

Abstract
Using SELEX (systematic evolution of ligands by exponential enrichment), we serendipitously discovered a ssDNA aptamer that binds selectively to the anti-FLAG M2 antibody. The aptamer consisted of two motifs (CCTTA and TGTCTWCC) separated by 2-3 bases, and the elimination of one or the other motif abrogated binding. The DNA aptamer and FLAG peptide competed for binding to the antigen-binding pocket of the M2 antibody. In addition, the aptamer eluted FLAG-tagged proteins from the antibody, suggesting a commercial application in protein purification. These findings demonstrate the feasibility of using SELEX to develop ssDNA aptamers that block the function of a specific antibody, a capability that could lead to the development of novel therapeutic modalities for patients with systemic lupus erythematosus, rheumatoid arthritis, and other autoimmune diseases.
1. Introduction
Antinuclear antibodies are diagnostic markers of systemic lupus erythematosus, rheumatoid arthritis, and other autoimmune diseases [1]. In these B lymphocyte disorders, a large variety of autoantibodies are made against nuclear self-antigens, including ribonucleoproteins, nucleosomes, chromatin, and polynucleotides (RNA, ssDNA, and dsDNA). Among these, anti-DNA antibodies have been the most extensively studied [2]. Anti-DNA antibodies bind with high-affinity to either single- or double-stranded DNA and many tend to favor association with pyrimidine bases [3, 4]. Several reports have also described antinuclear antibodies cross-reacting with peptide self-antigens and depositing in the brain, kidneys, and skin [5–9]. As proposed by several investigators, this deposition may be a cause of inflammation-mediated tissue damage, especially in the kidneys where nephritis is a major source of morbidity [1, 2]. In mouse models of systemic lupus erythematosus, attempts were made to block the function of these cross-reacting antibodies using peptide aptamers, derived either from their cognate peptide self-antigens or from phage display libraries [10, 11]. In some cases, the peptide aptamer competitively associated with the antinuclear autoantibodies, thereby preventing antibody-mediated tissue damage [10, 11]. Thus, direct antibody inhibition might be an effective therapy in patients with autoimmune diseases driven by the presence of antinuclear antibodies. Another viable approach to block antinuclear antibodies might be to use DNA aptamers, given the high-affinity of these antibodies for DNA and evidence of nucleotide base specificity. But this approach has clearly been underexplored, perhaps due to the lack of reports on the feasibility of developing DNA aptamers to block the function of specific antibodies.
An adaptive technique employed to define the sequence specificity of DNA/RNA-binding proteins is SELEX (systematic evolution of ligands by exponential enrichment). In SELEX, the protein of interest is used as a selection matrix to capture high-affinity DNA binding sites from a pool of randomized DNA molecules [12, 13]. This pool is comprised of an oligonucleotide that contains a randomized core (up to 35 bases in size) flanked by PCR priming sequences. The randomized core is made during chemical synthesis using a mixture of all four nucleoside phosphoramidites at each of the random positions. Following their capture, the selected DNA molecules are reamplified by PCR and then further enriched through successive rounds of selection. After 4–6 rounds, the selected DNA molecules are cloned and sequenced to identify any common DNA motifs recognized by the protein of interest. SELEX can be applied to the selection of ssDNA, dsDNA, or even RNA molecules [12, 13]. It is a powerful tool that has been used to optimize nucleic acid ligands for a multitude of proteins, even some which do not normally interact with DNA or RNA. As an example, SELEX was utilized to develop RNA aptamers that bind to blood coagulation factors, including thrombin [14], Von Willebrand factor [15], and Factor IXa [16]. In all three cases, the selected RNA aptamers interacted selectively with their corresponding protein targets and, in the process, inhibited their blood coagulation activities. A second generation of aptamers was developed, and, among these, some have entered clinical trials in patients with blood coagulation disorders [15].
Using SELEX, we serendipitously discovered a ssDNA sequence that binds selectively to the M2 antibody, a commonly used reagent that recognizes the Flag epitope (DYKDDDDK). The DNA aptamer and Flag peptide competed for binding to the M2 antibody, thereby allowing the aptamer to elute Flag-tagged proteins from an immobilized M2 antibody, a commonly employed procedure in protein purification. Aside from this immediate application in protein purification, identification of this DNA aptamer demonstrates the feasibility of using SELEX to develop aptamers that block specific antibodies. Applying this approach to antinuclear autoantibodies could lead to the development of novel therapeutic strategies for patients with systemic lupus erythematosus, rheumatoid arthritis, and other autoimmune diseases.
2. Materials and Methods
2.1. Materials
Oligonucleotides were synthesized by the Eppley Core Facility (University of Nebraska Medical Center, Omaha, NE). Plasmid pTetFLAGhTRF245-501 was a gift from Dr. Titia de Lange (Rockefeller University, New York, NY) [17]. The polynucleotide kinase and the Platinum Pfx and Taq DNA polymerases were purchased from Invitrogen (Carlsbad, CA). All other enzymes were obtained from Fermentas (Hanover, MD), New England BioLabs (Beverly, MA), Promega (Madison, WI), or Invitrogen (Carlsbad, CA). The TnT Quick coupled Transcription/Translation System was purchased from Promega (Madison, WI). The γ-[32P]-ATP (4500 Ci/mmol) was purchased from MP Biologicals (Solon, OH), and the L-[35S]-Methionine (1000 Ci/mmol) was obtained from PerkinElmer (Boston, MA). M-450 magnetic beads coated with a sheep anti-mouse IgG antibody were received from Dynal Biotech, Inc., (Lake Success, NY). 3XFLAG peptide and all other chemicals were from Sigma-Aldrich (St. Louis, MO). The Mini-PROTEAN 4%–20% SDS-PAGE gels were purchased from Bio-Rad (Hercules, CA).
2.2. Antibodies
Mouse monoclonal antibodies against the Flag tag (IgG1 clone M2) and StnI/OBFC1 (IgG2ak clone 3G12-1B7) were purchased from Sigma-Aldrich (St. Louis, MO). Normal mouse IgG (cat. # sc-2025) and anti-vimentin mouse monoclonal antibody (IgG1 clone sc-6260) were obtained from Santa Cruz (Santa Cruz, CA), as was the rabbit polyclonal antibody against TRF2 (cat. # H-300). Also purchased were mouse monoclonal antibodies against PTOP (IgG1 clone 1D8-1B6; Novus Biologicals, Littleton, CO) and TIN2 (IgG1 clone 59B388, AbCam, Cambridge, MA).
2.3. Expression Vectors
The plasmid pcDNA3.1-Flag-Stn1 was made by insertion of the human Stn1 sequences into plasmid pcDNA3.1-Flag. The coding sequence of Stn1 was amplified from plasmid pOTB7-Stn1 (GenBank # BC017400; American Type Culture Collection, Manassas, VA) using the Platinum Pfx DNA polymerase and primers 5′-GACTGACAATTGGGTGGTATGCAGCCTGGATCCAG-CC-3′and 5′-GACTGAAGATCTTCAGAACGCTGTGTAGTAGTG-3′. The PCR product was then cut with MfeI/BglII and inserted into the EcoRI/BamHI sites of pcDNA3.1-Flag, in frame with the Flag tag. pcDNA3.1-Flag is a pcDNA3.1(−) vector encoding a Flag epitope located downstream of a T7 promoter and immediately followed by an EcoRI site. Plasmid pcDNA3.1-Flag-TRF2ΔB was made from its pCMV1 equivalents by transfer of its TRF2 cassette to vector pcDNA3.1(−) (Invitrogen, Carlsbad, CA). pCMV1-Flag-TRF2ΔB was in turn made by the transfer to pCMV1 of a SacII/BamHI fragment from plasmid pTetFLAGhTRF245-501 (a gift from Titia de Lange, Rockefeller University, NY) [17].
2.4. Protein Expression
The Flag-Stn1 and [35S]-Flag-TRF2ΔB proteins were made by in vitro transcription/translation in a rabbit reticulocyte lysate. In a final volume of 50 μL, one microgram of pcDNA3.1-Flag-Stn1 was transcribed/translated using the TnT Quick Coupled system, according to the manufacturer's instructions (Promega, Madison, WI). A water-programmed lysate (mock) was produced in parallel to serve as a negative control. After translation, aliquots of the two reactions were analyzed by western blotting using both the Stn1/OBFC1 antibody and anti-Flag M2 antibody. A single species of 45 kDa was detected by the two antibodies in the Stn1-programmed lysate but not the Mock lysate (See Figure S1 in Supplementary Material available online at doi: 10.4061/2011/720798). The [35S]-labeled Flag-TRF2ΔB protein was produced similarly with the exception that the unlabeled methionine was replaced with 2 μL of L-[35S]-methionine (1000 Ci/mmol, 10 μCi/μL).
2.5. Preparation of Beads Coated with the Anti-Flag M2 Antibody
M2 antibody-coated beads were prepared by mixing 200 μL of M-450 magnetic beads coated with a sheep anti-mouse IgG antibody (Dynal Biotech, Inc., Lake Success, NY) with 15 μg of anti-Flag antibody (M2 mouse monoclonal, Stratagene, La Jolla, CA) in 5 mL of PBS containing 0.1% each of NP-40 and BSA. After overnight rotation at 4°C, M2-coated beads were washed 3 times in PBS containing 0.1% each of NP-40 and BSA, after which beads were suspended and stored in 200 μL of the same buffer. Before use, beads were washed 3 times with ice-cold 1X binding buffer containing 0.1% BSA. Control beads coated with normal mouse IgG (cat. # sc-2025, Santa Cruz) were prepared following the same exact protocol.
2.6. SELEX
An oligonucleotide containing a random core of 35 nucleotides flanked by PCR priming sequences was made: 5′-GCGTCGACAAGCTTTCTAGA(N)35GAATTCGGATCCCTCGAGCG-3′. In the first round of selection, 5 μg of this randomized oligo (215 pmoles, 130 trillion molecules) was incubated with 12.5 μL of rabbit reticulocyte lysate (programmed with either water or Flag-Stn1) and 5 μg of sonicated denatured E. coli genomic DNA in a 50 μL reaction containing 1X binding buffer (4% glycerol, 1 mM MgCl2, 1 mM DTT, 50 mM NaCl, 10 mM Tris-HCl pH 7.5). After 30 minutes at room temperature, 10 μL of M2-coated beads were added and the samples were rotated at room temperature for 1 hour. Magnetic beads were washed 3 times with ice-cold 1X binding buffer containing 0.1% BSA, after which the selected oligos were eluted for 10 minutes at room temperature in the presence of an excess of 3XFLAG peptide (34 μM in 1X binding buffer). Next, the eluted DNA was reamplified by PCR using Platinum Taq DNA polymerase and primers 5′-GCGTCGACAAGCTTTCTAGA-3′ (forward) and 5′-CGCTCGAGGGATCCGAATTC-3′ (reverse). Aliquots taken after 10, 15, 20, and 25 cycles of PCR were resolved on a 3% agarose gel. The most optimally amplified aliquot (no smear, no supershift, within the exponential range) was selected, cut, and gel purified using the GENECLEAN III kit (MP Biologicals, Solon, OH). Next, the gel-purified products were subjected to asymmetric PCR to regenerate ssDNA molecules needed for the following rounds of SELEX. Sixteen cycles of asymmetric PCR were performed using Platinum Taq DNA polymerase and the forward primer, after which the PCR products were extracted with phenol : chloroform (1 : 1), chloroform only, and then ethanol precipitated. The ssDNA pellet was dissolved in water and was ready to be used in the next round of SELEX. Additional round of SELEX were done identically, except that the randomized oligo was replaced with the previously selected and reamplified ssDNA.
After the sixth round of selection, the symmetrically amplified PCR product was gel purified and then prepared for TA-cloning into vector pCR2.1-Topo (Invitrogen, Carlsbad, CA). Chemically transformed TOP10 E. coli cells were spread onto plates containing kanamycin (25 μg/mL). A total of 50 white colonies were picked and sent for sequencing. Over 30 sequences were obtained for each of the two SELEX procedures performed (mock versus Flag-Stn1).
2.7. Radiolabeling of DNA Probes
In 20 μL of forward reaction buffer (10 mM MgCl2, 5 mM DTT, and 70 mM Tris-HCl, pH 7.6), 30 pmoles of oligonucleotide were labeled for 15 minutes at 37°C with 75 μCi of γ-[32P]-ATP (4500 Ci/mmole) and 10 units of T4 polynucleotide kinase (Invitrogen, Carlsbad, CA). Next, the probes were resolved by electrophoresis on an 8% polyacrylamide gel and were gel purified. Lastly, the eluted probes were desalted on a G50 spun column (GE Healthcare, Piscataway, NJ).
2.8. Electrophoretic Mobility Shift Assay (EMSA)
In 25 μL of 1X binding buffer, reactions contained 1 μg of the indicated antibody and 80,000 cpm of the designated [32P]-labeled oligonucleotide. Reactions were incubated at room temperature for 30 minutes, and were then loaded onto a 4% native polyacrylamide gel containing TBE buffer (45 mM Tris-borate, 2 mM EDTA, pH 8.3). Gels were run at 4°C for 1 hour at 180 Volts. Gels were then transferred to a DE81 anion exchange chromatography paper (Whatman International, Maidstone, England), dried, and exposed to a PhosphorImager screen (Molecular Dynamics, Sunnyvale, CA). In competition experiments, the indicated competitors were added 5 minutes prior to the addition of the [32P]-labeled ABA probe. The competitors used included an increasing concentration of 3XFLAG peptide (2, 5, 10, 20, 50, 100, 200, or 500 nM) or an excess of either the ABA or CTR oligo (500 nM each).
2.9. Binding Isotherm of the ABA Probe
Experiments were performed with [32P]-labeled ABA as described above, except that the M2 antibody was added at an increasing concentration (0, 50, 100, 200, 350, 500, 750, 1000, 2000, and 4000 ng). After exposure of the gel to a PhosphorImager screen, the amount of free [32P]-ABA and amount of [32P]-ABA bound to the M2 antibody were quantified by volume integration using ImageQuant program (Molecular Dynamics, Sunnyvale, CA). The amount of [32P]-ABA bound, expressed as a fraction of the total, was plotted as a function of total amount of M2 antibody. The resulting curves were fitted by nonlinear regression to a “one-site” saturation binding curve ([32P]-ABA bound = [M2]/(KD + [M2])). Fitting, performed using SigmaPlot version 11.0, allowed calculation of the apparent dissociation constant (KD).
2.10. Release of the Captured [32P]-ABA Oligo by the 3XFLAG Peptide
The [32P]-labeled ABA oligo was first immobilized onto the M2-coated beads. Briefly, 150 μL of M2-coated beads were added to 375 μL of 1X binding buffer containing 600,000 cpm of [32P]-ABA oligo. Capture of the oligo was allowed to proceed for one hour at room temperature, after which the beads were washed 3 times with 500 μL of ice-cold 1X binding buffer (containing 0.1% BSA). The radioactive beads were then resuspended in 450 μL of the same buffer. To assess the ability of the 3XFLAG peptide to elute the captured oligo, an increasing concentration of 3XFLAG peptide (0, 2, 5, 10, 20, 50, 100, 200, 500, 1000, 2000, and 5000 nM) was added to 30 μL of beads carrying the [32P]-ABA oligo. After 20 minutes at room temperature, the beads were pulled down and the amounts of radioactivity remaining on the beads and in the supernatant were counted by scintillation and expressed as a fraction of the total.
2.11. Inhibition of the Capture of [35S]-Labeled Flag-TRF2ΔB
Prior to these experiments, beads were blocked at 4°C for 30 minutes with 5% milk in 1X binding buffer to reduce nonspecific binding. In 1X binding buffer, M2-coated beads were first incubated with the indicated competitor (30 μM each 3XFLAG, 3XABA, or 3XCTR) or with no competitor (No Comp). After 10 minutes at room temperature, 2–10 μL of [35S]-labeled FLAG-TRF2ΔB were added. After 1 hour of rotation at room temperature, each reaction was washed 3 times with 500 μL of ice-cold 1X binding buffer (containing 0.1% BSA), after which the amount of [35S]-Flag-TRF2ΔB captured was quantified. In one experiment, the amount captured was quantified by SDS-PAGE electrophoresis and exposure to a PhosphorImager screen. In a second experiment done in triplicate, the amount of [35S]-Flag-TRF2ΔB captured was quantified by scintillation counting. In both experiments, beads coated with normal mouse IgG were included as a negative control for the capture (IgG).
2.12. Release of Precaptured [35S]-Labeled Flag-TRF2ΔB
The [35S]-labeled Flag-TRF2ΔB protein was first immobilized onto the M2-coated beads. Briefly, 80 μL of M2-coated beads were added to 800 μL of 1X binding buffer containing 32 μL of [35S]-Flag-TRF2ΔB. Capture of the protein was allowed to proceed for one hour at room temperature, after which the beads were washed 3 times with 500 μL of ice-cold 1X binding buffer (containing 0.1% BSA). The radioactive beads were then resuspended in 80 μL of the same buffer. To assess the ability of the 3XABA oligo to elute the captured protein, five microliters of these beads were mixed with 45 μL of 1X binding buffer containing the indicated competitor (30 μM each 3XFLAG, 3XABA, or 3XCTR) or no competitor (No Comp). After 20 minutes at room temperature, the beads were pulled down and the amount of radioactivity in the supernatants was quantified by SDS-PAGE electrophoresis and exposure to a PhosphorImager screen. In an experiment done in triplicate, the amount of [35S]-Flag-TRF2ΔB released was also quantified by scintillation counting. In both experiments, beads boiled to release all of the captured [35S]-labeled protein were included as positive control for the elution (Total).
3. Results
3.1. SELEX Driven by the Anti-Flag M2 Antibody
SELEX was used to characterize the DNA-binding specificity of a human Flag-tagged Stn1 protein [18, 19], in this case produced by in vitro translation in a rabbit reticulocyte cell-free system. The in vitro translated Flag-Stn1 was mixed with a pool of randomized ssDNA molecules and then, the anti-Flag M2 antibody was used to immunoprecipitate the Flag-Stn1/DNA complexes to select for high-affinity targets. After six successive rounds of selection, the selected ssDNA molecules were cloned and sequenced. Analysis of a related unselected random pool revealed a slight bias for G-rich sequences, but no evidence of any recurrent motifs [20]. In contrast, analysis of the Flag-Stn1 selected oligonucleotides revealed a bipartite consensus motif made of two distinct elements: CCTTA and TGTCTWCC (where W = A/T; Figure 1(a)). These elements were separated by 2-3 bases of poorly conserved sequences. However, the same bipartite consensus motif was also produced when SELEX was performed with the same antibody but using a mock-programmed reticulocyte lysate as a control source of proteins (Figure 1(b)). Based on these results, we concluded that the bipartite consensus had been selected, not by the Flag-Stn1 protein, but by the M2 antibody itself or proteins of the lysate that the antibody cross-reacted with.
Figure 1.

SELEX with or without the Flag-Stn1 protein yields the same bipartite consensus. A randomized pool of ssDNA molecules was incubated with an in vitro translation system programmed with either a Flag-Stn1 plasmid (Flag-Stn1 present) or water (Flag-Stn1 absent), after which protein/DNA complexes were recovered by immunoprecipitation using the anti-Flag M2 antibody. After six successive rounds of selection, the remaining oligonucleotides were cloned and sequenced. (a) Oligonucleotides selected in the presence of the Flag-Stn1 protein. The selected ssDNA molecules share a common bipartite consensus, which consists of a CCTTA (pink) and TGTCTWCC (green) motifs. Nonnucleotide letters denote ambiguous sequencing reads (R = A/G, Y = C/T, S = C/G, W = A/T, K = T/G, M = A/C). (b) Oligonucleotides selected in the absence of the Flag-Stn1 protein. The same bipartite consensus is shared among the ssDNA molecules selected by the mock-programmed in vitro translation system.
3.2. The M2 Antibody Binds the Consensus ssDNA Motifs
To test the possibility of direct interactions between the bipartite consensus and M2 antibody, EMSA was performed. A series of equal length [32P]-labeled ssDNA probes containing or lacking each element of the consensus were made (Figure 2(a)). Probes AB, BA, and ABA carried both the CCTTA and TGTCTWCC motifs, whereas probe 2XB and 4XA consisted of multiple copies of one or the other element. The unrelated CTR probe was used as negative control. Figure 2(b) shows that the M2 antibody could form a stable complex with the ABA probe and to a lesser extent, the AB probe. None of the other probes interacted with the M2 antibody. Also noteworthy, neither the BA probe lacking the CCTTA element nor the AA probe lacking the TGTCTWCC element interacted with the antibody, thereby indicating that both elements are essential for binding. Figure 2(b) also shows that among 7 different antibodies, the M2 antibody only interacted with probe ABA. These results show that probe ABA binds very selectively to the M2 antibody, thereby suggesting that these interactions are involving the variable regions of the antibody, whether part of the light chain, heavy chain, or both.
Figure 2.
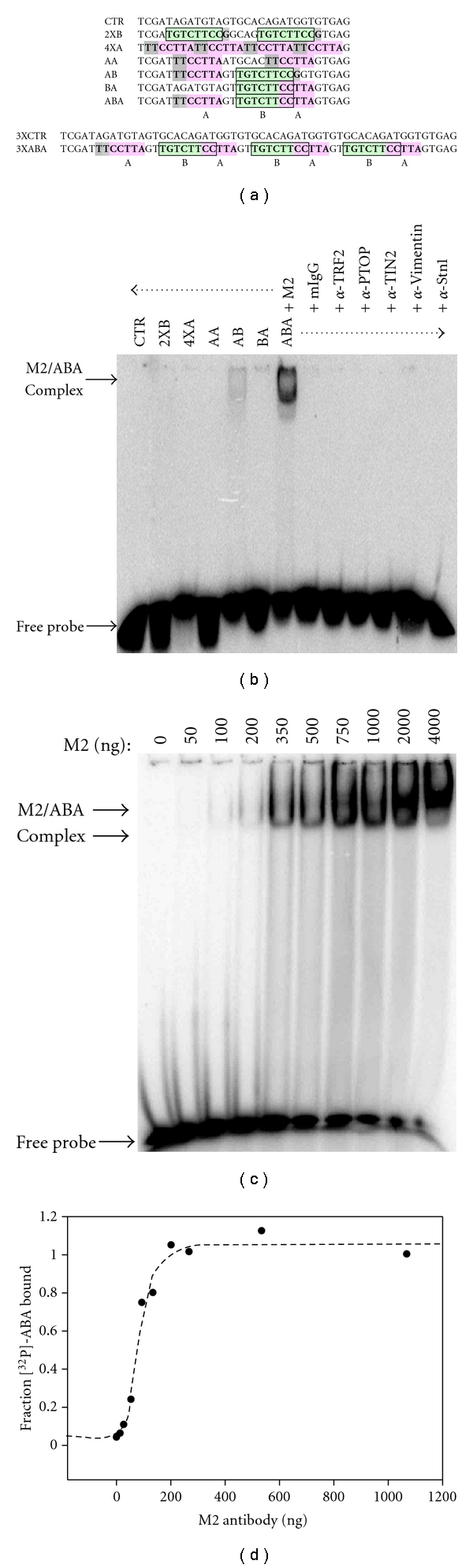
The bipartite ssDNA consensus binds directly to the anti-Flag M2 antibody. (a) Sequence and graphical representation of the probes used. All probes were labeled at the 5′-end with [32P]. Probes were designed to carry none, one, or both of the identified motifs. Motif A = CCTTA (pink). Motif B = TGTCTWCC (green). (b) The anti-Flag M2 antibody binds selectively to probe ABA. The indicated probes (80,000 cpm) were incubated with the listed antibodies (1 μg), after which protein/DNA complexes were resolved by electrophoresis in a native polyacrylamide gel. (c) Titration of the M2 antibody. A constant amount of probe ABA was incubated with an increasing concentration of M2 antibody, and the protein/DNA complexes were resolved by native electrophoresis. At the higher antibody concentrations, a larger protein/DNA complex is observed (top arrow), which may represent the product of antibody oligomerization. (d) Binding isotherm of probe ABA interacting with the anti-Flag M2 antibody. The scatter plot shows the amount of ABA bound (amount present in both protein/DNA complexes) as a function of antibody concentration. Dotted line shows fitting of the data to a “one site” saturation binding curve. Nonlinear regression of the data allowed for the determination of the dissociation constant (KD = 80 ± 7 nM). Error on the value of the KD represents the 95% confidence interval of the best-fit curve.
To measure the affinity of the M2 antibody for probe ABA, the apparent dissociation constant (KD) of the complex was determined. A binding isotherm was generated by EMSA using a constant amount of [32P]-ABA incubated with an increasing concentration of M2 antibody (Figure 2(c)). Plotting the amount of ABA bound as a function of the total concentration of antibody generated a binding isotherm. Fitting of this isotherm to a “one site” saturation binding curve allowed for calculation of the KD. In this case, the apparent KD was determined to have a value of 80 ± 7 nM (Figure 2(d)). The value is comparable to the apparent KD of other previously reported DNA aptamer/antibody complexes [21]. This result shows that the M2 antibody binds with high affinity to its preferred DNA ligand, the ABA probe.
3.3. The Flag Peptide Competes with the Binding of the ABA Oligo to the M2 Antibody
The M2 antibody binds to both the Flag peptide (DYKDDDDK) and the ABA probe, so we sought to determine whether the two ligands could compete for each other. In a first series of experiments, we asked if an excess of 3XFLAG peptide (MDYKDHDGDYKDHDIDYKDDDDK) could block the binding of [32P]-labeled ABA to the M2 antibody. In the EMSA shown in Figure 3(a), an increasing concentration of 3XFLAG peptide was incubated with the M2 antibody prior to adding the [32P]-ABA. At concentrations of 50 nM and higher, the 3XFLAG peptide could completely block the binding of ABA to the M2 antibody. A similar inhibition was produced following the inclusion of an excess of unlabeled ABA oligo, but not after the addition of the CTR oligo. These results show that the 3XFLAG peptide competes with the binding of probe ABA to the M2 antibody.
Figure 3.
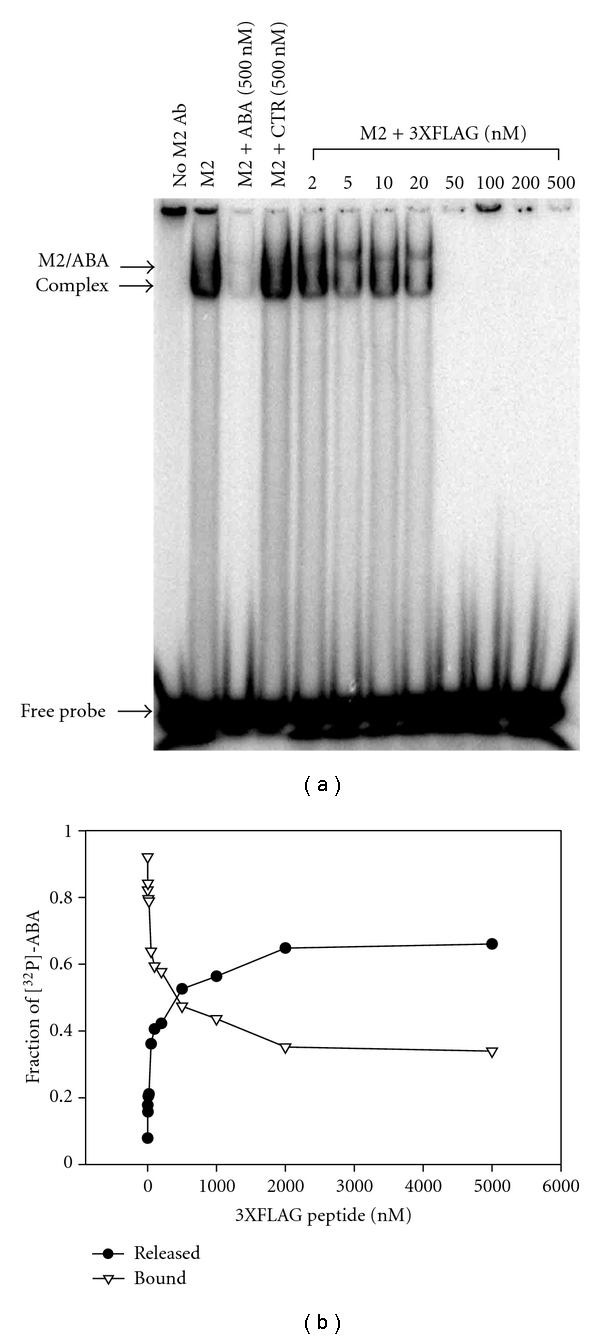
The 3XFLAG peptide blocks the binding of ABA to the M2 antibody. (a) The 3XFLAG peptide prevents formation of the ABA/M2 complex. The M2 antibody was incubated with an increasing concentration of 3XFLAG peptide after which the [32P]-ABA probe was added. Protein/DNA complexes were resolved by electrophoresis in a native polyacrylamide gel. (b) The 3XFLAG peptide elutes the ABA probe already bound to the M2 antibody. The [32P]-ABA was first captured by magnetic beads coated with the M2 antibody. The beads were then incubated with an increasing concentration of 3XFLAG peptide and the amount of [32P]-ABA released and remaining on the beads was counted by scintillation (n = 1).
In a second series of experiments, we asked whether an excess of 3XFLAG peptide could elute a [32P]-ABA oligo already bound to the M2 antibody. For this purpose, [32P]-ABA was first captured by magnetic beads coated with the M2 antibody, after which an increasing concentration of 3XFLAG peptide was added. In Figure 3(b), the amount of ABA bound to the beads and the amount released from the beads were plotted as a function of the concentration of the 3XFLAG peptide. The data show that the ABA oligo was very efficiently released by the addition of the 3XFLAG peptide. A small fraction of oligo, representing 36%, could not be displaced, but the other fraction was almost completely released by 2000 nM of 3XFLAG peptide. Fitting the data to a “one site” competition curve allowed for the calculation of an IC50. In agreement with the values of the KD, the IC50 was determined to be 96 nM (95% confidence interval: 72–127 nM). These results show that the 3XFLAG peptide can elute the ABA oligo when bound to the M2 antibody.
3.4. The 3XABA Oligo Blocks the Association of Flag-Tagged Proteins with the M2 Antibody
If the ABA oligo and the Flag peptide compete for binding to the M2 antibody, then an excess of ABA oligo could potentially allow the elution of Flag-tagged proteins from the M2-coated beads, a commonly used procedure in protein purification. To address this possibility, we first determined whether an excess of ABA oligo could block binding of [S35]-labeled Flag-TRF2ΔB to M2-coated beads. In these experiments, the M2-coated beads were first incubated with the competitors (30 μM of 3XFLAG peptide, 3XABA or 3XCTR oligos) or with no competitor (No Comp). Then, the [S35]-labeled Flag-TRF2ΔB was added and the amount of [S35] captured by the beads was measured by either SDS-PAGE electrophoresis (Figure 4(a)) or scintillation counting (Figure 4(b)). As Figures 4(a)–4(b) show, the 3XFLAG peptide abolished the capture of the [S35]-labeled protein whereas the 3XCTR oligo had no effect. Although not as active as the 3XFLAG peptide, the 3XABA oligo inhibited the capture of Flag-TRF2ΔB by 62%. The shorter ABA aptamer was also tested but found to be less active (Figure S2), with ABA blocking the capture by 45% only.
Figure 4.
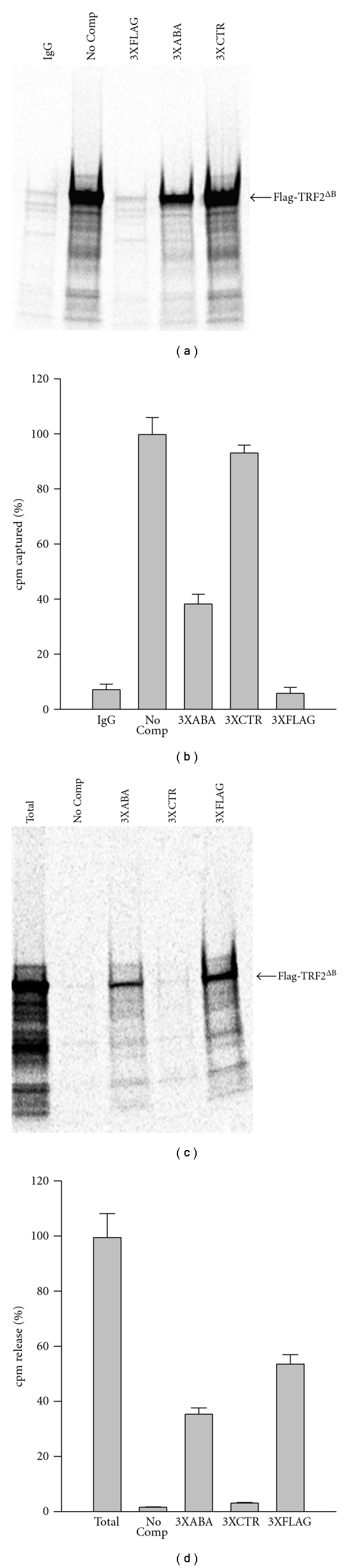
3XABA oligonucleotide blocks the interaction of Flag-tagged proteins with the M2 antibody. (a)-(b) The 3XABA oligo blocks the binding of Flag-TRF2ΔB to M2-coated beads. Magnetic beads coated with the M2 antibody were incubated in the absence (No Comp) or presence of the indicated competitor (3XABA, 3XCTR, 3XFLAG). In vitro translated [35S]-labeled Flag-TRF2ΔB was then added and the amount captured by the beads was determined by SDS-PAGE electrophoresis and exposure to a PhophorImager cassette (a). In a second experiment done in triplicate, the amount of [35S]-labeled protein captured was counted by scintillation (b). The amount of [35S]-labeled protein captured in the absence of competitor (No Comp) was arbitrarily set to 100%. In both experiments, beads coated with normal mouse IgG were included as negative control for the capture (IgG). Data represent the mean ± S.D. (n = 3). (c)-(d) The 3XABA oligo elutes the Flag-TRF2ΔB proteins already bound to M2-coated beads. The [35S]-Flag-TRF2ΔB protein was first captured by magnetic beads coated with the M2 antibody. The beads were then incubated in the absence (No comp) or presence of the indicated competitor (3XABA, 3XCTR, 3XFLAG). The amount of [35S]-Flag-TRF2ΔB released was determined by SDS-PAGE electrophoresis and exposure to a PhophorImager cassette (c). In a second experiment done in triplicate, the amount of [35S]-labeled protein released was counted by scintillation (d). The amount of [35S]-labeled protein released by the boiling (total) was arbitrarily set to 100%. In both experiments, beads boiled to release to all of the captured [35S]-labeled protein were included as positive control for the elution (Total). Data represent the mean ± S.D. (n = 3).
Next, we asked whether an excess of 3XABA oligo could elute an [S35]-labeled Flag-TRF2ΔB protein already bound to the M2-coated beads. In these experiments, the [S35]-labeled protein was first captured by the M2-coated beads. Next, the radioactive beads were exposed to the competitors (30 μM of 3XFLAG peptide, 3XABA or 3XCTR oligos) or to no competitor (No Comp), and the amount of [S35] released was quantified by either SDS-PAGE electrophoresis (Figure 4(c)) or scintillation counting (Figure 4(d)). As Figures 4(c)-4(d) show, the 3XFLAG peptide caused the release of 54% of the captured protein whereas the 3XCTR oligo had no activity. The 3XABA oligo was almost as effective as the 3XFLAG peptide, causing the release of 36% of the bound protein.
4. Discussion
The initial intent of this study was to use SELEX to characterize the ssDNA-binding specificity of Flag-tagged Stn1, a subunit of the CST complex [18, 19]. Instead, the SELEX procedure enriched for ssDNA molecules that bind with high affinity to the anti-Flag M2 antibody. How was this selection by the antibody even possible? First, it may be that the in vitro translated Flag-Stn1 protein did not possess the minimum required DNA-binding activity. Improper folding could have contributed to this lack of activity. Alternatively, it may be that the OB-fold domain (oligonucleotide/oligosaccharide binding) of Stn1 is involved in mediating protein-protein interactions rather than DNA-binding [18, 19]. Second, the Flag epitope (DYKDDDDYK) is highly negatively charged, making it possible for an optimized DNA molecule to mimic the electrostatic signature of the peptide. Third, ssDNA is far more flexible than dsDNA, and in that respect, could more readily adapt to the positively charged surface of the antigen-binding pocket of the M2 antibody [22]. In other words, we propose that selection mediated by the antibody would not have been possible if the Flag epitope had been neutral or positively charged, if SELEX had been performed using dsDNA, or if the Flag-tagged Stn1 protein had exhibited a higher affinity for ssDNA. In fact, we have successfully used SELEX under similar conditions to define the ssDNA binding specificity of POT1, a protein that recognizes telomeric DNA. This SELEX, also performed using the M2 antibody, almost exclusively selected for telomeric sequences with no evidence of the bipartite motif detected (Choi, K. H. and Ouellette, M. M., manuscript in preparation). The selection by SELEX of off-target aptamers is not uncommon, especially when selecting single-stranded nucleic acids [23]. Off-target aptamers that bind to the selection matrix (e.g. avidin, M2 antibody), as we have observed here, have also been reported by others [23]. Hence, caution and experimental controls are necessary, especially when selecting ssDNA molecules for their binding to a protein of unknown functionality. Under these conditions, an important control to perform, as we have done here, is a mock selection to verify that the molecules were indeed selected by the protein of interest.
Our findings show that the identified bipartite consensus binds selectively and with high affinity to the M2 antibody. An important finding was the specificity of this interaction. For example, the consensus did not bind to any of the other antibodies tested, many of which belonging to the same isotype as the M2 antibody (mouse IgG1). This specificity implied that the interactions involved the variable regions of the antibody, which together form the antigen-binding pocket. This assumption was confirmed by the results of the competition experiments. As Figure 4 shows, the ssDNA consensus could very efficiently displace the Flag peptide from the antigen-binding pocket of the M2 antibody. Conversely, the Flag peptide could similarly displace the DNA aptamer from the antibody as well (see Figure 3). This competition between the Flag peptide and DNA aptamer implies that the two are recognized by overlapping binding determinants at the surface of the antibody. A shared feature of the Flag peptide and ABA aptamer is their negative electrostatic charges. On the surface of the antibody, these negative charges on the two ligands are likely to interact with some of the same binding determinants. The binding pocket of the M2 antibody appears to be specific only for the first 4 amino acids of the Flag peptide, namely, DYKD [22]. Two clusters of positively charged residues (lysines, arginine, histidine) are present in the binding pocket, each capable of coordinating an aspartate within the DYKD motif. The same two clusters could potentially interact with phosphate groups present within the backbone of the ABA oligo. Also important for the binding are two tyrosine residues, each one capable of forming stacking interactions with the tyrosine of DYKD. When bound to the ABA oligo, the same tyrosines could provide stacking interactions to some of the nucleotide bases. Yet, these limited interactions would not suffice to explain the sequence specificity observed. The selected consensus consisted of two separate motifs (CCTTA and TGTCTWCC) and mutating one or the other was sufficient to abrogate binding. To allow recognition of this large bipartite consensus, other residues must be implicated that make contact with several of the nucleotide bases. Hence, it must be that the Flag peptide interacts with a subset only of all the residues implicated in the binding of ABA. If so, then how could the Flag peptide be so efficient at displacing ABA from the M2 antibody? It is possible that the binding determinants that the two ligands share are especially critical for the binding of ABA. A second possibility would be that the binding of the Flag peptide causes changes in the conformation of the antibody, and that these allosteric alterations are incompatible with the binding of ABA. How the ABA aptamer is able to mimic the structure of the Flag peptide remains to be determined, but this form of mimicry between peptide and DNA has previously been reported to play an important role in autoimmune diseases and may also have important applications in drug design [5–9]. The ABA oligo and Flag peptide offer an ideal system with which to study the structural basis of this form of molecular mimicry.
One commonly employed procedure in protein purification is the capture of Flag-tagged proteins by the M2 antibody and their subsequent release by incubation with an excess of 3XFLAG peptide [24]. This procedure allows the elution of the purified proteins under nondenaturing conditions. However, the procedure leaves the eluted protein in solution with an excess of 3XFLAG peptide, which can pose a problem if the Flag tag needs to remain functional. A DNA aptamer capable of eluting the Flag-tagged proteins, such as 3XABA, could alleviate these potential drawbacks. Once the protein is eluted, the DNA aptamer could conveniently be inactivated by an antisense oligo or eliminated using trace amounts of DNAse. Alternatively, if the aptamer is biotinylated, its removal could be accomplished using streptavidin-coated beads. Finally, a DNA aptamer would also offer the possibility of using mutated oligos as negative controls or for the differential elution of captured proteins.
These results described here may also have implications for the treatment of autoimmune diseases driven by antinuclear antibodies. Previous attempts to block the function of these autoantibodies have been made with peptide mimics, either isolated from phage display libraries or derived from peptide self-antigens with which these antinuclear antibodies cross-react [10, 11]. These results described therein show that SELEX can be used to identify DNA aptamers that block the function of a particular antibody. Could this approach be used to develop aptamers that block the function of antinuclear autoantibodies? In certain autoimmune diseases, blocking the function of antinuclear autoantibodies has been shown to limit tissue damage [10, 11]. The sequence specificity of antinuclear autoantibodies has not been systematically investigated [3, 4]. SELEX would represent an ideal method for characterizing the sequence specificity of antinuclear autoantibodies. The SELEX data could serve as starting point for the design of second-generation aptamers that might be useful to block the function of these autoantibodies. The development of these DNA aptamers could therefore represent an alternative approach to the treatment of patients afflicted with systemic lupus erythematosus, rheumatoid arthritis, and other autoimmune diseases.
5. Conclusion
In summary, SELEX identified a DNA aptamer that binds directly to the antigen-binding pocket of the anti-Flag M2 antibody. The DNA aptamer and Flag peptide competed for binding to the M2 antibody and the aptamer eluted Flag-tagged proteins from an immobilized M2 antibody. This new reagent therefore offers an alternative method for the elution of Flag-tagged proteins bound to the M2 antibody, a commonly employed procedure in protein purification. Aside from this immediate application in protein purification, identification of this bipartite consensus demonstrates the feasibility of using SELEX to develop DNA aptamers that block specific antibodies. Applying this approach to autoantibodies, particularly the antinuclear antibodies, could lead to the development of novel therapeutic strategies for patients with systemic lupus erythematosus, rheumatoid arthritis, and other autoimmune diseases.
Supplementary Material
Figure S1: Western blot analysis of the in vitro Flag-Stn1 protein.
Figure S2: Relative efficacy with which the different aptamers block the interaction of Flag-tagged proteins with the M2 antibody.
Acknowledgments
The authors wish to thank Titia de Lange (Rockefeller University, New York, NY) for the pTetFLAGhTRF245-501 plasmid. Appreciation is extended to Asserewou Honoré Etekpo for the quality of his technical assistance. They also wish to acknowledge the UNMC Eppley Cancer Center Molecular Biology Core and the UNMC DNA Sequencing Core Facility. This paper was supported by Grants from the National Institute of Health (P50CA127297, P30CA036727) and a GAANN fellowship from US Department of Education (P200A090064) to A. S. Lakamp.
References
- 1.Lyons R, Narain S, Nichols C, Satoh M, Reeves WH. Effective use of autoantibody tests in the diagnosis of systemic autoimmune disease. Annals of the New York Academy of Sciences. 2005;1050:217–228. doi: 10.1196/annals.1313.023. [DOI] [PubMed] [Google Scholar]
- 2.Jang YJ, Stollar BD. Anti-DNA antibodies: aspects of structure and pathogenicity. Cellular and Molecular Life Sciences. 2003;60(2):309–320. doi: 10.1007/s000180300026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pisetsky DS, Caster SA. Binding specificity of a monoclonal anti-DNA antibody. Molecular Immunology. 1982;19(5):645–650. doi: 10.1016/0161-5890(82)90364-9. [DOI] [PubMed] [Google Scholar]
- 4.Sasaki T, Muryoi T, Sekiguchi Y, Tamate E, Yoshinaga K, Kitagawa Y. Monoclonal human anti-DNA antibodies from EB virus-transformed lymphocytes of systemic lupus erythematosus (SLE) patients. Journal of Clinical Immunology. 1985;5(4):246–253. doi: 10.1007/BF00929459. [DOI] [PubMed] [Google Scholar]
- 5.DeGiorgio LA, Konstantinov KN, Lee SC, Hardin JA, Volpe BT, Diamond B. A subset of lupus anti-DNA antibodies cross-reacts with the NR2 glutamate receptor in systemic lupus erythematosus. Nature Medicine. 2001;7(11):1189–1193. doi: 10.1038/nm1101-1189. [DOI] [PubMed] [Google Scholar]
- 6.Deocharan B, Qing X, Beger E, Putterman C. Antigenic triggers and molecular targets for anti-double-stranded DNA antibodies. Lupus. 2002;11(12):865–871. doi: 10.1191/0961203302lu308rr. [DOI] [PubMed] [Google Scholar]
- 7.Faust TW, Chang EH, Kowal C, et al. Neurotoxic lupus autoantibodies alter brain function through two distinct mechanisms. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(43):18569–18574. doi: 10.1073/pnas.1006980107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lefkowith JB, Kiehl M, Rubenstein J, et al. Heterogeneity and clinical significance of glomerular-binding antibodies in systemic lupus erythematosus. Journal of Clinical Investigation. 1996;98(6):1373–1380. doi: 10.1172/JCI118924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rice JS, Kowal C, Volpe BT, DeGiorgio LA, Diamond B. Molecular mimicry: anti-DNA antibodies bind microbial and nonnucleic acid self-antigens. Current Topics in Microbiology and Immunology. 2005;296:137–151. doi: 10.1007/3-540-30791-5_8. [DOI] [PubMed] [Google Scholar]
- 10.Gaynor B, Putterman C, Valadon P, Spatz L, Scharff MD, Diamond B. Peptide inhibition of glomerular deposition of an anti-DNA antibody. Proceedings of the National Academy of Sciences of the United States of America. 1997;94(5):1955–1960. doi: 10.1073/pnas.94.5.1955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee HB, Diamond BA, Blaufox MD. In vivo detection of deposition of radiolabeled lupus antikidney antibody and its inhibition by soluble antigen. Journal of Nuclear Medicine. 2001;42(1):138–140. [PubMed] [Google Scholar]
- 12.Ouellette MM, Wright WE. Use of reiterative selection for defining protein-nucleic acid interactions. Current Opinion in Biotechnology. 1995;6(1):65–72. doi: 10.1016/0958-1669(95)80011-5. [DOI] [PubMed] [Google Scholar]
- 13.Zimmermann B, Bilusic I, Lorenz C, Schroeder R. Genomic SELEX: a discovery tool for genomic aptamers. Methods. 2010;52(2):125–132. doi: 10.1016/j.ymeth.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tasset DM, Kubik MF, Steiner W. Oligonucleotide inhibitors of human thrombin that bind distinct epitopes. Journal of Molecular Biology. 1997;272(5):688–698. doi: 10.1006/jmbi.1997.1275. [DOI] [PubMed] [Google Scholar]
- 15.Becker RC, Oney S, Becker KC, Sullenger B. Antidote-controlled antithrombotic therapy targeting factor IXa and von willebrand factor. Annals of the New York Academy of Sciences. 2009;1175:61–70. doi: 10.1111/j.1749-6632.2009.05017.x. [DOI] [PubMed] [Google Scholar]
- 16.Rusconi CP, Scardino E, Layzer J, et al. RNA aptamers as reversible antagonists of coagulation factor IXa. Nature. 2002;419(6902):90–94. doi: 10.1038/nature00963. [DOI] [PubMed] [Google Scholar]
- 17.van Steensel B, Smogorzewska A, de Lange T. TRF2 protects human telomeres from end-to-end fusions. Cell. 1998;92(3):401–413. doi: 10.1016/s0092-8674(00)80932-0. [DOI] [PubMed] [Google Scholar]
- 18.Price CM, Boltz KA, Chaiken MF, Stewart JA, Beilstein MA, Shippen DE. Evolution of CST function in telomere maintenance. Cell Cycle. 2010;9(16):3157–3165. doi: 10.4161/cc.9.16.12547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Miyake Y, Nakamura M, Nabetani A, et al. RPA-like mammalian Ctc1-Stn1-Ten1 complex binds to single-stranded DNA and protects telomeres independently of the Pot1 pathway. Molecular Cell. 2009;36(2):193–206. doi: 10.1016/j.molcel.2009.08.009. [DOI] [PubMed] [Google Scholar]
- 20.Prakash A, Natarajan A, Marky LA, Ouellette MM, Borgstahl GE. Identification of the DNA-binding domains of human replication protein A that recognize G-quadruplex DNA. Journal of Nucleic Acids. 2011;2011:14 pages. doi: 10.4061/2011/896947. Article ID 896947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mendonsa SD, Bowser MT. In vitro evolution of functional DNA using capillary electrophoresis. Journal of the American Chemical Society. 2004;126(1):20–21. doi: 10.1021/ja037832s. [DOI] [PubMed] [Google Scholar]
- 22.Roosild TP, Castronovo S, Choe S. Structure of anti-FLAG M2 Fab domain and its use in the stabilization of engineered membrane proteins. Acta Crystallographica Section F. 2006;62(9):835–839. doi: 10.1107/S1744309106029125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Joyce GF. Directed evolution of nucleic acid enzymes. Annual Review of Biochemistry. 2004;73:791–836. doi: 10.1146/annurev.biochem.73.011303.073717. [DOI] [PubMed] [Google Scholar]
- 24.Einhauer A, Jungbauer A. The FLAG peptide, a versatile fusion tag for the purification of recombinant proteins. Journal of Biochemical and Biophysical Methods. 2001;49:455–465. doi: 10.1016/s0165-022x(01)00213-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1: Western blot analysis of the in vitro Flag-Stn1 protein.
Figure S2: Relative efficacy with which the different aptamers block the interaction of Flag-tagged proteins with the M2 antibody.