

Abstract
Background. Disability in multiple sclerosis (MS) is related to neuroaxonal degeneration. A reliable blood biomarker for neuroaxonal degeneration is needed. Objectives. To explore the relationship between cerebrospinal fluid (CSF) and serum concentrations of a protein biomarker for neuroaxonal degeneration, the neurofilaments heavy chain (NfH). Methods. An exploratory cross-sectional (n = 51) and longitudinal (n = 34) study on cerebrospinal fluid (CSF) and serum NfH phosphoform levels in patients with MS. The expanded disability status scale (EDSS), CSF, and serum levels of NfH-SMI34 and NfH-SMI35 were quantified at baseline. Disability progression was assessed at 3-year followup. Results. At baseline, patients with primary progressive MS (PPMS, EDSS 6) and secondary progressive MS (SPMS, EDSS 6) were more disabled compared to patients with relapsing remitting MS (RRMS, EDSS 2, P < .0001). Serum and CSF NfH phosphoform levels were not correlated. Baseline serum levels of the NfH-SMI34 were significantly (P < .05) higher in patients with PPMS (2.05 ng/mL) compared to SPMS (0.03 ng/mL) and RRMS (1.56 ng/mL). In SPMS higher serum than CSF NfH-SMI34 levels predicted disability progression from baseline (ΔEDSS 2, P < .05). In RRMS higher CSF than serum NfH-SMI35 levels predicted disability progression (ΔEDSS 2, P < .05). Conclusion. Serum and CSF NfH-SMI34 and NfH-SMI35 levels did not correlate with each other in MS. The quantitative relationship of CSF and serum NfH levels suggests that neuroaxonal degeneration of the central nervous system is the likely cause for disability progression in RRMS. In more severely disabled patients with PP/SPMS, subtle pathology of the peripheral nervous system cannot be excluded as an alternative source for blood NfH levels. Therefore, the interpretation of blood protein biomarker data in diseases of the central nervous system (CNS) should consider the possibility that pathology of the peripheral nervous system (PNS) may influence the results.
1. Introduction
In multiple sclerosis, irreversible disability progression is anatomically associated with neuroaxonal degeneration [1–3]. Using cerebral microdialysis, it was shown that as a result of neuroaxonal degeneration, protein biomarkers were released into the extracellular fluid (ECF) [4, 5]. Once released into the ECF of the brain, these brain-specific proteins diffuse into the cerebrospinal fluid (CSF) [6]. A protein biomarker specific for neuroaxonal degeneration are neurofilaments [7, 8]. Of the various neurofilament proteins (Nf), the light (NfL) and heavy (NfH) chains were successfully quantified from the CSF and found to be of prognostic value in patients with MS (reviewed in [7, 8] and newer references [9–12]). The Nf proteins diffuse from the CSF into the blood stream from where different NfH phosphoforms have been quantified by different groups [13–15]. Because of the relative ease of blood sampling compared to a spinal tap, it is highly desirable to have a reliable blood biomarker for neuroaxonal degeneration.
There is a need to better understand the relationship between CSF and serum protein biomarkers for neuroaxonal degeneration. The situation in the CSF is relatively straightforward, because any increase can with a considerable degree of confidence be associated with damage to the brain. Interpretation of serum data is more complex. One potentially confounding issue is that Nf are also expressed in the peripheral nervous system [7].
The relationship between CSF and serum NfH levels in MS is not known. In this study, we hypothesized that the well-recognized neuroaxonal degeneration of the central nervous system would lead to a higher concentration in the CSF compared to the serum. To test this hypothesis, we quantified NfH heavy chain phosphoforms from the serum and CSF in a cohort of MS patients we have been published on before [16].
2. Methods
2.1. Patients
This study was approved by the local ethics committee, and informed written consent was obtained from the patients. All patients with MS were from a previously published Dutch cohort [17] and were classified into having relapsing remitting MS (RRMS, n = 21), secondary progressive MS (SPMS, n = 22), or primary progressive MS (PPMS, n = 9) according to published criteria [18]. Blood and CSF samples were taken at the same time. Matched aliquots of CSF and serum samples were coded and stored in polypropylene tubes as described [19].
2.2. Clinical Assessment
Disability was recorded on the expanded disability status scale score (EDSS) [20]. Progression of disability was calculated over the 3-year interval as ΔEDSS = followup EDSS − baseline-EDSS. Significant disability progression was defined as worsening on the EDSS scale by at least 1 point for an EDSS < 5.5 or at least 0.5 point for an EDSS ≥ 5.5.
2.3. Neurofilament Analysis
CSF and serum Nf levels were measured using a sensitive sandwich ELISA which allows to quantify various NfH phosphoforms by exchanging the capturing monoclonal antibodies [21]. This ELISA gives the best analytical performance for the monoclonal antibodies SMI34 and SMI35 (originally from Sternberger Monoclonals, now sold through Covance). Adhering to a previously proposed nomenclature NfH captured by SMI34 is indicated as NfHSMI34 and NfH captured by SMI35 as NfHSMI35. The precise binding epitopes of these antibodies are not known. Binding of SMI34 is phosphate dependent which is correlated with but not identical to the degree of NfH phosphorylation. SMI35 binds more specifically to phosphorylated NfH. Nonmeasurable samples were reported as 0 ng/mL.
2.4. Data Analysis
Because of non-Gaussian data distribution the median and interquartile range (IQR) are shown. Nonparametric statistics were used for comparison throughout. We used general linear models for comparison of three variables. The concentration of NfH was compared between CSF and serum for each patient individually. If the concentration of NfH was higher in the CSF compared to the matched serum sample, this was indicated as “C > S”, otherwise as “S ≥ C”. The relationship between higher CSF or serum NfH levels with clinical progression on the EDSS was analyzed using the Kruskall-Wallis test. Correlation analyzes were performed using Spearman's R. The Bonferroni method was used to correct for multiple correlations. All statistical analyzes were performed in SAS (version 9.1).
3. Results
3.1. Baseline
The demographic data of the MS patients is summarized in Table 1. At baseline, the groups differed in their demographic data for age (F 2,48 = 4.87, P < .05), EDSS (F 2,48 = 19,26, P < .0001), and disease duration (F 2,48 = 7.33, P < .01). None of the biomarkers was correlated with age, age at onset, disease duration, or the EDSS in the MS groups (data not shown).
Table 1.
Patient characteristics at baseline. The median (range) is shown.
| Characteristic | PPMS | SPMS | RRMS |
|---|---|---|---|
| Number | 9 | 22 | 20 |
| Age | 52 (46–55) | 46 (29–65) | 40 (27–55) |
| Gender (female : male) | 6 : 3 | 11 : 11 | 11 : 9 |
| Disease duration (years) | 16 (6–27) | 18 (6–35) | 8 (4–13) |
| EDSS | 6 (2–8) | 6 (4–7) | 2 (1–8) |
| CSF NfHSMI34 (ng/mL) | 0.01 (0–0.05) | 0.11 (0–0.04) | 0.01 (0–0.04) |
| CSF NfHSMI35 (ng/mL) | 0.10 (0.01–0.15) | 0.04 (0.01–0.24) | 0.08 (0–1.39) |
| Serum NfHSMI34 (ng/mL) | 2.05 (0–3.08) | 0.03 (0.19–2.44) | 1.56 (0–2.05) |
| Serum NfHSMI35 (ng/mL) | 0 (0–0.49) | 0 (0–0.16) | 0 (0–0.27) |
The only biomarker distinguishing the MS groups were serum NfHSMI34 levels (F 2,48 = 3.63, P < .05). Significance was missed for CSF NfHSMI34 (P = .62), CSF NfHSMI35 (P = .59), and serum NfHSMI35 (P = .83).
There was no correlation between either the CSF and serum NfHSMI35 or NfHSMI34 concentration in any of the MS groups (data not shown). The post hoc analysis showed that serum NfHSMI34 levels were higher in PPMS patients compared to SPMS patients (P = .0128).
The concentration for NfHSMI35 was higher in the CSF compared to the serum in 8/9 (89%) of PPMS, 18/22 (82%) of SPMS, and 14/20 (70%) of RRMS patients. Surprisingly, for NfHSMI34 the CSF concentration was higher in 2/9 (22%) of PPMS, 11/22 (50%) of SPMS, and 5/20 (25%) of RRMS patients.
3.2. 3-Year Followup
The dropout rate for the followup clinical assessment was 4/9 (44%) for PPMS, 4/22 (18%) for SPMS, and 9/20 (45%) for RRMS patients.
At 3-year followup most of the MS patients had progressed clinically on the EDSS scale (Table 2). At followup, there was a difference between the MS groups for the EDSS (F 2,31 = 9.63, P < .001) and also for the individual progression on the EDSS (F 2,31 = 3.69, P < .05).
Table 2.
Patient characteristics at 3-year followup. The median (range) are shown.
| Characteristic | PPMS | SPMS | RRMS |
|---|---|---|---|
| Number | 5 | 18 | 11 |
| Gender (female : male) | 2 : 3 | 9 : 9 | 4 : 7 |
| EDSS | 7 (4–8) | 6 (3–8) | 4 (0–5) |
| ΔEDSS | 0 (−1–1.0) | 0 (−2–3) | 2 (−1–4) |
Tables 3–5 summarizes the demographic data of the MS patients according to their individual CSF and serum NfHSMI35 and NfHSMI34 levels.
Table 3.
Primary progressive MS. The patients were classified according to the relationship of CSF and blood NfH levels. If the concentration was higher in the CSF compared to the blood, this was indicated by “C > S” and “S ≥ C” otherwise. The data is presented for each of the two NfH phosphoforms quantified (NfHSMI34 and NfHSMI35).
| Characteristic | NfHSMI35 | NfHSMI34 | ||
|---|---|---|---|---|
| C > S | S ≥ C | C > S | S ≥ C | |
| Number | 4 | 1 | 2 | 3 |
| Age | 51 (46–55) | 52 | 48 (46–49) | 54 (52–54) |
| Gender (female : male) | 1 : 3 | 1 : 0 | 0 : 2 | 2 : 1 |
| Disease duration (years) | 13 (8–20) | 17 | 14 (8–20) | 16 (10–17) |
| EDSS | 5 (3–8) | 7 | 7 (7–8) | 3 (3–7) |
| ΔEDSS | 1 (−1–1) | 0 | 0 (−1–0) | 1 (0–1) |
Table 5.
Relapsing remitting MS. The patients were classified according to the relationship of CSF and blood NfH levels. If the concentration was higher in the CSF compared to the blood this was indicated by “C > S” and “S ≥ C” otherwise. The data is presented for each of the two NfH phosphoforms quantified (NfHSMI34 and NfHSMI35). ⋆ P < .05.
| Characteristic | NfHSMI35 | NfHSMI34 | ||
|---|---|---|---|---|
| C > S | S ≥ C | C > S | S ≥ C | |
| Number | 8 | 3 | 5 | 6 |
| Age | 41 (34–53) | 38 (29–40) | 40 (34–53) | 39 (29–48) |
| Gender (female : male) | 4 : 4 | 0 : 3 | 2 : 3 | 2 : 4 |
| Disease duration (years) | 10 (4–19) | 3 (1–10) | 8 (4–13) | 9 (1–19) |
| EDSS | 2 (0–4) | 2 (1–2) | 1 (1–4) | 2 (0–2) |
| ΔEDSS | 2 (0–4)⋆ | 0 (−1–1) | 2 (0–3) | 1 (−1–4) |
The low number of patients with PPMS at followup (n = 5) precluded any meaningful statistical analyzes. Disability progression (ΔEDSS) appeared to be associated with higher CSF than serum levels for NfHSMI35 and higher serum than CSF levels for NfHSMI34 (Table 3).
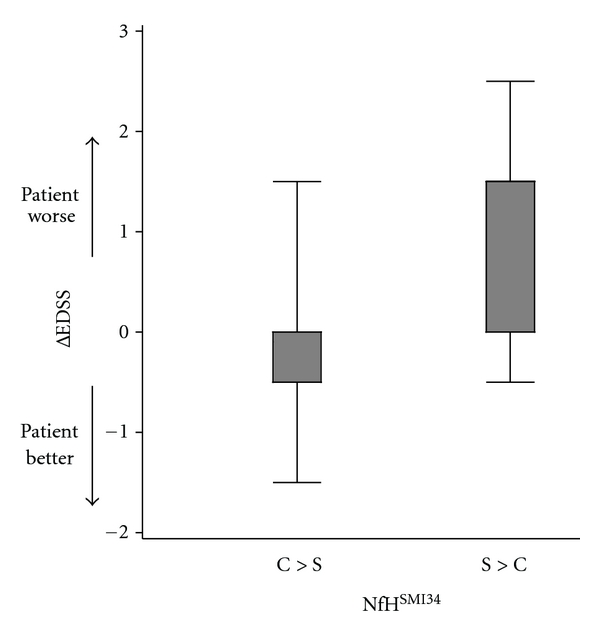
In patients with SPMS, NfHSMI34 predicted disability progression (ΔEDSS) from baseline if higher in the serum compared to the CSF (P = .0358, Table 4, Figure 2).
Table 4.
Secondary progressive MS. The patients were classified according to the relationship of CSF and blood NfH levels. If the concentration was higher in the CSF compared to the blood, this was indicated by “C > S” and “S ≥ C” otherwise. The data is presented for each of the two NfH phosphoforms quantified (NfHSMI34 and NfHSMI35). ⋆ P < .05.
| Characteristic | NfHSMI35 | NfHSMI34 | ||
|---|---|---|---|---|
| C > S | S ≥ C | C > S | S ≥ C | |
| Number | 15 | 3 | 11 | 7 |
| Age | 45 (29–55) | 57 (32–65) | 49 (32–65) | 44 (29–50) |
| Gender (female : male) | 6 : 9 | 3 : 0 | 7 : 4 | 2 : 5 |
| Disease duration (years) | 16 (6–28) | 22 (15–24) | 19 (6–28) | 16 (7–22) |
| EDSS | 5 (1–8) | 7 (6–8) | 6 (3–8) | 3 (1–7) |
| ΔEDSS | 0 (−1–3) | 0 (−2–0) | 0 (−1.5–1.5) | 2 (−1–3)⋆ |
Figure 2.
Disability progression in patients with SPMS is associated with higher serum NfHSMI34 levels compared to CSF data (P = .0358) suggesting that some degree of damage to the peripheral nervous system may exist in these patients.
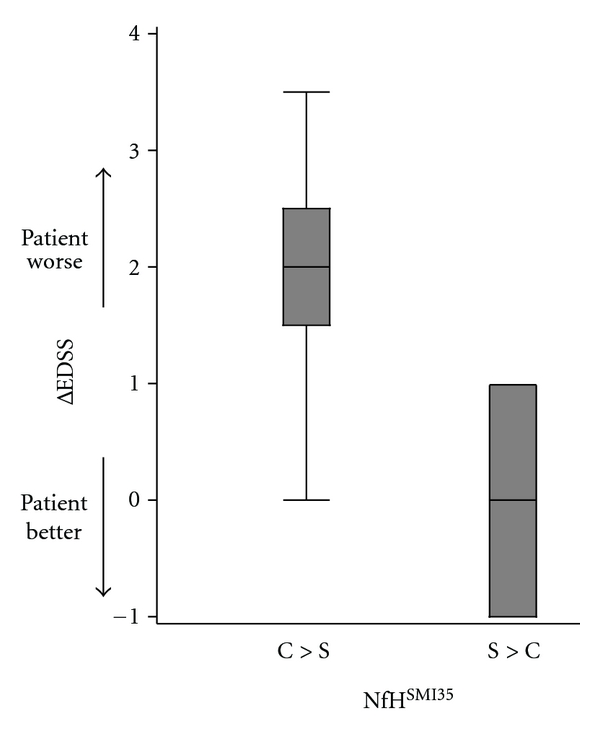
In patients with RRMS, NfHSMI35 predicted disability progression (ΔEDSS) from baseline if higher in the CSF compared to the serum (P = .0298, Table 5, Figure 1).
Figure 1.
Disability progression in patients with RRMS is associated with higher CSF NfHSMI35 levels compared to the corresponding serum concentrations (P = .0298) likely indicating neuroaxonal degeneration of the central nervous system. The median (thick horizontal bar), IQR (boxes), and range (whiskers) are shown.
4. Discussion
The main finding at baseline was that the concentration of NfH phosphoforms was not correlated between matched CSF and serum samples. Unexpectedly, a proportion of patients with MS had higher concentration of NfH phosphoforms in the serum compared to the CSF which was considerable for NfHSMI34 (50–88%). Furthermore the absolute concentration of serum NfHSMI34 was highest in patients with PPMS (Table 1). This is consistent with the notion that neurodegeneration may be more severe and predominating over inflammation in PPMS [22].
At baseline, there were also demographic differences between patients with RRMS, SPMS and PPMS. Patients with PPMS, and SPMS tended to be older and have a longer disease duration compared to patients with RRMS. These demographic differences did not appear to be related to CSF or blood NfH phosphoform levels, because no correlations were found. This is consistent with other NfH studies on CSF [23] and blood samples [13, 24] based on the same ELISA technique. The present study is underpowered to reveal weak correlations which remain statistical possible. Examining a larger cohort with aid of a newer and more sensitive ECL-based technique compared to our ELISA, age was found to correlated with NfHSMI35 levels [12].
Another weakness of this study was the high dropout of patients from baseline to followup. This was likely due to the requirement of a second spinal tap from this community rather than hospital-based cohort of MS patients [16, 25].
At 3-year followup, most patients had progressed on the EDSS, but some did improve (Table 2). Sustained progression on the EDSS was highest for patients with RRMS. Consistent with previous data on this [16] and other cohorts [9–12] of patients with MS, high CSF NfHSMI35 levels were of prognostic value (Figure 1). Because in these patients the concentration of CSF NfHSMI35 levels was higher compared to the matched serum (Table 5), it is suggested that the NfHSMI35 measured was of central origin.
The followup data on patients with PPMS and SPMS showed a prognostic value for higher concentration of NfHSMI34 in the serum compared to the CSF. This was significant for patients with SPMS (Figure 2). Among the number of different explanations, we tentatively suggest that it may be possible that the source for serum NfHSMI34 could at least in part originate from the peripheral nervous system. There is some clinical literature supporting this idea. Subtle alterations on routine electrophysiological measurements in patients with MS are found by some [26] but not by others [27]. More sophisticated measurements using nerve excitability measures [28] show changes in the motor nerve recovery cycle, providing indirect evidence for Na+/K+ ATPase pump dysfunction [29–31], a feature of MS pathology [32]. In addition, teased fibre studies from sural nerve biopsies in MS patients showed more disorganized axonal cytoskeleton (Figure 6 in [33]) similar to what is seen in the brain [34, 35]. In view of this data, we speculate that subtle neuroaxonal degeneration of the peripheral nervous system may be present in more severely disabled PPMS and SPMS patients.
An important limitation of our study is that in absence of specific tests [26–28], there is no direct evidence for damage to the peripheral nervous system. Therefore, this hypothesis will need to be investigated prospectively to be substantiated or defeated. We think this is important in order to ensure that attempts to find a blood-based biomarker for central neuroaxonal degeneration in MS are not contaminated by possible pathology of the peripheral nervous system.
It is noted that the results for NfHSMI34 are different to NfHSMI35. For NfHSMI34, serum levels were frequently higher than CSF levels compared to NfHSMI35. Trapp et al. reported changes of NfH phosphorylation, particularly dephosphorylation of demyelinated axons in the MS brain [1]. As MS progresses from RR to SP disease, the burden of altered NfH phosphorylation increases [34–36]. Because NfHSMI34 binds to a wider range of NfH phosphoforms than NfHSMI35, it may be that serum NfHSMI34 levels are more sensitive in detecting axonal damage in MS than serum NfHSMI35 levels. This argumentation would be consistent with the finding that a higher concentration of NfHSMI34 in the serum compared to the CSF was predictive of disease progression in SPMS.
Could inflammation-related impairment of the blood brain barrier (BBB) function in MS explain higher blood than CSF NfH levels? We do not think so. Historically, the concept of the BBB originated from the observation that certain compounds did not diffuse freely into the central nervous system (CNS), but they would lead to dramatic symptoms if injected intracerebrally, intraventricularly, or intrathecally [37]. For over a century, research on the BBB has focused on diffusion of compounds into the brain. It is now well established that assessment of BBB integrity requires quantification of compounds on both sides of the barrier (reviewed in [38]). In fact, two barriers need to be considered: the morphologically defined BBB and the functionally defined blood CSF barrier (BCB) [6, 38, 39]. Large molecules (e.g., IgM with a molecular weight of 800 kD) can pass the barriers in very small quantities (e.g., IgM serum : IgM CSF = 3000 : 1). Smaller molecules pass through the barriers more easily because of molecular size-dependent diffusion (QAlb = 1 : 200; QIgG = 1 : 500). Starling's principle applies, and an increase of QAlb can be caused by a reduced CSF flow rate without any leakage in the morphological structures [6, 38]. The concept of using a biomarker for parenchymal brain damage (e.g., NfH) for a “BBB leakage” model is incompatible with Starling's principle and the well-established physiology of the BBB/BCB function. Brain-derived proteins in blood can indicate brain damage, as consistently reported by a number of groups, but “leakage” of the BBB/BCB is not a precondition of increased blood concentrations.
Could the localization of MS lesion formation in the CNS influence whether products of damage are predominantly released into CSF or blood? This certainly is a possibility. The CSF flow dynamics are such that biomarkers released from cortical pathology are likely to diffuse through the cortical arachnoid villi into the blood stream and only a fraction may reach the lumbar CSF [40]. In contrast, pathology of the spinal cord is more likely to be reflected in lumbar CSF. Neurofilaments are one of the few CNS protein biomarkers with a higher lumbar spinal CSF concentration compared to ventricular CSF [41]. The likely reason for this anatomical: there is a rostrocaudal gradient of the parenchymal Nf protein concentration with the lowest concentration in cortical neurons and the highest concentration in spinal cord axons [34, 42, 43]. It could, therefore, be that a small amount of spinal cord damage may mask more extensive cortical damage if investigated from lumbar CSF alone. Conversely, one may hypothesize that blood NfH levels may be better suited for investigating cortical pathology. This hypothesis is tempting, because of the relative ease to obtain serial blood samples as opposed to CSF samples. Precisely for testing, this hypothesis it will be important to ensure that there is no data contamination by pathology of the PNS as tentatively suggested by the present study.
Acknowledgments
This study was in part funded by the Dutch MS Research Foundation (JE). The MS Center VUMC is partially funded by a program grant of the Dutch MS Research Foundation.
References
- 1.Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mörk S, Bö L. Axonal transection in the lesions of multiple sclerosis. The New England Journal of Medicine. 1998;338(5):278–285. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- 2.Ferguson B, Matyszak MK, Esiri MM, Perry VH. Axonal damage in acute multiple sclerosis lesions. Brain. 1997;120(3):393–399. doi: 10.1093/brain/120.3.393. [DOI] [PubMed] [Google Scholar]
- 3.Seewann A, Vrenken H, van der Valk P, et al. Diffusely abnormal white matter in chronic multiple sclerosis: imaging and histopathologic analysis. Archives of Neurology. 2009;66(5):601–609. doi: 10.1001/archneurol.2009.57. [DOI] [PubMed] [Google Scholar]
- 4.Petzold A, Tisdall MM, Girbes AR, et al. In vivo monitoring of neuronal loss in traumatic brain injury: a microdialysis study. Brain. 2011;134(2):464–483. doi: 10.1093/brain/awq360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lakshmanan R, Loo JA, Drake T, et al. Metabolic crisis after traumatic brain injury is associated with a novel microdialysis proteome. Neurocritical Care. 2010;12:324–336. doi: 10.1007/s12028-010-9342-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Petzold A. CSF biomarkers for improved prognostic accuracy in acute CNS disease. Neurological Research. 2007;29(7):691–708. doi: 10.1179/016164107X240080. [DOI] [PubMed] [Google Scholar]
- 7.Petzold A. Neurofilament phosphoforms: surrogate markers for axonal injury, degeneration & loss. Journal of the Neurological Sciences. 2005;233(1-2):183–198. doi: 10.1016/j.jns.2005.03.015. [DOI] [PubMed] [Google Scholar]
- 8.Teunissen CE, Dijkstra PC, Polman C. Biological markers in CSF and blood for axonal degeneration in multiple sclerosis. Lancet Neurology. 2005;4(1):32–41. doi: 10.1016/S1474-4422(04)00964-0. [DOI] [PubMed] [Google Scholar]
- 9.Norgren N, Sundström P, Svenningsson A, Rosengren L, Stigbrand T, Gunnarsson M. Neurofilament and glial fibrillary acidic protein in multiple sclerosis. Neurology. 2004;63(9):1586–1590. doi: 10.1212/01.wnl.0000142988.49341.d1. [DOI] [PubMed] [Google Scholar]
- 10.Teunissen CE, Iacobaeus E, Khademi M, et al. Combination of CSF N-acetylaspartate and neurofilaments in multiple sclerosis. Neurology. 2009;72(15):1322–1329. doi: 10.1212/WNL.0b013e3181a0fe3f. [DOI] [PubMed] [Google Scholar]
- 11.Salzer J, Svenningsson A, Sundström P. Neurofilament light as a prognostic marker in multiple sclerosis. Multiple Sclerosis. 2010;16(3):287–292. doi: 10.1177/1352458509359725. [DOI] [PubMed] [Google Scholar]
- 12.Kuhle J, Regeniter A, Leppert D, et al. A highly sensitive electrochemiluminescence immunoassay for the neurofilament heavy chain protein. Journal of Neuroimmunology. 2010;220(1-2):114–119. doi: 10.1016/j.jneuroim.2010.01.004. [DOI] [PubMed] [Google Scholar]
- 13.Petzold A, Rejdak K, Plant GT. Axonal degenaration and inflammation in acute optic neuritis. Journal of Neurology, Neurosurgery and Psychiatry. 2004;75(8):1178–1180. doi: 10.1136/jnnp.2003.017236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shaw G, Yang C, Ellis R, et al. Hyperphosphorylated neurofilament NF-H is a serum biomarker of axonal injury. Biochemical and Biophysical Research Communications. 2005;336(4):1268–1277. doi: 10.1016/j.bbrc.2005.08.252. [DOI] [PubMed] [Google Scholar]
- 15.Siman R, Toraskar N, Dang A, et al. A panel of neuron-enriched proteins as markers for traumatic brain injury in humans. Journal of Neurotrauma. 2009;26(11):1867–1877. doi: 10.1089/neu.2009.0882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Petzold A, Eikelenboom MJ, Keir G, et al. Axonal damage accumulates in the progressive phase of multiple sclerosis: three year follow up study. Journal of Neurology, Neurosurgery and Psychiatry. 2005;76(2):206–211. doi: 10.1136/jnnp.2004.043315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Petzold A, Eikelenboom MJ, Gveric D, et al. Markers for different glial cell responses in multiple sclerosis: clinical and pathological correlations. Brain. 2002;125(7):1462–1473. doi: 10.1093/brain/awf165. [DOI] [PubMed] [Google Scholar]
- 18.Lublin FD, Reingold SC. Defining the clinical course of multiple sclerosis: results of an international survey. National Multiple Sclerosis Society (USA) Advisory Committee on Clinical Trials of New Agents in Multiple Sclerosis. Neurology. 1996;46(4):907–911. doi: 10.1212/wnl.46.4.907. [DOI] [PubMed] [Google Scholar]
- 19.Teunissen CE, Petzold A, Bennett JL, et al. A consensus protocol for the standardization of cerebrospinal fluid collection and biobanking. Neurology. 2009;73(22):1914–1922. doi: 10.1212/WNL.0b013e3181c47cc2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kurtzke JF. Rating neurologic impairment in multiple sclerosis: an expanded disability status scale (EDSS) Neurology. 1983;33(11):1444–1452. doi: 10.1212/wnl.33.11.1444. [DOI] [PubMed] [Google Scholar]
- 21.Petzold A, Keir G, Green AJE, Giovannoni G, Thompson EJ. A specific ELISA for measuring neurofilament heavy chain phosphoforms. Journal of Immunological Methods. 2003;278(1-2):179–190. doi: 10.1016/s0022-1759(03)00189-3. [DOI] [PubMed] [Google Scholar]
- 22.Thompson AJ, Polman CH, Miller DH, et al. Primary progressive multiple sclerosis. Brain. 1997;120(6):1085–1096. doi: 10.1093/brain/120.6.1085. [DOI] [PubMed] [Google Scholar]
- 23.Rejdak K, Petzold A, Stelmasiak Z, Giovannoni G. Cerebrospinal fluid brain specific proteins in relation to nitric oxide metabolites during relapse of multiple sclerosis. Multiple Sclerosis. 2008;14(1):59–66. doi: 10.1177/1352458507082061. [DOI] [PubMed] [Google Scholar]
- 24.Petzold A, Brassat D, Mas P, et al. Treatment response in relation to inflammatory and axonal surrogate marker in multiple sclerosis. Multiple Sclerosis. 2004;10(3):281–283. doi: 10.1191/1352458504ms1021sr. [DOI] [PubMed] [Google Scholar]
- 25.Eikelenboom MJ, Petzold A, Lazeron RHC, et al. Multiple sclerosis: neurofilament light chain antibodies are correlated to cerebral atrophy. Neurology. 2003;60(2):219–223. doi: 10.1212/01.wnl.0000041496.58127.e3. [DOI] [PubMed] [Google Scholar]
- 26.Gartzen K, Katzarava Z, Diener H-C, Putzki N. Peripheral nervous system involvement in multiple sclerosis. doi: 10.1111/j.1468-1331.2010.03149.x. European Journal of Neurology. In press. [DOI] [PubMed] [Google Scholar]
- 27.Antonini G, Millefiorini E, Borsellino G, Morino S, Rasura M, Pozzilli C. Subclinical peripheral nervous system involvement in multiple sclerosis. Muscle and Nerve. 1995;18(10):1216–1217. [PubMed] [Google Scholar]
- 28.Bostock H. Nerve excitability studies: past, present, future? Supplements to Clinical Neurophysiology. 2004;57:85–90. doi: 10.1016/s1567-424x(09)70346-0. [DOI] [PubMed] [Google Scholar]
- 29.Eisen A, Paty D, Hoirch M. Altered supernormality in multiple sclerosis peripheral nerve. Muscle and Nerve. 1982;5(5):411–414. doi: 10.1002/mus.880050513. [DOI] [PubMed] [Google Scholar]
- 30.Burke D, Kiernan MC, Bostock H. Excitability of human axons. Clinical Neurophysiology. 2001;112(9):1575–1585. doi: 10.1016/s1388-2457(01)00595-8. [DOI] [PubMed] [Google Scholar]
- 31.Boërio D, Créange A, Hogrel JY, Lefaucheur JP. Alteration of motor nerve recovery cycle in multiple sclerosis. Clinical Neurophysiology. 2007;118(8):1753–1758. doi: 10.1016/j.clinph.2007.04.025. [DOI] [PubMed] [Google Scholar]
- 32.Trapp BD, Stys PK. Virtual hypoxia and chronic necrosis of demyelinated axons in multiple sclerosis. The Lancet Neurology. 2009;8(3):280–291. doi: 10.1016/S1474-4422(09)70043-2. [DOI] [PubMed] [Google Scholar]
- 33.Pollock M, Calder C, Allpress S. Peripheral nerve abnormality in multiple sclerosis. Annals of Neurology. 1977;2(1):41–48. doi: 10.1002/ana.410020107. [DOI] [PubMed] [Google Scholar]
- 34.Petzold A, Gveric D, Groves M, et al. Phosphorylation and compactness of neurofilaments in multiple sclerosis: indicators of axonal pathology. Experimental Neurology. 2008;213(2):326–335. doi: 10.1016/j.expneurol.2008.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dutta R, McDonough J, Yin X, et al. Mitochondrial dysfunction as a cause of axonal degeneration in multiple sclerosis patients. Annals of Neurology. 2006;59(3):478–489. doi: 10.1002/ana.20736. [DOI] [PubMed] [Google Scholar]
- 36.Schirmer L, Antel JP, Brück W, Stadelmann C. Axonal loss and neurofilament phosphorylation changes accompany lesion velopment and clinical progression in multiple sclerosis. doi: 10.1111/j.1750-3639.2010.00466.x. Brain Pathology. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Biedel A, Kraus R. Über einer bisher unbekannte toxische wirkung der gallensäure auf das zentralnervensystem. Zentralblatt für Innere Medizin. 1898;19:1185–1200. [Google Scholar]
- 38.Reiber H. Proteins in cerebrospinal fluid and blood: barriers, CSF flow rate and source-related dynamics. Restorative Neurology and Neuroscience. 2003;21(3-4):79–96. [PubMed] [Google Scholar]
- 39.Reiber H. Flow rate of cerebrospinal fluid (CSF)—a concept common to normal blood-CSF barrier function and to dysfunction in neurological diseases. Journal of the Neurological Sciences. 1994;122(2):189–203. doi: 10.1016/0022-510x(94)90298-4. [DOI] [PubMed] [Google Scholar]
- 40.Petzold A. The CSF Analysis in Dementia. 1st edition. New York, NY, USA: NOVA Science Publishers; 2008. Biomarker for early diagnosis of Alzheimer’s disease; pp. 123–156. [Google Scholar]
- 41.Tarnaris A, Toma AK, Chapman MD, et al. Rostrocaudal dynamics of CSF biomarkers. Neurochemical Research. 2011;36(3):528–532. doi: 10.1007/s11064-010-0374-1. [DOI] [PubMed] [Google Scholar]
- 42.Anderson KJ, Scheff SW, Miller KM, et al. The phosphorylated axonal form of the neurofilament subunit NF-H (pNF-H) as a blood biomarker of traumatic brain injury. Journal of Neurotrauma. 2008;25(9):1079–1085. doi: 10.1089/neu.2007.0488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marszalek JR, Williamson TL, Lee MK, et al. Neurofilament subunit NF-H modulates axonal diameter by selectively slowing neurofilament transport. The Journal of Cell Biology. 1996;135(3):711–724. doi: 10.1083/jcb.135.3.711. [DOI] [PMC free article] [PubMed] [Google Scholar]