
Figure 1.
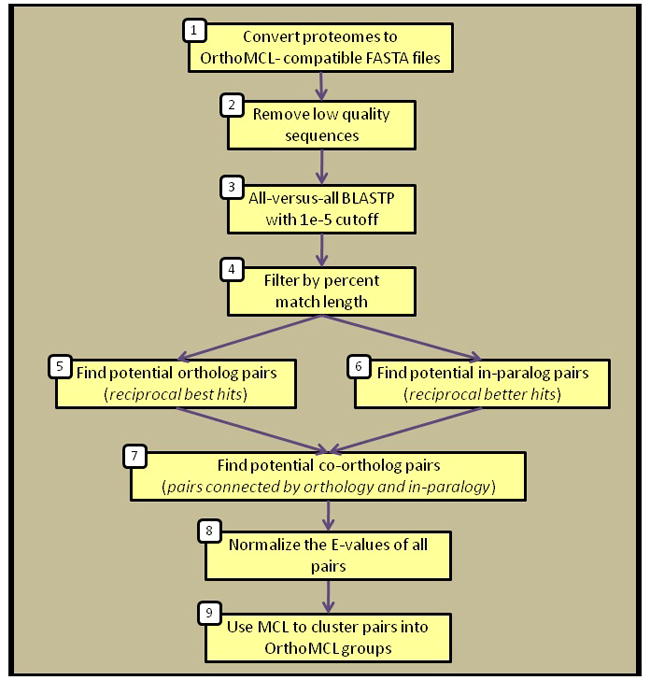
Overview of the OrthoMCL algorithm. (1) Proteomes must each be in FASTA format where the file name and definition lines comply with simple requirements. (2) The proteome files are filtered to remove low quality sequences based on length and percent stop codons. (3) The proteomes are all compared to each other using BLASTP. They are masked with seg and an E-value cutoff of 1e-5 is applied. (4) For each pair of sequences that match, compute the “percent match length” score: count the number of amino acids in the shorter sequence that participate in any HSP, divide that by the length of the shorter sequence and multiply by 100. Filter away matches with percent match < 50%. (5) For all pairs of proteomes, find all pairs of proteins across them that have hits as good as or better than any other hits between these proteins and other proteins in those species. (6) Find all pairs of proteins within a species that have mutual E-values that are better or equal to all of those proteins’ hits to proteins in other species. (7) Find all pairs of proteins across two species that are connected through orthology and in-parology. (8) Normalize in-paralog E-values by averaging all qualifying in-paralog pairs in a genome and divide each pair by the average. Within a genome, in-paralog pairs qualify if either of the proteins in the pair has an ortholog in any genome. If no in-paralogs within a genome have any orthologs, all in-paralogs in that genome qualify. Normalize ortholog and co-ortholog pairs for any two species by averaging the E-values across them, and normalize using that average. (9) Pass all ortholog, in-paralog and co-ortholog pairs, with their normalized E-values, to the MCL program for clustering.