

We found 11 wide mutants, seven of which affect the activation of Cdc42. Through epistasis analysis and protein retargeting, we showed that a guanine nucleotide exchange factor and a GTPase-activating protein of Cdc42 each affects cell width independently from different cellular domains. We propose that these proteins set up a spatially restricted gradient of activated Cdc42 that directs cell growth.
Abstract
The fission yeast Schizosaccharomyces pombe is a rod-shaped cell that grows by linear extension at the cell tips, with a nearly constant width throughout the cell cycle. This simple geometry makes it an ideal system for studying the control of cellular dimensions. In this study, we carried out a near-genome-wide screen for mutants wider than wild-type cells. We found 11 deletion mutants that were wider; seven of the deleted genes are implicated in the control of the small GTPase Cdc42, including the Cdc42 guanine nucleotide exchange factor (GEF) Scd1 and the Cdc42 GTPase-activating protein (GAP) Rga4. Deletions of rga4 and scd1 had additive effects on cell width, and the proteins localized independently of one another, with Rga4 located at the cell sides and Scd1 at the cell tips. Activated Cdc42 localization is altered in rga4Δ, scd1Δ, and scd2Δ mutants. Delocalization and ectopic retargeting experiments showed that the localizations of Rga4 and Scd1 are crucial for their roles in determining cell width. We propose that the GAP Rga4 and the GEF Scd1 establish a gradient of activated Cdc42 within the cellular tip plasma membrane, and it is this gradient that determines cell growth-zone size and normal cell width.
INTRODUCTION
The overall morphology of a nonisotropically growing cell is determined by two processes: the first is the polarization of growth, and the second is the determination of the size and shape of the polarized growth zones. Polarization of growth involves several steps: an initial cue to polarize, changes in the cytoskeleton, polarized secretion, and continued signaling to maintain polarized growth (reviewed in Pruyne et al., 2004b, for yeast; and in St Johnston and Ahringer, 2010, for metazoans). In many different cells, small GTPases are important for growth polarization. Small GTPases are effective growth regulators because they can act as molecular switches with an active GTP-bound state that interacts with effector proteins, and an inactive GDP-bound state. They have intrinsic GTPase activity and can hydrolyze GTP to GDP, but this hydrolysis is usually inefficient. It is facilitated when the small GTPase binds to a GTPase-activating protein (GAP), which catalyzes the transition from the active to inactive state. After hydrolysis to the GDP-bound state, reactivation is stimulated by binding to a guanine nucleotide exchange factor (GEF), which facilitates the release of GDP. Since the cellular concentration of GTP is higher than that of GDP, the GTPase will usually rebind GTP and return it to its active state (Perez and Rincon, 2010).
The importance of Rho-GTPases, especially Cdc42, in establishing polarized growth has been most fully explored in budding yeast. The budding yeast transition from isotropic to polarized growth during late G1 is triggered by the localized activation of Cdc42, which then activates both Rho1 to stimulate cell wall growth and Rho3 to direct secretory vesicles (Adams et al., 1990; Lew and Reed, 1993). Activated Cdc42 also recruits actin cables through the formin Bni1, activates the Arp2/3 complex via Bee1/Las17, and is required for polarized localization of members of the exocyst, such as Sec3 (Lechler et al., 2001; Zhang et al., 2001; Pruyne et al., 2004a). In a wild-type cell, the location of polarized growth is determined by the GTP-binding protein Rsr1. But in the absence of Rsr1, cells can still polarize through the stochastic enrichment of activated Cdc42, which depends on the scaffold Bem1. The need for Bem1 can be overcome by a fusion protein that joins the GEF Cdc24 and the downstream kinase Cla4 (Kozubowski et al., 2008). The importance of small GTPases is conserved in metazoan cells, as shown by a fibroblast migration assay that identified the small GTPases Rho, Rac, Cdc42, and Ras as playing roles in directing cell growth (Nobes and Hall, 1999).
Although the establishment of polarized growth has been studied extensively, much less work has been done on the determinants of the size and shape of the subsequent polarized growth zones. The fission yeast cell provides a good model for investigating this problem, because it has well-defined polarized-tip growth zones where the size and shape of the growth zone is stably maintained. The cell is rod-shaped and grows by length extension at the cell tips, where actin patches, glucan synthases, exocytosis, and endocytosis are concentrated. After cell division, cells begin growth at one end of the cell before transitioning to bipolar growth. Bipolar growth continues until cell division, when the growth machinery is relocated to form a septum in the middle of the cell. Throughout this growth cycle, these cylindrical cells maintain a near-constant diameter of approximately 4 μm. We will refer to this dimension as the cell width (Hayles and Nurse, 2001)
Two mutants that are still polarized but have altered cell widths have been previously described. These mutants are a deletion of the GAP rga4, which increases the width of the cell by 10% (Das et al., 2007), and a deletion of the GAP rga2, which decreases the width of the cell by 9% (Villar-Tajadura et al., 2008). These mutants establish that specific genes can influence cell width and implicate small GTPases as key factors in cell-width control. They have relatively small phenotypic effects and so would be difficult to identify using traditional genetic screens. Both mutants were identified because of their homology to known GAPs and were then found to have altered cell widths. But the development of a whole-genome gene deletion collection (Kim et al., 2010) allowed the systematic examination of mutant strains for identification of genes that determine cell width. To achieve this, we performed a visual screen for mutants that had increased cell width from a near-genome-wide collection of the viable gene deletions. We found 11 mutants, seven of which are implicated in the regulation of the small GTPase Cdc42, and we proceeded to investigate a Cdc42 GEF and a Cdc42 GAP identified in the screen to determine how they contribute to the control of cell width.
RESULTS
Eleven gene deletions increase cell width
To identify gene deletions that increase cell width, we used a subset of the whole-genome gene deletion collection to conduct a genetic screen for viable wide mutants. The screen was designed to find cells with an increased cell width that still exhibited polarized growth. We screened only viable haploid deletion mutants because we did not expect the increase in cell width, a more subtle change in morphology than the complete loss of cell polarity, to be lethal, although genes that are essential may also contribute to the control of cell width. The viable haploid deletion mutants available at that time covered 85% of the final haploid deletion library (Kim et al., 2010). A primary screen was carried out by observing morphology phenotypes in microcolonies on plates during spore germination and subsequent initial cell divisions (J. Hayles, unpublished data), and identified a group of 97 viable morphology mutants as candidates for increased cell width. This group included mutants that were wide, irregular in shape, or almost completely depolarized. To help distinguish between unpolarized, spherical mutants and polarized mutants with a wider growth zone, we performed a secondary screen, where we elongated cells by blocking cell-cycle progression, but not growth, with the ribonucleotide reductase inhibitor hydroxyurea (HU; Sazer and Sherwood, 1990) and then measured the width of each polarized mutant near the tip of the cell (Figure 1A). Wild-type cells, when arrested in HU for 5 h, increase in width slightly, from an average of 3.9 μm to an average of 4.1 μm, so mutants grown in HU were compared with wild-type cells grown in HU. The widths measured in the secondary screen are plotted in ascending order in Figure 1A and can be found in Supplemental Table S1. Because the objective of this screen was to isolate of mutants with a specific phenotype—normal polarity but an altered width—mutants that had other phenotypic defects, such as a grossly irregular shape, were excluded, but will be described in a separate manuscript (J. Hayles, unpublished data). Another morphology mutant, tea1Δ, was previously described as wide (Foethke et al., 2009), but was not found to be wide in this screen (Table S1), perhaps due to differences in measurement protocol, since Foethke and coworkers measured cells by tubulin fluorescence, not cell shape in bright-field images.
FIGURE 1:

A set of genes that determine cell width. (A) Secondary screen of 97 selected deletion mutants. Average widths of deletion mutants grown in HU for 5 h are plotted in ascending order, error bars represent one standard deviation. Magenta arrow and magenta diamond indicate WT. Green line indicates the cutoff for mutants selected for tertiary screening. Green diamonds indicate mutants that were ultimately found to be wide. At least 40 cells were measured for each strain. (B) Box-and-whiskers plot of the exponentially growing cell widths of wild-type and 11 wide deletion mutants isolated from the gene deletion collection. At least 100 cells were measured for each strain. Boxes indicate one quartile of the distribution in each direction. Asterisks indicate genes implicated in Cdc42 regulation. All mutant widths are significantly different from wild-type by Student's t test with p < 0.0001. (C) Of the 11 deletion mutants identified, seven have been implicated in the control of Cdc42 (see text). Arrows indicate positive regulation; T indicates negative regulation.
Using a cutoff of > 4.6 μm (0.5 μm wider than wild-type cells), we identified 26 wide mutants that we rescreened by measuring exponentially growing cells. This tertiary screen identified 11 mutants that were at least 10% wider than wild-type cells in both exponential growth and HU treatment conditions (Figure 1B). Three criteria were used to exclude mutants that were wide in the secondary screen from the final group of wide mutants: 1) some mutant strains were sickly and grew very poorly in exponential culture, 2) some mutant strains that were wide in the initial measurement in HU were not wide in exponential culture, and 3) two of the deletion mutants proved to be diploid.
Seven of the 11 gene deletions that increase cell width (scd1Δ, scd2Δ, rga4Δ, ras1Δ, cpp1Δ, ral2Δ, and efc25Δ) are linked to the regulation of the small GTPase Cdc42 (Figure 1C). Scd1 is the main GEF for Cdc42 (Fukui and Yamamoto, 1988; Chang et al., 1994), and binds Cdc42 in a complex with the scaffold Scd2 (Chang et al., 1999). Ras1 acts upstream of Scd1, presumably in its activation (Chang et al., 1994), and the Ras1 activity related to cell morphology is regulated by Efc25 (Tratner et al., 1997; Papadaki et al., 2002) and Ral2 (Fukui et al., 1989). Ras1 is probably farnesylated by Cpp1 (Yang et al., 2000). Rga4 is a predicted GAP that can affect the activation state of Cdc42 (Das et al., 2007; Tatebe et al., 2008). Our near-genome-wide screen was not guided by previous studies of gene function, so the fact that seven of the 11 wide deletion mutants are known to affect Cdc42 regulation suggests that this pathway is a major mechanism for the determination of cell width.
To characterize the mutants further, we first investigated whether the changes in cell width could be due to defects in the cell wall. Fungal cells, like plant cells, maintain an osmotic differential with the surrounding liquid, which results in a constant internal turgor pressure. A weakened cell wall might not withstand the internal osmotic pressure, and the cell could swell, increasing its width. Some morphology mutants with defects in cell wall composition can be rescued by the addition of 1.2 M sorbitol to the media, decreasing the relative internal pressure in the cell by increasing the external osmolarity (Ribas et al., 1991). But only one of the width mutants, asl1Δ, was well corrected by addition of sorbitol (Table 1). Asl1 was initially identified by homology as a putative adhesin (Linder and Gustafsson, 2008), but has not been further studied. Our results indicate that this deletion might affect either cell wall integrity or turgor pressure. Deletion of ras1, which alters cell wall composition (Harmouch et al., 1995), was partially corrected by addition of sorbitol. But the mutant phenotype could not be completely rescued by sorbitol, so Ras1 may have both cell wall–dependent and cell wall–independent effects on width. Since 10 of our original 11 wide deletion mutants are not well rescued by sorbitol, we conclude they are likely to play roles in directly determining the size of the polarized growth zone.
TABLE 1:
Wide mutant correction by sorbitol.
| Width in EMM4S + 1.2 M sorbitol, μm | % Wild-type width in EMM4S + 1.2 M sorbitol | |
|---|---|---|
| Wild-type | 3.81 | 100 |
| asl1Δ | 3.90 | 102 |
| spy1Δ | 4.50 | 118 |
| asp1Δ | 4.57 | 120 |
| rga4Δ | 4.18 | 110 |
| ral2Δ | 4.54 | 119 |
| cpp1Δ | 4.40 | 115 |
| efc25Δ | 4.41 | 115 |
| myo52Δ | 5.36 | 141 |
| scd2Δ | 4.70 | 123 |
| ras1Δ | 4.48 | 118 |
| scd1Δ | 5.28 | 139 |
Cells were grown in EMM4S + 1.2 M sorbitol for at least two generations. The % wild-type width compares the width of the mutant strain grown in sorbitol to the width of the wild-type strain grown in sorbitol.
Wide mutants can polarize growth
Among the 11 wide mutants, six (scd1Δ, scd2Δ, ral2Δ, efc25Δ, ras1Δ, cpp1Δ) had been previously described as spherical, implying they were depolarized (Fukui and Yamamoto, 1988; Chang et al., 1994; Tratner et al., 1997; Yang et al., 2000). We were primarily interested in mutants that were wider than wild-type cells but still exhibited polarized growth, so we used three different assessments of growth polarity to test if the exemplary wide mutants scd1Δ and scd2Δ were truly spherical. As in the secondary screen, we grew these mutants in HU to arrest the cell cycle, and we found that these mutants are actually polarized and wide (Figure 2A). We grew all the wide mutants in HU; all of the wide mutants were able to polarize growth and none were spherical. To confirm that scd1Δ and scd2Δ cells were growing at the cell tips, we examined cell wall staining with the polysaccharide stain Blankophor, which selectively stains newly added cell wall. The scd1Δ cells were grown in HU to elongate cells, which facilitates the identification of cell tips. In both of these mutants the cell tips were stained more brightly than the rest of the cell (Figure 2B). Actin-patch localization is correlated with growth and endocytosis in fission yeast, as in many other eukaryotic cells (Knust, 2000; Evangelista et al., 2002; Engqvist-Goldstein and Drubin, 2003). To determine whether growth was still polarized in the wide mutants, we elongated cells by growth in HU and then stained for actin. As in wild-type cells, actin structures were enriched at the cell tips in scd1Δ and scd2Δ mutants (Figure 2C). This result also shows that actin organization is not grossly disrupted in either mutant. All three methods of assessing polarized growth show that scd1Δ and scd2Δ mutants, which had previously been described as spherical, are actually polarized and grow at the cell tips with an increased cell width.
FIGURE 2:

Wide mutants still exhibit polarized growth. (A) Wild-type, scd1Δ, and scd2Δ cells in exponential growth conditions (top) and grown in 11 mM HU for 5 h to elongate the cells (bottom). DIC microscopy. (B) Blankophor, which preferentially stains newly added cell wall, showed brighter staining at the cell tips in wild-type and scd1Δ and scd2Δ mutants. Images show single focal planes with inverted look-up tables (LUTs). Wild-type and scd2Δ cells are growing exponentially; scd1Δ cells are shown after 5 h growth in HU to elongate cells. (C) Actin patches, stained with Alexa Fluor 488 phalloidin, were enriched at cell tips in WT, scd1Δ, and scd2Δ cells grown in HU for 5 h to elongate the cells. Images are deconvolved maximum projections of the full cell width, with inverted LUTs. Scale bars: 5 μm.
Two independent pathways act to control cell width
Deletions of a Cdc42 GEF (Scd1) and its scaffold (Scd2), which are predicted to reduce cellular levels of Cdc42-GTP, and deletion of a Cdc42 GAP (Rga4), which is predicted to increase cellular levels of Cdc42-GTP, all produced wider cells. This paradoxical result can be explained if the GEF and GAP operate independently in different parts of the cell, as suggested by previous studies of the proteins' localizations. Scd2-green fluorescent protein (GFP) and overexpressed Scd1-GFP localize to growing cell tips (Sawin et al., 1999; Hirota et al., 2003), and Rga4-GFP forms puncta at the cell cortex and is excluded from the cell tips (Das et al., 2007; Tatebe et al., 2008). On the basis of localization and function, we hypothesized that these two sets of regulators were acting to control Cdc42 independently, and that their spatially separated localizations were important for cell width determination. We tested these possibilities through genetic epistasis analysis and retargeting.
Epistasis analysis predicts that proteins that function in parallel independent pathways should have additive genetic interactions, while proteins that function together, either in a linear pathway or in a complex, should not. To test the genetic relationships of the GEF, GAP, and scaffold, we made double mutants and measured their widths. When both the GAP Rga4 and the scaffold Scd2 were deleted, the cells were still polarized but wider than either of the single mutants (Figure 3, A and B). Similarly, the scd1Δ rga4Δ strain, lacking both the GEF and the GAP, was wider than any single mutant, supporting the hypothesis that the GEF and GAP work in parallel to control width (Figure 3, A and B). In fission yeast, there is another Cdc42 GEF, Gef1, that acts primarily in septum formation but has a partially overlapping function with Scd1, as evidenced by the synthetic lethality of these two deletions (Coll et al., 2003). The deletion mutant gef1Δ does not have an increased cell width (exponential cell width = 3.83 μm ± 0.25), but like scd1Δ and scd2Δ, it is additive with rga4Δ (Figure 3, A and B). This additive interaction may occur because Gef1 is also involved in the activation of Cdc42 at cell tips. Gef1, which normally localizes to the cell tips and septum (Coll et al., 2003), can activate inappropriate cell growth when it is mislocalized to cell sides in orb6 loss-of-function mutants (Das et al., 2009). In contrast to the additive interactions with rga4Δ, double mutant scd1Δ scd2Δ cells, with both the GEF and the scaffold deleted, resembled scd1Δ alone (Figure 3, A and B). The scaffold Scd2 is known to bind Scd1 and Cdc42 (Endo et al., 2003; Wheatley and Rittinger, 2005), so the primary effect of deleting scd2 may be the disruption of this complex (Endo et al., 2003). In this case, a nonadditive genetic interaction would be expected.
FIGURE 3:

At least two independent pathways act to control cell width. (A) Cell-width defects of double mutants scd2Δ rga4Δ and scd1Δ rga4Δ and gef1Δ rga4Δ are additive, but the scd1Δ scd2Δ double mutant resembles scd1Δ. Box-and-whiskers plot of cell widths for each strain in exponential growth. All differences are statistically significant by a Student's t test with p < 0.0001; at least 50 cells were measured for each genotype. (B) Exponentially growing cells, bright-field microscopy. Scale bars: 5 μm.
If Rga4 and Scd1/Scd2 act in parallel pathways controlling width, and if their cellular localizations are important to their functions, then their localizations should be independent of one another. To test this, we determined the localizations of Scd1, Scd2, and Rga4 by GFP-tagging the proteins in the endogenous genomic locations in wild-type and mutant cells. Rga4-3×GFP is primarily localized to the cell sides in wild-type, scd1Δ, and scd2Δ cells (Figure 4A). Neither Scd1-3×GFP nor Scd2-3×GFP showed dependence on Rga4 for their localization, as they were unchanged in the rga4Δ strain, localizing to the wider cell ends (Figure 4B). This comports well with the genetic epistasis experiments, which suggested Rga4 acts in a separate pathway from Scd1 and Scd2. This resolves the apparent paradox—deletion of both positive and negative regulators of Cdc42 can increase cell width, since the two proteins act in different locations.
FIGURE 4:

Both pathways can localize independently. (A) Rga4-3×GFP does not require Scd1 or Scd2 for localization to cell sides. (B) Neither Scd1-3×GFP nor Scd2-3×GFP is dependent on Rga4 for localization to cell tips. (C) Scd1-3×GFP is lost from the cell tips in scd2Δ. (D) Scd2-3×GFP is mislocalized in scd1Δ. Images for all are representative, deconvolved, single focal planes with inverted LUTs. Scale bars: 5 μm. Unless marked, cells were in exponential growth at the time of imaging. scd1Δ is also shown after growth for 5 h in HU to elongate the cells.
The deletions of scd1 and scd2 were not additive. To see whether this was because Scd2 was required for the localization of Scd1, we GFP-tagged the endogenous Scd1 in wild-type and scd2Δ cells. In wild-type cells, Scd1-3×GFP localized to the growing cell ends, as has been reported for the overexpressed protein (Hirota et al., 2003). In the scd2Δ mutant, the cell-end localization of Scd1-3×GFP was lost, and most of the protein was found in the cytoplasm and nucleus (Figure 4C), indicating that Scd2 is required for the localization of Scd1-3×GFP. Similarly, we found that Scd2-3×GFP is poorly localized to the cell tips in scd1Δ (Figure 4D), and we therefore conclude that these proteins are mutually dependent for localization. It had been previously suggested that Scd2 acts upstream of Scd1, since Scd2 overexpression could not rescue a scd1Δ mutant (Chang et al., 1994). However, our results indicate that the genetic relationship is not that simple—there is at least a mutual dependence for localization.
Cdc42-GTP distribution is altered in rga4Δ, scd1Δ, and scd2Δ mutants
The localization of activated Cdc42 at the membrane can be assessed using a GFP-tagged Cdc42/Rac-interactive binding (CRIB) domain (Brown et al., 1997), which acts as a marker for Cdc42-GTP in fission yeast (Tatebe et al., 2008). CRIB-GFP is enriched at cell tips and the septum, where Cdc42 is activated by Gef1 and Scd1. CRIB-GFP localization to cell tips is reduced in scd2Δ (Figure 5, A and B) and largely lost in scd1Δ (Figure 5A; similar to results from Tatebe et al., 2008). Quantitative linescan analysis of CRIB-GFP distribution at the tip of the cell revealed that the scd2Δ mutant has lower peak fluorescence than wild-type cells, and higher fluorescence at the cell sides (Figure 5B). This may be because Scd1 is no longer localized to cell tips in the scd2Δ mutant. In a gef1Δ mutant, linescan analysis showed that the localization of CRIB-GFP was not significantly altered from the wild-type distribution (Supplemental Figure S1), which indicates that Gef1 has a more minor role in activating Cdc42 at cell tips than does Scd2 or Scd1.
FIGURE 5:

Cdc42-GTP localization is altered in rga4Δ, scd2Δ, and scd1Δ cells. (A) CRIB-GFP in indicated genotypes. Images are single focal planes, shown with constant, inverted LUTs. (B) Quantification of the distribution of CRIB-GFP in wild-type, rga4Δ, and scd2Δ genetic backgrounds. Linescans were done on half tips, aligned by the cell center, as shown by the curved white line in the image. Plots show the average intensity of linescans along the cell tip in the indicated genetic backgrounds. Each line is the average of 50 linescans (25 total randomly selected cell tips) of each genotype. Error bars represent 95% confidence intervals. Scale bars: 5 μm.
The localization of Rga4-GFP to cell sides suggested that it might act as a boundary element to restrict the spread of Cdc42-GTP. To investigate this possibility, we analyzed the distribution of CRIB-GFP in rga4Δ cells. It has been previously shown that rga4Δ does not disrupt overall CRIB-GFP localization (Tatebe et al., 2008), but we used linescan analysis to investigate whether there was a quantitative difference in the distribution of Cdc42 within the cell tip in this mutant. There is some increase in the intensity of CRIB-GFP at the distal portion of the cell tip in the rga4Δ mutant, which is consistent with boundary element activity (Figure 5B).
For Rga4 and Scd1, localization is crucial for proper control of cell width
The genetically independent, mutually exclusive localizations of Rga4 and Scd1 led us to hypothesize that these proteins establish spatially separate domains that determine the width of the cell. If these domains of Cdc42 activation and inhibition are important, then the localization of Scd1 to the cell tip and Rga4 to the cell sides will be necessary for their functions in determining cell width. We tested this model by disrupting and then restoring the localizations of Scd1 and Rga4.
Scd2 is necessary for the localization of Scd1-3×GFP (Figure 4C) and is thought to act as a scaffold stabilizing the interaction between Scd1 and Cdc42, as well as that between Cdc42 and downstream effectors, including the essential kinase Pak1 (Endo et al., 2003). If the primary function of Scd2 is to localize Scd1, then the mislocalization of Scd1 in scd2Δ cells could be responsible for the increased width of this mutant. To test whether the localization of Scd1 is important for the control of cell width, we added an extra domain to target the protein to the cell tips in scd2Δ. We fused the N-terminal targeting domain of For3, which is sufficient for cell-tip localization (Martin and Chang, 2006), to Scd1 under an inducible promoter. This fusion protein, where the For3N domain replaces Scd2 as a tip-targeted membrane tether, localized to the cell tips even in the scd2Δ strain, while the induced Scd1-GFP was located diffusely in the cytoplasm and in the nucleus (Figure 6A). For3N-Scd1-GFP complemented the cell-width phenotype of scd2Δ, but Scd1-GFP did not (Figure 6B). Overexpression of For3N alone did not affect the width of the scd2Δ mutant (unpublished data). The effect of the fusion protein is not due to increased protein levels, because For3N-Scd1 was not expressed at higher levels than Scd1-GFP (Figure S2A). Therefore tip-localized Scd1 acts to set the normal width of the cell whether it is localized there by Scd2 or by an artificial fusion with For3N. In budding yeast, the deletion of the Cdc42-scaffold homologue of Scd2, Bem1p, can be complemented by a fusion of the GEF to a downstream kinase (Kozubowski et al., 2008). Numerous fusion proteins in that study showed that the connection between the GEF and the kinase was an essential part of the function of the scaffold Bem1. However, in our case, we have rescued the width defect of scd2Δ simply by restoring the localization of the GEF Scd1 to the cell tips, without restoring the interaction of Scd1 with the homologous essential kinase Pak1.
FIGURE 6:

Retargeting of Scd1 to the cell tips can complement scd2Δ. (A) For3N-Scd1 fusion protein is targeted to cell tips. Images show scd2Δ cells expressing For3N-Scd1-GFP or Scd1-GFP, as indicated, induced from the nmt41 promoter for 15 h. Images are deconvolved single focal planes, with inverted LUTs. Scale bars: 5 μm. (B) Box-and-whiskers plot of cell width in the same strains, under the same expression conditions. (C) Linescan analysis of Pak1-mCherry distribution in WT, scd2Δ, and scd2Δ expressing For3N-scd1-GFP. Plots show the average intensity of linescans along the cell tip in the indicated genetic backgrounds. Each line is the average of 50 linescans (25 total randomly selected cell tips) of each genotype. Error bars represent 95% confidence intervals.
Pak1 is one of the major downstream effectors of Cdc42, and interacts with both activated Cdc42 (via a CRIB domain) and Scd2 (Endo et al., 2003). These interactions localize Pak1, which is also known as Shk1, to growing cell tips and the cell septum (Qyang et al., 2002). In a two-hybrid system, Scd2 overexpression increases Pak1 binding to Cdc42 (Chang et al., 1999), but in vitro Scd2 and Pak1 compete for binding to Cdc42-GTP, with Pak1 binding more tightly (Endo et al., 2003). To test how well the targeting of Scd1 to the cell tip restored the functional interactions of activated Cdc42 with effectors, we used linescans to analyze the localization of Pak1-mCherry in wild-type cells, the scd2Δ mutant, and scd2Δ mutant with the overexpressed For3N-Scd1-GFP (Figure 6C). Perhaps due to the loss of the interaction with Scd2, Pak1-mCherry levels at the cell tips of scd2Δ cells are even more reduced than CRIB-GFP levels. However, retargeting Scd1 to the cell tips restored Pak1 localization to cell tips (Figure 6C). This restoration of Pak1 localization reflects a restoration of activated Cdc42 at cell tips.
To parallel the Scd1 loss of localization in scd2Δ, we expressed a version of Rga4 that localizes to the cytoplasm (Tatebe et al., 2008). This cytoplasmic Rga4 (cytoRga4) has an internal deletion of 120 amino acids that mediates localization to the cell periphery. cytoRga4 only partially rescued the rga4Δ phenotype when expressed either under the endogenous rga4 promoter or at higher levels under the inducible nmt41 promoter (Figure 7B). When overexpressed, cytoRga4-GFP also formed aggregates in some cells, usually one to two per cell, but when cytoRga4 was expressed as the sole copy from the endogenous promoter, it did not form aggregates, and the cells were still wider than wild-type cells. The inability to fully correct the deletion phenotype could be due to mislocalization or impaired protein function. To test whether mislocalization was the primary defect, we relocalized cytoRga4 back to the cell sides by fusion with the cortically localized protein Blt1 (Moseley et al., 2009). Blt1-cytoRga4-GFP localized throughout the cell periphery, including to the cell sides and cell tips (Figure 7A). Despite this broader localization, Blt1-Rga4-GFP fully corrected the width phenotype of rga4Δ (Figure 7B). When Rga4 is overexpressed, it makes cells thinner (Das et al., 2007), so differences in expression level could potentially cause differential effects on cell width, but cytoRga4 is expressed at higher levels than Blt1-cytoRga4, as measured by Western blotting (Figure S2B). When Blt1 was expressed alone, it actually slightly increased the width of rga4Δ cells (unpublished data) and so is not responsible for the decrease in cell width by the fusion protein. We conclude that the localization of Rga4 to the cell cortex is important for its function, and like Scd1, that Rga4 cannot influence the width of the cell unless it is localized to the cell membrane.
FIGURE 7:
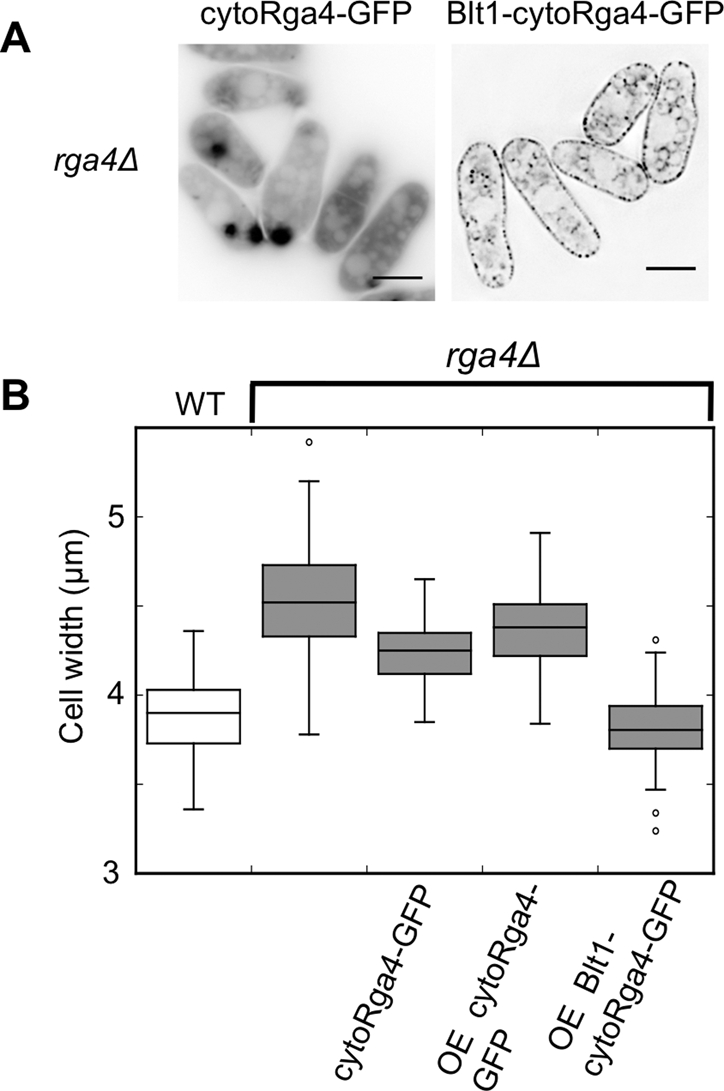
Proper localization of Rga4 is crucial for width control. (A) Blt1-cytoRga4 is relocalized to the cell cortex. Fluorescence images of GFP-tagged cytoRga4 and Blt1-cytoRga4 as indicated after induction for 18 h. For localization of the full-length Rga4-GFP, see Figure 4A. All images are deconvolved single planes, with inverted LUTs. Scale bars: 5 μm. (B) Box-and-whiskers plot of the cell widths of the indicated strains. Only the wild-type strain retains full-length Rga4 in the endogenous location.
Scd1 and Scd2 are dependent on actin for localization, although Rga4 is not
To test the role of actin and microtubules in determining the localization of these Cdc42 regulators, we incubated cells in either 50 μg/ml carbendazim (MBC) to inhibit microtubule polymerization or 100 μM latrunculin A (LatA) to depolymerize actin. After 1 h of growth in MBC, both Scd1-3×GFP and Scd2-3×GFP were still localized to cell tips (Figure 8A). In contrast, treatment with 100 μM LatA caused Scd1-3×GFP to become diffuse throughout the cytoplasm within 10 min and Scd2-3×GFP to move away from the cell tips within 30 min (Figure 8A). Similar results were previously noted for Scd2-GFP after treatment with MBC and thiabendazole (TBZ), but the broader spectrum of defects associated with TBZ made these experiments more difficult to interpret (Sawin and Snaith, 2004). These results clearly show that, in addition to being mutually dependent, both Scd1-3×GFP and Scd2-3×GFP are dependent on actin, although not microtubules, for their localization. In contrast, localization of fluorescently tagged Rga4 is not affected by the inhibition of actin polymerization (Das et al., 2007), microtubule polymerization, or even inhibition of both at the same time (Figure 8B).
FIGURE 8:
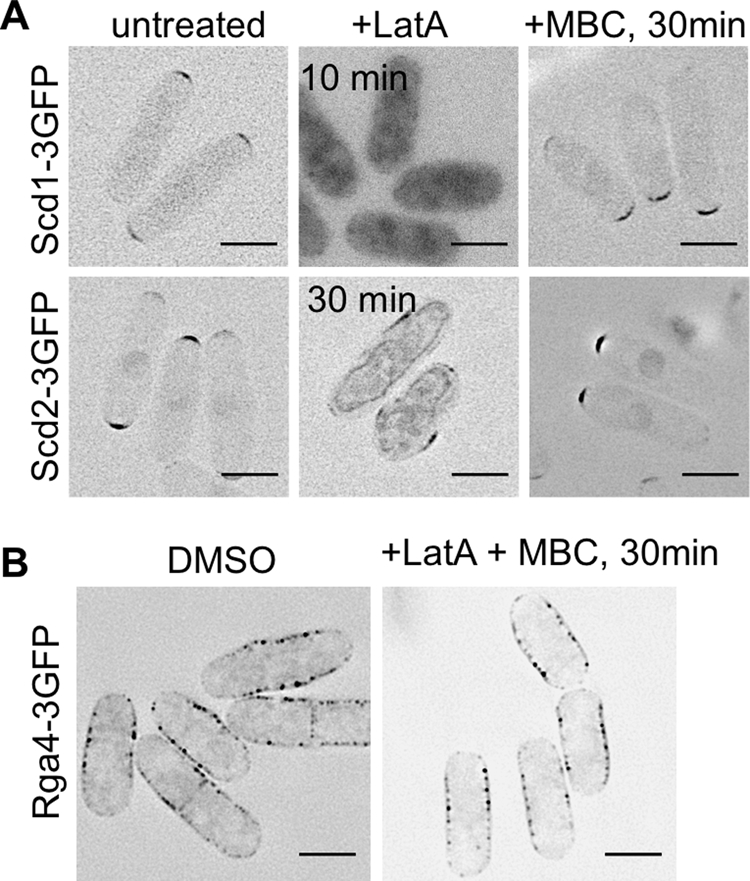
Scd1 and Scd2, but not Rga4, are dependent on actin for localization. (A) Scd1-3×GFP and Scd2-3×GFP in DMSO, 100 μM Latrunculin A to depolymerize actin, or 50 μg/ml carbendazim (MBC). Images are deconvolved, single focal planes with inverted LUTs. (B) Rga4-3×GFP in DMSO or 100 μM LatA and 50 μg/ml MBC. Images are representative, deconvolved sums of the fluorescence across the middle 1.2 μm of the cell with inverted LUTs. Scale bars: 5 μm.
Partial disruption of actin can increase cell width
In fission yeast, Cdc42 controls actin-cable polarization, at least in part through its regulation of the cable-promoting formin For3. Severe actin-cable defects are evident in temperature-sensitive mutants of Cdc42 that disrupt the cell-tip localization of For3 (Martin et al., 2007; Rincon et al., 2009). To investigate whether actin organization affects cell width, we treated cells with a range of concentrations of the actin-depolymerizing drug LatA. At a concentration of 10 μM LatA, which is 10-fold less than what is used to completely depolymerize actin, actin cables were largely depolymerized, but there were no visible effects on the more resistant actin patches assessed by phalloidin staining (unpublished data) and the localization of a GFP-tagged calponin-homology domain (CHD; Martin and Chang, 2006), as shown in Figure 9A. Higher levels of LatA blocked growth, but at a concentration of 10 μM, cells continued exponential growth for about 2 h (2.3 h generation time in 10 μM LatA vs. 2.2 h in the control culture). Within 1 h of 10 μM LatA addition, cells became 0.5 μm wider (Figure 9B).
FIGURE 9:

Partial disruption of actin increases cell width. (A) Cells treated with 10 μM LatA have reduced actin cables while retaining polarized actin patches, as shown by CHD-GFP binding to actin in growing cells. Top, images are deconvolved maximum sums across the cell width, with inverted LUTs. Bottom, bright-field images of growing cells, which are wider in 10 μM LatA. (B) Cell width is increased after 1 h of growth in 10 μM LatA. Box-and-whiskers plot, with 50 cells measured for each condition, p < 0.001. (C) Low-level LatA effects on the localization of Scd1, Scd2, and CRIB. Images are deconvolved, single focal planes with inverted LUTs. (D) Low-level LatA reduces CRIB-GFP concentration at the cell tips. Linescan quantification of the distribution of CRIB-3×GFP with and without low-level LatA. Plots show the average intensity of linescans along the cell tip. The scans were done on half tips, aligned by the cell center. Each line is the average of 50 linescans (25 total cell tips) of each genotype. Scale bars: 5 μm.
Although Scd1 and Scd2 were delocalized upon complete depolymerization of actin (Figure 8A), they still localized to cell tips in 10 μM LatA (Figure 9C). Therefore low-level LatA affected the width of the cell without affecting the localization of these Cdc42 regulators. To see whether there might be a resultant change in Cdc42 activation, we looked at the localization of CRIB-GFP (a marker for activated Cdc42) in 10 μM LatA by linescan analysis. This analysis showed that the level of CRIB-GFP at the cell tip is significantly reduced after 1 h of incubation in 10 μM LatA, though the level at the cell sides is not increased (Figure 9D). We conclude that actin-cable disruption increases cell width and reduces Cdc42 activation at cell tips. This nongenetic manipulation of localized Cdc42 activation forms another link between the precise spatial control of Cdc42 localization and the control of cell width. When activated Cdc42 is not concentrated at cell tips, or is inappropriately activated at the cell sides, the fission yeast cell becomes wider.
DISCUSSION
Cell-width control is distinct from growth polarization
Cell width in fission yeast is determined in a two-stage process. Polarization is the first stage, and is an on–off switch—either growth is polarized or it is not. The second stage is the determination of the size of the growth zone, and the near-genome-wide screen in this study was designed to identify the genes important for this second stage. The size of the tip-localized growth zone will determine the width of the cell, so we screened for mutants that are wider than wild-type, but still polarized. Even the widest mutants we have identified can polarize growth, as shown by actin-patch distribution, newly growing cell wall staining, and cell shape.
Polarization and growth-zone size are controlled by the Cdc42 regulatory network
Of the genes found in this screen, seven of 11 are involved in the control of Cdc42 activation, establishing its importance in determining growth-zone size. Cdc42 has been previously shown to be important in growth polarization, but this study provides an additional role for the Cdc42 regulatory network. In fission yeast, cdc42 is essential and spores that lack the gene cannot form germination tubes, because they cannot polarize growth (Miller and Johnson, 1994). In addition, mutations in the essential kinase Orb6 lead to the mislocalization of Cdc42 to cell sides (Das et al., 2009), and render cells round (Verde et al., 1998). But we have shown here that even when the basic shape of the cell is polarized, the spatial control of Cdc42 affects the width of the growth zone.
Using CRIB-GFP as a marker for activated Cdc42, we have quantitatively shown that there is a gradient of activation, with maximum activation near the center of the cell tip. Deletion of either the GEF scd1 or the scaffold scd2 reduces the peak of Cdc42 activation, and makes cells wider. Scd1, Scd2, and Cdc42 form a complex at the cell tip (Endo et al., 2003), and this complex may act to concentrate and focus growth by stabilizing activated Cdc42. We propose that Cdc42 can still be activated at the cell tip when the complex is disrupted by deletion of scd2, but that activation is not as well focused, as evidenced by the localization of the CRIB-GFP in scd2Δ mutant (Figure 5, A and B). We speculate that when there is not as much activated Cdc42 focused at the cell tips, the overall rate of cell growth will be similar, but directed addition at the center of the cell width will not be as efficient, and as a consequence growth will occur over a wider area, resulting in a wider cell. Scd1 regulates cell width through its localized activity at cell tips, which results in localized activation of Cdc42 (Figure 6, A and B). When scd1 or scd2 is deleted Rga4 becomes a major determinant of cell width, as demonstrated by the additive effects of rga4Δ scd1Δ and rga4Δ scd2Δ. The role of Rga4 may be to form a boundary preventing the spread of activated Cdc42 away from the cell tip. When Scd2 and Scd1 are present, Rga4 on the cell sides may be responsible for fine-tuning the boundaries of Cdc42 activation, since activated Cdc42 is distributed throughout the now-wider cell tip of rga4Δ cells (Figure 5), and cytoplasmic Rga4 cannot set the cell width (Figure 7).
Actin-based delivery may play a role in Cdc42 recruitment to the cell end
Our results indicate a role for actin polarization in determining the width of the fission yeast cell. Actin is essential for normal cell growth (Ishijima et al., 1999), and disruption of actin-cable formation and exocyst localization can result in isotropic growth (Bendezu and Martin, 2011). Both actin-cable formation and proper exocyst localization are dependent on the essential Cdc42 effector Pob1 (Nakano et al., 2011), and these two systems seem partially redundant for maintaining polarized growth. Though actin organization was not previously linked directly to cell-width control, we found that addition of 10 μM LatA caused cells to increase in width, with major actin-cable defects evidenced by the localization of a GFP-tagged calponin homology domain. Deletion of the S. pombe formin for3 (Feierbach and Chang, 2001) increases the width of the cell only slightly (for3Δ cell width is 4.1 μm ± 0.25, compared with 3.9 μm ± 0.15 for wild-type cells). Treatment with 10 μM LatA is probably a more severe actin disruption, and is eventually lethal, while the for3Δ mutant is viable. Activated Cdc42 is still localized to the cell tips when cells are grown in low-level LatA (Figure 9D), but the level at the cell tip is reduced. The low-level LatA effect on cell width could be acting through disruption of localized Cdc42 activation, and the higher-level LatA disruption of growth and Cdc42 localization (Bendezu and Martin, 2010) could occur via disruption of delivery and recruitment of Scd1 and Scd2. Both Scd1 and Scd2 are delocalized after the complete disruption of actin cables and patches, but not after actin-cable disruption, implying that they may be dependent on actin patches, but not cables, for their localizations.
In this study, we show that Scd1 and Scd2 are mutually dependent for localization, and that both mutants are somehow defective in concentrating activated Cdc42 at the site of growth. This mutual dependence implies a cooperative system, where Scd1, stabilized by Scd2, activates Cdc42, which then stabilizes the complex, leading to an accumulation of both Scd1 and Scd2. The interaction between the homologous budding yeast scaffold protein Bem1 and the GEF Cdc24 is necessary for the localization of Cdc24, but not for the localization of Bem1, since a mutant version of Bem1 that does not interact with Cdc24 can still localize to the bud neck and the bud cortex (Butty et al., 2002). However, localization of Bem1 is dependent on Cdc42 activation, and our results do not allow us to determine whether the loss of Scd2 from the tips of scd1Δ cells is due to the reduced levels of Cdc42-GTP or because of a direct interaction between Scd1 and Scd2 that is lost.
Domains of activation and inhibition of Cdc42 may be conserved
The small GTPase Cdc42 and its regulation by GEFs and GAPs are conserved from yeasts to humans. There are metazoan parallels for the role of GAPs and GEFs in determining Cdc42 localization, and perhaps for their role in controlling cellular dimensions. In Caenorhabditis elegans development, at the single-cell stage, the GAP CHIN-1 is required for the polarized distribution of Cdc42 on the membrane, and CHIN-1 is localized to the end of the embryo opposite activated Cdc42 (Kumfer et al., 2010). In the eight-cell C. elegans embryo, the GAP PAC-1 is restricted to inner surfaces of the embryo and is required for the exclusion of Cdc42 from those inner surfaces, even though the embryo is properly formed at that point (Anderson et al., 2008). Like the GEF Scd1 and the scaffold Scd2, the Cdc42 GEF βPIX is important in fibroblasts for the formation of leading-edge protrusions after wound healing, and the scaffold Scrib mediates βPIX localization to the leading edge (Osmani et al., 2006). These parallels with metazoa show that a system of a GEF, a scaffold, and a GAP controlling the localization of Cdc42 activation, and therefore growth, is conserved in a range of eukaryotic organisms, and so our model for determining the size of cellular growth zones in fission yeast may also be relevant to metazoan cells.
MATERIALS AND METHODS
Yeast strains and growth conditions
Yeast strains were grown as previously described (Moreno et al., 1991). For all experiments, cells were grown in Edinburgh Minimal Media (EMM) with supplements at 32°C, unless specified. For microscopy with fluorescent proteins, media was sterilized by filtration to reduce autofluorescence.
All strains used in this study are listed in the supplemental strain table (Table S2). Deletion strain genotypes were verified by colony PCR across the kanamycin resistance cassette. The scd1 gene was not completely deleted in the strain contained in the deletion collection, so this strain was reconstructed. The deletion of scd1 and GFP-tagging of other proteins was performed by homologous recombination as described (Bahler et al., 1998). Strains expressing GFP- and 3×GFP-tagged proteins showed growth rate, cell size, and cell width similar to untagged strains. All other strains were constructed by mating and tetrad dissection, where applicable. The deletion mutants scd1Δ and scd2Δ are sterile. To circumvent the sterility defects of the scd1Δ and scd2Δ strains, the mutants were complemented by transformation with a plasmid that contained the deleted gene under control of the high-level nmt1 promoter. The plasmid also contained the selectable marker LEU2, and was maintained by growth on selective minimal media (EMM-leu). After crossing the strains, dissecting tetrads, and selecting double mutants based on antibiotic resistance, we screened the double mutants by growing them on selective plates, and those that no longer grew on EMM-leu, and therefore had lost the complementing plasmid, were selected for further analyses.
Strains containing genes under the control of the thiamine-repressible nmt41 promoter were grown in the presence of 5 μg/ml thiamine. To induce expression, we washed cells three times by filtration with EMM4S, resuspended them in EMM4S, and grew them for the number of hours noted in the text, after which the cells were measured and photographed. The expression of the CHD-GFP construct was induced for 16 h prior to the addition of LatA, then LatA was added, and cells were imaged 1 h later, for 17 h of total induction.
Screen for wide mutants
The primary genetic screen was performed as previously described (Kim et al., 2010), by sporulating diploid heterozygous deletions and observing the morphology of the haploid mutants following germination. Secondary and tertiary screening was performed by observing growth of the haploid deletions identified in the primary screen. For the secondary screen measurements in HU, cells were grown for 5 h in EMM4S + 11 mM HU (Sigma-Aldrich, St. Louis, MO) from a 1 M stock in distilled water and photographed live. For the tertiary screen measurements of exponentially growing cells, cells were grown in liquid culture until midexponential-phase growth and then photographed live. For cell-width measurements and differential interference contrast (DIC) photographs, cells were imaged using an epifluorescence microscope (Axioplan 2, Carl Zeiss, Thornwood, NY) equipped with an αPlan Fluor 100× numerical aperture (NA) 1.4, oil objective (Zeiss) and a CoolSNAP HQ camera (Roper Scientific, Tucson, AZ), and analyzed in MetaMorph (MDS Analytical Technologies, Sunnyvale, CA) and ImageJ. Width measurements were made from photographs using the line tool in ImageJ. Cell width was measured on the cell body, approximately 2 μm from the cell tip. For width measurements in sorbitol, cells were grown exponentially in EMM4S + 1.2 M sorbitol for at least two generations.
Other microscopy and cytoskeletal analysis
Actin patches and cables were visualized by staining fixed cells with Alexa Fluor 488 phalloidin (Invitrogen, Carlsbad, CA) and imaging them on a DeltaVision Spectris (Applied Precision, Issaquah, WA) comprising an Olympus IX71 wide-field inverted fluorescence microscope, an Olympus UPlanSApo 100×, N.A. 1.4, oil immersion objective, and a Photometrics CoolSNAP HQ camera (Roper Scientific). Images were captured, processed by iterative constrained deconvolution using SoftWoRx (Applied Precision) where noted, and analyzed in ImageJ. This microscope setup was used for all of the fluorescent protein live-cell imaging, except in Figure 2B, where the previously described Zeiss microscope was used, and images were not deconvolved. In Figure 5, A and B, images were not deconvolved. To stain growing cell wall (Figure 2B), we added Blankophor (Frank, 1991) to the media (1:100,000 dilution; MP Biomedicals, Solon, OH).
For the CRIB-GFP and Pak1-mCherry linescan analyses, a series of images were taken spanning the width of the cell, and the image in the Z-stack with the highest intensity was chosen. In ImageJ, a segmented line was drawn along the cell edge from the center of the tip of the cell, following the cell edge, to ∼1 μm past the edge of the tip. The line was 0.3 μm wide and drawn with a spline fit. The intensity profile of the line was then plotted, and 50 of these profiles were averaged for each genotype.
Actin was disrupted by addition of either 10 or 100 μM LatA (Sigma) from a 1 mM stock. Microtubules were disrupted by treatment with MBC (Sigma) at a final concentration of 50 μg/ml, from a 1 mg/ml stock in dimethyl sulfoxide (DMSO).
Fusion protein construction and assessment
Fusion proteins and the control proteins under the control of the nmt41 promoter were constructed as follows. To GFP-tag scd1 and drive its expression under the control of the nmt41 promoter, we amplified the scd1 ORF from the genome with primers that contained a Sal1 (F) and an Xma1 (R) site. The PCR fragment and the vector pREP41 carrying a C-terminal enhanced GFP (eGFP) tag (Craven et al., 1998) were digested with Sal1 and Xma1 and ligated together to produce nmt41P-scd1-GFP-nmt41T (plasmid ARC2077). This construct was then moved into the integration vector pJK148 by amplification with primers for the nmt41 promoter- and terminator-containing restriction enzyme sites for Eag1 and Cla1, respectively and then ligated into the same sites of pJK148, creating plasmid ARC2078. The plasmid was then integrated into the leu1 locus as previously described (Keeney and Boeke, 1994). We made the For3N-Scd1 fusion by digesting the plasmid ARC2077 with Sal1 at the Scd1 N-terminus. The first 2106 nucleotides of for3 were amplified out of the genome using primers with homology to the sequences flanking the Sal1 cut site, and then the PCR product and digested vectors were ligated using the In-Fusion PCR Cloning Kit (Clontech, Mountain View, CA) to produce nmt41P-for3N-scd1-GFP-nmt41T (plasmid ARC2079). This construct was then subcloned into the integration vector pJK210 at the Eag1 site using the In-Fusion PCR Cloning Kit (Clontech), as described above to make plasmid ARC2080, and then integrated at ura4-294 (Keeney and Boeke, 1994).
To GFP-tag cytoRga4 and drive the expression under the control of the nmt41 promoter, we amplified cytoRga4 from the genome of the strain CA5544 (Tatebe et al., 2008) with primers that contained an Nde1 (F) and an Xma1 (R) site. The PCR fragment and the vector pREP41 carrying a C-terminal eGFP tag (Craven et al., 1998) were also digested with Nde1 and Xma1 and ligated together to produce nmt41P-cytoRga4-GFP-nmt41T (plasmid ARC2081). This construct was then moved into the integration vector pJK148 by amplification with primers that anneal at the nmt41 promoter and terminator and contain restriction enzyme sites for Eag1 and Cla1, respectively, and then ligated into the same sites of to create plasmid ARC2082. The plasmid was then integrated into the leu1 locus as described (Keeney and Boeke, 1994).
To generate nmt41P-blt1-cytoRga4-GFP-nmt41T, we added Nde1 and BamHI sites between the nmt41 promoter and cytoRga4 in plasmid ARC2081, and the blt1 open reading frame without stop codon was amplified by PCR using primers carrying NdeI and BamHI sites, digested, and inserted into the nmt41-cytoRga4 plasmid digested with the same enzymes. This created the plasmid ARC2083. The construct was then subcloned into pJK148, as described above, to create ARC 2084. This plasmid was then integrated into the genome in the leu1 locus as previously described (Keeney and Boeke, 1994).
The levels of the fusion proteins were assessed by a standard Western blotting procedure. Whole-protein extracts were obtained from ∼108 cells by rapid bead-beating in HB lysis buffer (100 mM NaCl, 25 mM MOPS, pH 7.5, 15 mM MgCl, 15 mM ethylene glycol tetraacetic acid, 1 mM dithiothreitol, 1% Triton X-100, complete protease inhibitor [Roche, Indianapolis, IN]). Western blots were probed with an anti-GFP antibody (Cristea et al., 2005) to assess fusion protein level and an anti-tubulin antibody to assess protein loading (anti-Tat1, gift of K. Gull, University of Oxford, UK).
Supplementary Material
Acknowledgments
We thank J. Hayles for sharing unpublished data on her characterizations of the deletion collection and for her thoughtful comments on the manuscript; the members of the Nurse Lab for critical readings of the manuscript; the Rockefeller University Bioimaging Resource Center for assistance with image collection and analysis; K. Shiozaki for the CRIB-GFP and cytoRga4 strains; and P. Perez for additional strains. This study was supported by the Breast Cancer Research Foundation, the Anderson Center for Cancer Research, and Rockefeller University.
Abbreviations used:
- CRIB
Cdc42/Rac-interactive binding
- cytoRga4
cytoplasmic Rga4
- DIC
differential interference contrast
- DMSO
dimethyl sulfoxide
- EMM
Edinburgh Minimal Media
- GAP
GTPase-activating protein
- GEF
guanine nucleotide exchange factor
- GFP
green fluorescent protein
- HU
hydroxyurea
- LatA
latrunculin A
- LUT
look-up table
- MBC
carbendazim
- NA
numerical aperture
- TBZ
thiabendazole
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E11-01-0057) on August 17, 2011.
REFERENCES
- Adams AE, Johnson DI, Longnecker RM, Sloat BF, Pringle JR. CDC42 and CDC43, two additional genes involved in budding and the establishment of cell polarity in the yeast Saccharomyces cerevisiae. J Cell Biol. 1990;111:131–142. doi: 10.1083/jcb.111.1.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson DC, Gill JS, Cinalli RM, Nance J. Polarization of the C. elegans embryo by RhoGAP-mediated exclusion of PAR-6 from cell contacts. Science. 2008;320:1771–1774. doi: 10.1126/science.1156063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahler J, Wu JQ, Shah NG, McKenzie A III, Steever AB, Wach A, Philippsen P, Pringle JR. Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast. 1998;14:943–951. doi: 10.1002/(SICI)1097-0061(199807)14:10<943::AID-YEA292>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Bendezu FO, Martin SG. Actin cables and the exocyst form two independent morphogenesis pathways in the fission yeast. Mol Biol Cell. 2011;22:44–53. doi: 10.1091/mbc.E10-08-0720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown JL, Jaquenoud M, Gulli MP, Chant J, Peter M. Novel Cdc42-binding proteins Gic1 and Gic2 control cell polarity in yeast. Genes Dev. 1997;11:2972–2982. doi: 10.1101/gad.11.22.2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butty AC, Perrinjaquet N, Petit A, Jaquenoud M, Segall JE, Hofmann K, Zwahlen C, Peter M. A positive feedback loop stabilizes the guanine-nucleotide exchange factor Cdc24 at sites of polarization. EMBO J. 2002;21:1565–1576. doi: 10.1093/emboj/21.7.1565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang E, Bartholomeusz G, Pimental R, Chen J, Lai H, Wang L-hL, Yang P, Marcus S. Direct binding and in vivo regulation of the fission yeast p21-activated kinase Shk1 by the SH3 domain protein Scd2. Mol Cell Biol. 1999;19:8066–8074. doi: 10.1128/mcb.19.12.8066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang EC, Barr M, Wang Y, Jung V, Xu H-P, Wigler MH. Cooperative interaction of S. pombe proteins required for mating and morphogenesis. Cell. 1994;79:131–141. doi: 10.1016/0092-8674(94)90406-5. [DOI] [PubMed] [Google Scholar]
- Coll PM, Trillo Y, Ametzazurra A, Perez P. Gef1p, a new guanine nucleotide exchange factor for Cdc42p, regulates polarity in Schizosaccharomyces pombe. Mol Biol Cell. 2003;14:313–323. doi: 10.1091/mbc.E02-07-0400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craven RA, Griffiths DJ, Sheldrick KS, Randall RE, Hagan IM, Carr AM. Vectors for the expression of tagged proteins in Schizosaccharomyces pombe. Gene. 1998;221:59–68. doi: 10.1016/s0378-1119(98)00434-x. [DOI] [PubMed] [Google Scholar]
- Cristea IM, Williams R, Chait BT, Rout MP. Fluorescent proteins as proteomic probes. Mol Cell Proteomics. 2005;4:1933–1941. doi: 10.1074/mcp.M500227-MCP200. [DOI] [PubMed] [Google Scholar]
- Das M, Wiley DJ, Chen X, Shah K, Verde F. The conserved NDR kinase Orb6 controls polarized cell growth by spatial regulation of the small GTPase Cdc42. Curr Biol. 2009;19:1314–1319. doi: 10.1016/j.cub.2009.06.057. [DOI] [PubMed] [Google Scholar]
- Das M, Wiley DJ, Medina S, Vincent HA, Larrea M, Oriolo A, Verde F. Regulation of cell diameter, For3p localization, and cell symmetry by fission yeast Rho-GAP Rga4p. Mol Biol Cell. 2007;18:2091–101. doi: 10.1091/mbc.E06-09-0883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo M, Shirouzu M, Yokoyama S. The Cdc42 binding and scaffolding activities of the fission yeast adaptor protein Scd2. J Biol Chem. 2003;278:843–852. doi: 10.1074/jbc.M209714200. [DOI] [PubMed] [Google Scholar]
- Engqvist-Goldstein AEY, Drubin DG. Actin assembly and endocytosis: from yeast to mammals. Annu Rev Cell Dev Biol. 2003;19:287–332. doi: 10.1146/annurev.cellbio.19.111401.093127. [DOI] [PubMed] [Google Scholar]
- Evangelista M, Pruyne D, Amberg DC, Boone C, Bretscher A. Formins direct Arp2/3-independent actin filament assembly to polarize cell growth in yeast. Nat Cell Biol. 2002;4:260–269. doi: 10.1038/ncb770. [DOI] [PubMed] [Google Scholar]
- Feierbach B, Chang F. Roles of the fission yeast formin for3p in cell polarity, actin cable formation and symmetric cell division. Curr Biol. 2001;11:1656–1665. doi: 10.1016/s0960-9822(01)00525-5. [DOI] [PubMed] [Google Scholar]
- Foethke D, Makushok T, Brunner D, Nedelec F. Force- and length-dependent catastrophe activities explain interphase microtubule organization in fission yeast. Mol Syst Biol. 2009;5:241. doi: 10.1038/msb.2008.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank V. The use of some fluorescent stains for studying morphogenesis of micromycetes. Folia Microbiol (Praha) 1991;36:92–96. doi: 10.1007/BF02935829. [DOI] [PubMed] [Google Scholar]
- Fukui Y, Miyake S, Satoh M, Yamamoto M. Characterization of the Schizosaccharomyces pombe ral2 gene implicated in activation of the ras1 gene product. Mol Cell Biol. 1989;9:5617–5622. doi: 10.1128/mcb.9.12.5617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukui Y, Yamamoto M. Isolation and characterization of Schizosaccharomyces pombe mutants phenotypically similar to ras1−. Mol Gen Genet. 1988;215:26–31. doi: 10.1007/BF00331298. [DOI] [PubMed] [Google Scholar]
- Harmouch N, Pichova A, Coulon J, Streiblova E, Bonaly R. Changes in cell wall composition of deformed ras1− cells of Schizosaccharomyces pombe. Folia Microbiol (Praha) 1995;40:519–527. doi: 10.1007/BF02814736. [DOI] [PubMed] [Google Scholar]
- Hayles J, Nurse P. A journey into space. Nat Cell Biol. 2001;2:647–656. doi: 10.1038/35089520. [DOI] [PubMed] [Google Scholar]
- Hirota K, Tanaka K, Ohta K, Yamamoto M. Gef1p and Scd1p, the two GDP-GTP exchange factors for Cdc42p, form a ring structure that shrinks during cytokinesis in Schizosaccharomyces pombe. Mol Biol Cell. 2003;14:3617–3627. doi: 10.1091/mbc.E02-10-0665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishijima SA, Konomi M, Takagi T, Sato M, Ishiguro J, Osumi M. Ultrastructure of cell wall of the cps8 actin mutant cell in Schizosaccharomyces pombe. FEMS Microbiol Lett. 1999;180:31–37. doi: 10.1111/j.1574-6968.1999.tb08774.x. [DOI] [PubMed] [Google Scholar]
- Keeney JB, Boeke JD. Efficient targeted integration at leu1–32 and ura4–294 in Schizosaccharomyces pombe. Genetics. 1994;136:849–856. doi: 10.1093/genetics/136.3.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim DU, et al. Analysis of a genome-wide set of gene deletions in the fission yeast Schizosaccharomyces pombe. Nat Biotechnol. 2010;28:617–623. doi: 10.1038/nbt.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knust E. Control of epithelial cell shape and polarity. Curr Opin Genet Dev. 2000;10:471–475. doi: 10.1016/s0959-437x(00)00115-5. [DOI] [PubMed] [Google Scholar]
- Kozubowski L, Saito K, Johnson JM, Howell AS, Zyla TR, Lew DJ. Symmetry-breaking polarization driven by a Cdc42p GEF-PAK complex. Curr Biol. 2008;18:1719–1726. doi: 10.1016/j.cub.2008.09.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumfer KT, Cook SJ, Squirrell JM, Eliceiri KW, Peel N, O'Connell KF, White JG. CGEF-1 and CHIN-1 regulate CDC-42 activity during asymmetric division in the Caenorhabditis elegans embryo. Mol Biol Cell. 2010;21:266–277. doi: 10.1091/mbc.E09-01-0060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechler T, Jonsdottir GA, Klee SK, Pellman D, Li R. A two-tiered mechanism by which Cdc42 controls the localization and activation of an Arp2/3-activating motor complex in yeast. J Cell Biol. 2001;155:261–270. doi: 10.1083/jcb.200104094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lew DJ, Reed SI. Morphogenesis in the yeast cell cycle: regulation by Cdc28 and cyclins. J Cell Biol. 1993;120:1305–1320. doi: 10.1083/jcb.120.6.1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linder T, Gustafsson CM. Molecular phylogenetics of ascomycotal adhesins—a novel family of putative cell-surface adhesive proteins in fission yeasts. Fungal Genet Biol. 2008;45:485–497. doi: 10.1016/j.fgb.2007.08.002. [DOI] [PubMed] [Google Scholar]
- Martin SG, Chang F. Dynamics of the formin For3p in actin cable assembly. Curr Biol. 2006;16:1161–1170. doi: 10.1016/j.cub.2006.04.040. [DOI] [PubMed] [Google Scholar]
- Martin SG, Rincon SA, Basu R, Perez P, Chang F. Regulation of the formin For3p by Cdc42p and Bud6p. Mol Biol Cell. 2007;18:4155–4167. doi: 10.1091/mbc.E07-02-0094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller PJ, Johnson DI. Cdc42 GTPase is involved in controlling polarized cell growth in Schizosaccharomyces pombe. Mol Cell Biol. 1994;14:1075–1083. doi: 10.1128/mcb.14.2.1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno S, Klar A, Nurse P. Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods Enzymol. 1991;194:795–823. doi: 10.1016/0076-6879(91)94059-l. [DOI] [PubMed] [Google Scholar]
- Moseley JB, Mayeux A, Paoletti A, Nurse P. A spatial gradient coordinates cell size and mitotic entry in fission yeast. Nature. 2009;459:857–860. doi: 10.1038/nature08074. [DOI] [PubMed] [Google Scholar]
- Nakano K, Toya M, Yoneda A, Asami Y, Yamashita A, Kamasawa N, Osumi M, Yamamoto M. Pob1 ensures cylindrical cell shape by coupling two distinct Rho signaling events during secretory vesicle targeting. Traffic. 2011;12:726–739. doi: 10.1111/j.1600-0854.2011.01190.x. [DOI] [PubMed] [Google Scholar]
- Nobes CD, Hall A. Rho GTPases control polarity, protrusion, and adhesion during cell movement. J Cell Biol. 1999;144:1235–1244. doi: 10.1083/jcb.144.6.1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osmani N, Vitale N, Borg JP, Etienne-Manneville S. Scrib controls Cdc42 localization and activity to promote cell polarization during astrocyte migration. Curr Biol. 2006;16:2395–2405. doi: 10.1016/j.cub.2006.10.026. [DOI] [PubMed] [Google Scholar]
- Papadaki P, Pizon V, Onken B, Chang EC. Two Ras pathways in fission yeast are differentially regulated by two Ras guanine nucleotide exchange factors. Mol Cell Biol. 2002;22:4598–4606. doi: 10.1128/MCB.22.13.4598-4606.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez P, Rincon SA. Rho GTPases: regulation of cell polarity and growth in yeasts. Biochem J. 2010;426:243–253. doi: 10.1042/BJ20091823. [DOI] [PubMed] [Google Scholar]
- Pruyne D, Gao L, Bi E, Bretscher A. Stable and dynamic axes of polarity use distinct formin isoforms in budding yeast. Mol Biol Cell. 2004a;15:4971–4989. doi: 10.1091/mbc.E04-04-0296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruyne D, Legesse-Miller A, Gao L, Dong Y, Bretscher A. Mechanisms of polarized growth and organelle segregation in yeast. Annu Rev Cell Dev Biol. 2004b;20:559–591. doi: 10.1146/annurev.cellbio.20.010403.103108. [DOI] [PubMed] [Google Scholar]
- Qyang Y, Yang P, Du H, Lai H, Kim H, Marcus S. The p21-activated kinase, Shk1, is required for proper regulation of microtubule dynamics in the fission yeast, Schizosaccharomyces pombe. Mol Microbiol. 2002;44:325–334. doi: 10.1046/j.1365-2958.2002.02882.x. [DOI] [PubMed] [Google Scholar]
- Ribas JC, Diaz M, Duran A, Perez P. Isolation and characterization of Schizosaccharomyces pombe mutants defective in cell wall (1-3)β-D-glucan. J Bacteriol. 1991;173:3456–3462. doi: 10.1128/jb.173.11.3456-3462.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rincon SA, Ye Y, Villar-Tajadura MA, Santos B, Martin SG, Perez P. Pob1 participates in the Cdc42 regulation of fission yeast actin cytoskeleton. Mol Biol Cell. 2009;20:4390–4399. doi: 10.1091/mbc.E09-03-0207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawin KE, Nasser Hajibagheri MA, Nurse P. Mis-specification of cortical identity in a fission yeast PAK mutant. Curr Biol. 1999;9:1335–1338. doi: 10.1016/s0960-9822(00)80058-5. [DOI] [PubMed] [Google Scholar]
- Sawin KE, Snaith HA. Role of microtubules and tea1p in establishment and maintenance of fission yeast cell polarity. J Cell Sci. 2004;117:689–700. doi: 10.1242/jcs.00925. [DOI] [PubMed] [Google Scholar]
- Sazer S, Sherwood SW. Mitochondrial growth and DNA synthesis occur in the absence of nuclear DNA replication in fission yeast. J Cell Sci. 1990;97:509–516. doi: 10.1242/jcs.97.3.509. [DOI] [PubMed] [Google Scholar]
- St Johnston D, Ahringer J. Cell polarity in eggs and epithelia: parallels and diversity. Cell. 2010;141:757–774. doi: 10.1016/j.cell.2010.05.011. [DOI] [PubMed] [Google Scholar]
- Tatebe H, Nakano K, Maximo R, Shiozaki K. Pom1 DYRK regulates localization of the Rga4 GAP to ensure bipolar activation of Cdc42 in fission yeast. Curr Biol. 2008;18:322–330. doi: 10.1016/j.cub.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tratner I, Fourticq-Esqueoute A, Tillit J, Baldacci G. Cloning and characterization of the S. pombe gene efc25+, a new putative guanine nucleotide exchange factor. Gene. 1997;193:203–210. doi: 10.1016/s0378-1119(97)00115-7. [DOI] [PubMed] [Google Scholar]
- Verde F, Wiley DJ, Nurse P. Fission yeast orb6, a ser/thr protein kinase related to mammalian rho kinase and myotonic dystrophy kinase, is required for maintenance of cell polarity and coordinates cell morphogenesis with the cell cycle. Proc Natl Acad Sci USA. 1998;95:7526–7531. doi: 10.1073/pnas.95.13.7526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villar-Tajadura MA, Coll PM, Madrid M, Cansado J, Santos B, Perez P. Rga2 is a Rho2 GAP that regulates morphogenesis and cell integrity in S. pombe. Mol Microbiol. 2008;70:867–881. doi: 10.1111/j.1365-2958.2008.06447.x. [DOI] [PubMed] [Google Scholar]
- Wheatley E, Rittinger K. Interactions between Cdc42 and the scaffold protein Scd2: requirement of SH3 domains for GTPase binding. Biochem J. 2005;388:177–184. doi: 10.1042/BJ20041838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Urano J, Tamanoi F. Protein farnesylation is critical for maintaining normal cell morphology and canavanine resistance in Schizosaccharomyces pombe. J Biol Chem. 2000;275:429–438. doi: 10.1074/jbc.275.1.429. [DOI] [PubMed] [Google Scholar]
- Zhang X, Bi E, Novick P, Du L, Kozminski KG, Lipschutz JH, Guo W. Cdc42 interacts with the exocyst and regulates polarized secretion. J Biol Chem. 2001;276:46745–46750. doi: 10.1074/jbc.M107464200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.