A), making it a point of convergence between cell surface and Golgi mechanisms. Unlike plasma membrane receptors, which tend to recycle if MVB sorting is subverted, misfolded Wsc1p traffics to the vacuole even if the pathway cannot be utilized. This suggests that the signals required for vacuolar transport and MVB sorting are separable. This made it possible to demonstrate that the route is essential for complete substrate degradation. Diversion of misfolded Wsc1p to the alternative vacuolar transport route caused the accumulation of partially degraded molecules still embedded in the vacuolar membrane (Fig. 8B). This result provided evidence that the normal transport pathways used by resident vacuolar membrane proteins are not suitable for protein quality control. Remarkably, we discovered that the resulting “luminally sheared” form of Wsc1p is highly toxic. This form disrupts vacuolar membrane integrity and might indirectly affect lipid homeostasis. This showed that the efficient whole-molecule degradation provided by the MVB pathway could also provide a protective function for the cell (Fig. 8). Taken together, these data provide a physiological basis for trafficking misfolded proteins through the MVB pathway. Because peripheral quality control substrates ultimately use the same pathway, the principles gleaned from the Wsc1* studies are likely to be more generally applicable.
FIGURE 8.

Model of the MVB-dependent pathway for the transport of misfolded Wsc1p. A, normally, misfolded Wsc1p exits the ER and transits through the Golgi apparatus. It is next sorted to the MVB pathway and degraded in its entirety within the vacuolar lumen. This mechanism requires the ubiquitin ligase Rsp5p and ubiquitination of the Wsc1p cytoplasmic domain. B, misfolded GQC substrates missorted to the vacuolar limiting membrane. Degradation proceeds but is constrained by the membrane, leading to partial degradation products still integrated in the membrane. These aberrant products disrupt vacuolar membranes, which can lead to cell death.
With the MVB pathway being a convergence point, why are two (or more) post-ER surveillance sites needed? The studies from the Lukacs and Gardner laboratories (4, 5, 77, 91) may have already provided the answer. In both cases, the groups examined the fate of proteins where folding states can be controlled by temperature. At low temperatures, the proteins are folded and functional. After shifting to restrictive temperatures, the mutant proteins change conformations to sufficiently pique the attention of local quality control mechanisms at the plasma membrane and nucleus. These studies suggest that quality control mechanisms are at play anywhere there are proteins, folded or unfolded. Thus, the other role of GQC may be to continuously monitor the integrity of folded resident Golgi proteins. The discovery of its role in capturing misfolded proteins undetected by ERAD could be explained simply by the fact that such model molecules were easier to generate. However, there are also clear differences between plasma membrane and Golgi systems that might impact their client range. In GQC, misfolded luminal domains like those in Wsc1* are detected by Golgi cargo-sorting factors, such as Vps10p (soluble substrates are entirely luminal) (10, 26, 29). By contrast, misfolded soluble proteins that can make it to the plasma membrane are secreted, suggesting the absence of a surveillance mechanism on the exterior of the cell, which is the topologic equivalent of the organelle lumen (10, 92). Indeed, the ΔF508 lesion of cystic fibrosis transconductance regulator affects the conformation of its cytoplasmic domain, and the known recognition factors are cytosolic (5). Although it is tempting to suggest a division of labor between these pathways, the mutations in the pma1-7 allele that make it a substrate of GCQ probably cause conformational changes to its cytosolic domains, suggesting that GQC is not restricted to luminal abnormalities (9).
In this paper, we report the transport mechanism used by GCQ for the obligate substrate Wsc1p. Major questions remain. The most immediate is how misfolded proteins are detected in the Golgi lumen. Although Vps10p is an important factor, how it and other yet to be identified factors differentiate folded and unfolded proteins remains mysterious. Another question is the molecular mechanism of toxicity for Wsc1*-6R degradation by-products. Although the severity of their effects on vacuolar membrane may be sufficient to explain their toxicity, it remains to be determined how it manifests simply through the degradation of the luminal domain. To our knowledge, this is the first instance in which such a phenomenon has been observed in vivo. Although many questions remain, recent studies demonstrate that cellular protein homeostasis relies on a complex network of quality control systems, some of which remain to be discovered.
Supplementary Material
Acknowledgments
We thank Dr. Chris Kaiser for providing the rsp5-1 strain. We are grateful to the TLL/NUS core facilities, especially Dr. Graham Wright and Dr. Meredith Calvert for providing excellent technical support for imaging studies and Dr. Xuezhi Ouyang for transmission electron microscopy sample preparation. We also acknowledge Chengchao Xu and Alisha Chakrabarti for critical reading of the manuscript.
This work was supported by funds from the Temasek Trust and by grants from the Singapore Millennium Foundation (predoctoral fellowship (to S. W.) and postdoctoral fellowship (to G. T.)).

The on-line version of this article (available at http://www.jbc.org) contains supplemental Tables S1 and S2 and Figs. S1–S8.
S. Wang, G. Thibault, and D. T. W. Ng, unpublished results.
- ERAD
- endoplasmic reticulum-associated degradation
- ER
- endoplasmic reticulum
- GQC
- Golgi quality control
- MVB
- multivesicular body
- CPY
- carboxypeptidase Y
- PGK
- 3-phosphoglyceric phosphokinase
- DIC
- differential interference contrast
- ESCRT
- endosomal sorting complexes required for transport.
REFERENCES
- 1. Vembar S. S., Brodsky J. L. (2008) Nat. Rev. Mol. Cell Biol. 9, 944–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Spear E. D., Ng D. T. (2003) Mol. Biol. Cell 14, 2756–2767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ashok A., Hegde R. S. (2009) PLoS Pathog. 5, e1000479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Apaja P. M., Xu H., Lukacs G. L. (2010) J. Cell Biol. 191, 553–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Okiyoneda T., Barrière H., Bagdány M., Rabeh W. M., Du K., Höhfeld J, Young J. C., Lukacs G. L. (2010) Science 329, 805–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Arvan P., Zhao X., Ramos-Castaneda J., Chang A. (2002) Traffic 3, 771–780 [DOI] [PubMed] [Google Scholar]
- 7. Anelli T., Sitia R. (2008) EMBO J. 27, 315–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hegde R. S., Ploegh H. L. (2010) Curr. Opin. Cell Biol. 22, 437–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chang A., Fink G. R. (1995) J. Cell Biol. 128, 39–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hong E., Davidson A. R., Kaiser C. A. (1996) J. Cell Biol. 135, 623–633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jenness D. D., Li Y., Tipper C., Spatrick P. (1997) Mol. Cell Biol. 17, 6236–6245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Coughlan C. M., Walker J. L., Cochran J. C., Wittrup K. D., Brodsky J. L. (2004) J. Biol. Chem. 279, 15289–15297 [DOI] [PubMed] [Google Scholar]
- 13. Kincaid M. M., Cooper A. A. (2007) Mol. Biol. Cell 18, 455–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Buchberger A., Bukau B., Sommer T. (2010) Mol. Cell 40, 238–252 [DOI] [PubMed] [Google Scholar]
- 15. Hicke L., Dunn R. (2003) Annu. Rev. Cell Dev. Biol. 19, 141–172 [DOI] [PubMed] [Google Scholar]
- 16. Raymond C. K., Howald-Stevenson I., Vater C. A., Stevens T. H. (1992) Mol. Biol. Cell 3, 1389–1402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Piper R. C., Cooper A. A., Yang H., Stevens T. H. (1995) J. Cell Biol. 131, 603–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rieder S. E., Banta L. M., Köhrer K., McCaffery J. M., Emr S. D. (1996) Mol. Biol. Cell 7, 985–999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Babst M., Sato T. K., Banta L. M., Emr S. D. (1997) EMBO J. 16, 1820–1831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Piper R. C., Bryant N. J., Stevens T. H. (1997) J. Cell Biol. 138, 531–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Odorizzi G., Babst M., Emr S. D. (1998) Cell 95, 847–858 [DOI] [PubMed] [Google Scholar]
- 22. Cowles C. R., Snyder W. B., Burd C. G., Emr S. D. (1997) EMBO J. 16, 2769–2782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Stepp J. D., Huang K., Lemmon S. K. (1997) J. Cell Biol. 139, 1761–1774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kunz J. B., Schwarz H., Mayer A. (2004) J. Biol. Chem. 279, 9987–9996 [DOI] [PubMed] [Google Scholar]
- 25. Dubouloz F., Deloche O., Wanke V., Cameroni E., De Virgilio C. (2005) Mol. Cell 19, 15–26 [DOI] [PubMed] [Google Scholar]
- 26. Wang S., Ng D. T. (2010) Mol. Biol. Cell 21, 1153–1165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Verna J., Lodder A., Lee K., Vagts A., Ballester R. (1997) Proc. Natl. Acad. Sci. U.S.A. 94, 13804–13809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Jacoby J. J., Nilius S. M., Heinisch J. J. (1998) Mol. Gen. Genet. 258, 148–155 [DOI] [PubMed] [Google Scholar]
- 29. Jørgensen M. U., Emr S. D., Winther J. R. (1999) Eur. J. Biochem. 260, 461–469 [DOI] [PubMed] [Google Scholar]
- 30. Sambrook J., Fritsch E. M., Maniatis T. (1989) Molecular Cloning: A Laboratory Manual, 2nd Ed., Cold Spring Habor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 31. Malkus P., Jiang F., Schekman R. (2002) J. Cell Biol. 159, 915–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sawano A., Miyawaki A. (2000) Nucleic Acids Res. 28, E78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vida T. A., Emr S. D. (1995) J. Cell Biol. 128, 779–792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Spandl J., White D. J., Peychl J., Thiele C. (2009) Traffic 10, 1579–1584 [DOI] [PubMed] [Google Scholar]
- 35. Wright R. (2000) Microsc. Res. Tech. 51, 496–510 [DOI] [PubMed] [Google Scholar]
- 36. Ng D. T., Spear E. D., Walter P. (2000) J. Cell Biol. 150, 77–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kanehara K., Xie W., Ng D. T. (2010) J. Cell Biol. 188, 707–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Prasad R., Kawaguchi S., Ng D. T. (2010) Mol. Biol. Cell 21, 2117–2127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Burd C. G., Emr S. D. (1998) Mol. Cell 2, 157–162 [DOI] [PubMed] [Google Scholar]
- 40. Wooding S., Pelham H. R. (1998) Mol. Biol. Cell 9, 2667–2680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gillooly D. J., Morrow I. C., Lindsay M., Gould R., Bryant N. J., Gaullier J. M., Parton R. G., Stenmark H. (2000) EMBO J. 19, 4577–4588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Losev E., Reinke C. A., Jellen J., Strongin D. E., Bevis B. J., Glick B. S. (2006) Nature 441, 1002–1006 [DOI] [PubMed] [Google Scholar]
- 43. Hurley J. H., Hanson P. I. (2010) Nat. Rev. Mol. Cell Biol. 11, 556–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bilodeau P. S., Urbanowski J. L., Winistorfer S. C., Piper R. C. (2002) Nat. Cell Biol. 4, 534–539 [DOI] [PubMed] [Google Scholar]
- 45. Ren J., Kee Y., Huibregtse J. M., Piper R. C. (2007) Mol. Biol. Cell 18, 324–335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Shiflett S. L., Ward D. M., Huynh D., Vaughn M. B., Simmons J. C., Kaplan J. (2004) J. Biol. Chem. 279, 10982–10990 [DOI] [PubMed] [Google Scholar]
- 47. Azmi I., Davies B., Dimaano C., Payne J., Eckert D., Babst M., Katzmann D. J. (2006) J. Cell Biol. 172, 705–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Chu T., Sun J., Saksena S., Emr S. D. (2006) J. Cell Biol. 175, 815–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Curtiss M., Jones C., Babst M. (2007) Mol. Biol. Cell 18, 636–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Oestreich A. J., Davies B. A., Payne J. A., Katzmann D. J. (2007) Mol. Biol. Cell 18, 646–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rue S. M., Mattei S., Saksena S., Emr S. D. (2008) Mol. Biol. Cell 19, 475–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Davis N. G., Horecka J. L., Sprague G. F., Jr. (1993) J. Cell Biol. 122, 53–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bache K. G., Stuffers S., Malerød L., Slagsvold T., Raiborg C., Lechardeur D., Wälchli S., Lukacs G. L., Brech A., Stenmark H. (2006) Mol. Biol. Cell 17, 2513–2523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Baldys A., Raymond J. R. (2009) Biochemistry 48, 9321–9323 [DOI] [PubMed] [Google Scholar]
- 55. Glozman R., Okiyoneda T., Mulvihill C. M., Rini J. M., Barriere H., Lukacs G. L. (2009) J. Cell Biol. 184, 847–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Hayer A., Stoeber M., Ritz D., Engel S., Meyer H. H., Helenius A. (2010) J. Cell Biol. 191, 615–629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Lobert V. H., Brech A., Pedersen N. M., Wesche J., Oppelt A., Malerød L., Stenmark H. (2010) Dev. Cell 19, 148–159 [DOI] [PubMed] [Google Scholar]
- 58. Raiborg C., Stenmark H. (2009) Nature 458, 445–452 [DOI] [PubMed] [Google Scholar]
- 59. Spormann D. O., Heim J., Wolf D. H. (1992) J. Biol. Chem. 267, 8021–8029 [PubMed] [Google Scholar]
- 60. Reggiori F., Pelham H. R. (2002) Nat. Cell Biol. 4, 117–123 [DOI] [PubMed] [Google Scholar]
- 61. Katzmann D. J., Sarkar S., Chu T., Audhya A., Emr S. D. (2004) Mol. Biol. Cell 15, 468–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Helliwell S. B., Losko S., Kaiser C. A. (2001) J. Cell Biol. 153, 649–662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Soetens O., De Craene J. O., Andre B. (2001) J. Biol. Chem. 276, 43949–43957 [DOI] [PubMed] [Google Scholar]
- 64. Pizzirusso M., Chang A. (2004) Mol. Biol. Cell 15, 2401–2409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Risinger A. L., Kaiser C. A. (2008) Mol. Biol. Cell 19, 2962–2972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Amerik A. Y., Nowak J., Swaminathan S., Hochstrasser M. (2000) Mol. Biol. Cell 11, 3365–3380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Dupré S., Haguenauer-Tsapis R. (2001) Mol. Cell Biol. 21, 4482–4494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Losko S., Kopp F., Kranz A., Kölling R. (2001) Mol. Biol. Cell 12, 1047–1059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Nikko E., André B. (2007) Traffic 8, 566–581 [DOI] [PubMed] [Google Scholar]
- 70. Richter C., West M., Odorizzi G. (2007) EMBO J. 26, 2454–2464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Reggiori F., Pelham H. R. (2001) EMBO J. 20, 5176–5186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Springael J. Y., André B. (1998) Mol. Biol. Cell 9, 1253–1263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Katzmann D. J., Babst M., Emr S. D. (2001) Cell 106, 145–155 [DOI] [PubMed] [Google Scholar]
- 74. Roberts C. J., Pohlig G., Rothman J. H., Stevens T. H. (1989) J. Cell Biol. 108, 1363–1373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Listenberger L. L., Han X., Lewis S. E., Cases S., Farese R. V., Jr., Ory D. S., Schaffer J. E. (2003) Proc. Natl. Acad. Sci. U.S.A. 100, 3077–3082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Fei W., Wang H., Fu X., Bielby C., Yang H. (2009) Biochem. J. 424, 61–67 [DOI] [PubMed] [Google Scholar]
- 77. Gardner R. G., Nelson Z. W., Gottschling D. E. (2005) Cell 120, 803–815 [DOI] [PubMed] [Google Scholar]
- 78. Kaganovich D., Kopito R., Frydman J. (2008) Nature 454, 1088–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Heck J. W., Cheung S. K., Hampton R. Y. (2010) Proc. Natl. Acad. Sci. U.S.A. 107, 1106–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Heo J. M., Livnat-Levanon N., Taylor E. B., Jones K. T., Dephoure N., Ring J., Xie J., Brodsky J. L., Madeo F., Gygi S. P., Ashrafi K., Glickman M. H., Rutter J. (2010) Mol. Cell 40, 465–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Jakob C. A., Bodmer D., Spirig U., Battig P., Marcil A., Dignard D., Bergeron J. J., Thomas D. Y., Aebi M. (2001) EMBO Rep. 2, 423–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Quan E. M., Kamiya Y., Kamiya D., Denic V., Weibezahn J., Kato K., Weissman J. S. (2008) Mol. Cell 32, 870–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Clerc S., Hirsch C., Oggier D. M., Deprez P., Jakob C., Sommer T., Aebi M. (2009) J. Cell Biol. 184, 159–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Gong X., Chang A. (2001) Proc. Natl. Acad. Sci. U.S.A. 98, 9104–9109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Liu Y., Sitaraman S., Chang A. (2006) J. Biol. Chem. 281, 31457–31466 [DOI] [PubMed] [Google Scholar]
- 86. Cheng S. H., Gregory R. J., Marshall J., Paul S., Souza D. W., White G. A., O'Riordan C. R., Smith A. E. (1990) Cell 63, 827–834 [DOI] [PubMed] [Google Scholar]
- 87. Denning G. M., Anderson M. P., Amara J. F., Marshall J., Smith A. E., Welsh M. J. (1992) Nature 358, 761–764 [DOI] [PubMed] [Google Scholar]
- 88. Lukacs G. L., Mohamed A., Kartner N., Chang X. B., Riordan J. R., Grinstein S. (1994) EMBO J. 13, 6076–6086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Jensen T. J., Loo M. A., Pind S., Williams D. B., Goldberg A. L., Riordan J. R. (1995) Cell 83, 129–135 [DOI] [PubMed] [Google Scholar]
- 90. Ward C. L., Omura S., Kopito R. R. (1995) Cell 83, 121–127 [DOI] [PubMed] [Google Scholar]
- 91. Sharma M., Pampinella F., Nemes C., Benharouga M., So J., Du K., Bache K. G., Papsin B., Zerangue N., Stenmark H., Lukacs G. L. (2004) J. Cell Biol. 164, 923–933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Nakatsukasa K., Okada S., Umebayashi K., Fukuda R., Nishikawa S., Endo T. (2004) J. Biol. Chem. 279, 49762–49772 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.