

SUMMARY
Why do opiates make human beings itch ? Spinal opioid-induced itch, a prevalent side effect of pain management, has been considered to occur as a result of pain inhibition. We report that morphine-induced scratching (MIS) is abolished in mice lacking either gastrin-releasing peptide receptor (GRPR) or the μ opioid receptor (MOR). Using exon-specific knockdown, we identified the MOR1D isoform as essential for MIS, whereas MOR1 is important for morphine-induced analgesia (MIA) with no cross activity present. MOR1D and GRPR form constitutive heterodimers in the spinal cord and relay itch information upon morphine activation. Morphine induces internalization of both GRPR and MOR1D, whereas GRP induces that of GRPR but not MOR1D, when co-expressed. Moreover, GRP-induced scratching (GIS) is independent of MOR activation. These results suggest a unidirectional cross-activation of GRPR signaling by MOR1D via heterodimerization, and that opioid-induced itch is an active process concomitant with but independent of opioid analgesia.
INTRODUCTION
Itch and pain are two fundamental sensory perceptions evoked by distinct external inputs. They are encoded and transmitted by primary nociceptive fibers and dorsal horn neurons that activate contralateral supraspinal regions (Davidson and Giesler, 2010; Patel and Dong, 2010). The ability to discriminate between itch and pain allows animals to employ the proper motor response (scratching vs. withdrawal) so that potentially damaging stimuli from the environment can be avoided. Intriguingly, it has been well documented that itch and pain may counteract each other under some conditions; a wide range of noxious stimuli including thermal, mechanical, chemical and electrical stimuli are able to inhibit itch (Ikoma et al., 2006). Conversely, it is widely assumed that itch may be unmasked by pain reduction, and one of the most cited examples of this antagonistic relationship is opioid-induced itch, or pruritus (Davidson and Giesler, 2010; Ikoma et al., 2006; Paus et al., 2006). In fact, pruritus is one of the most prevalent acute side effects of the spinal or epidural use of opioids in patients who undergo pain treatment or in those who receive a cesarean section (Ballantyne et al., 1988; Chaney, 1995; Hales, 1980), which has hampered the use of opioids as an analgesic. The most influential theory offered to explain the antagonism of itch and pain is perhaps the “occlusion” or selectivity hypothesis, which stipulates that pruriceptors are a subpopulation of nociceptors and that an inactivation of the pain signaling centrally is a prerequisite for activation of the itch signaling (Carstens, 1997; McMahon and Koltzenburg, 1992). The occlusion hypothesis has gained more support from an analysis of mutant mice lacking vesicular glutamate transporter 2 in subsets of dorsal root ganglia (DRG) neurons that displayed attenuated pain but enhanced itch (Lagerstrom et al., 2010; Liu et al., 2010). In the spinal cord of primates, all lamina I spinothalamic track neurons that were responsive to capsaicin also responded to pruritic stimuli (Davidson et al., 2007). In addition, ablation of dorsal horn neurons expressing neurokinin 1 receptor attenuated both pain and itch in rats (Carstens et al., 2010; Nichols et al., 1999). In mice lacking neurons that express gastrin-releasing peptide receptor (GRPR), a molecular signature for the putative itch-specific labeled line in the spinal cord, scratching responses to a range of pruritic stimuli are nearly abolished, but nociceptive transmission is not altered (Sun and Chen, 2007; Sun et al., 2009). Conversely, mice lacking a subset of neurons expressing transcription factor Bhlhb5 during development display enhanced spontaneously scratching behavior but their pain behavior is not reduced (Ross et al., 2010), suggesting that removal of pain signaling is not a prerequisite for induction of itch and that the central itch signaling can be induced independently of nociceptive transmission. Collectively, convincing evidence in support of “occlusion” theory in the spinal cord is lacking.
Opioid-induced itch has been suggested to be mediated primarily through the μ opioid receptor (MOR), a key receptor for opiates (Kieffer, 1999). Intrathecal (i.t.) injection of morphine, a prototypical opiate agonist, produces dose-dependent scratching behavior (Ko and Naughton, 2000; Kuraishi et al., 2000). Consistently opioid antagonists have been found to reduce itch and concomitantly attenuate the analgesic effects of opiates (Ballantyne et al., 1988; Ko et al., 2004). MOR1 is activated by morphine without rapid internalization in several cell types including dorsal horn neurons (Alvarez et al., 2002; Keith et al., 1996; Trafton et al., 2000). Activation of MOR1 primarily inhibits adenylyl cyclase, and the cAMP/PKA signaling pathway (Law et al., 2000). Since opioid-induced itch/pruritus is most notable and severe when opioids are intrathecally applied, one tantalizing hypothesis is that opioids evoke itch sensation by activating GRPR signaling. The present study was designed to test this hypothesis and to determine whether activation of the itch signaling is due to a removal of pain inhibition.
RESULTS
Morphine-Induced Scratching (MIS) Occurs Independent of Morphine-Induced Analgesia (MIA)
To examine whether MIS and MIA are correlated to each other, we studied the dose-response curve and time course of MIS and MIA after i.t. injection of morphine and found that both MIA and MIS increased in a dose-dependent manner (Figure 1A). However, when the morphine dose increased from 0.3 nmol to 1.0 nmol, MIA effect was enhanced by 81%, while MIS only had a slight increase. In addition, time course analysis at 0.3 nmol of morphine revealed obvious segregation of MIA and MIS (Figure 1B). After i.t. morphine injection, MIS increased dramatically within 10 min and then quickly decreased, while MIA was maintained for at least 1 hr. To further examine whether opioid-induced itch is due to pain inhibition, we employed a morphine tolerance paradigm in which the degree of tolerance to morphine is measured by the latency of tail-flick (analgesic effect) (Fairbanks and Wilcox, 1999). If pain inhibition unmasks itch, MIS would attenuate in mice with morphine tolerance. Twenty-four hr after morphine pretreatment, tail-flick latencies of mice returned to their baseline (Figure 1C). As expected, mice pretreated with morphine developed morphine tolerance as measured by a significant reduction of MIA relative to the saline control (Figure 1D). To our surprise, despite reduced analgesic effect, MIS did not differ between the two groups (Figure 1E). Separation of MIS from MIA was also examined by chronic morphine tolerance model. Tail immersion assay showed gradually reduced amplitude of MIA during the five days of induction (Figure 1F), and morphine tolerance was evident on the sixth day (Figure 1G). Again, there was no significant difference of MIS between the control and tolerant mice (Figure 1H). Therefore, MIS occurs irrespective of the degree of MIA, indicating that MIS and MIA are mediated by distinct mechanisms.
Figure 1. MIS is not Correlated with MIA.
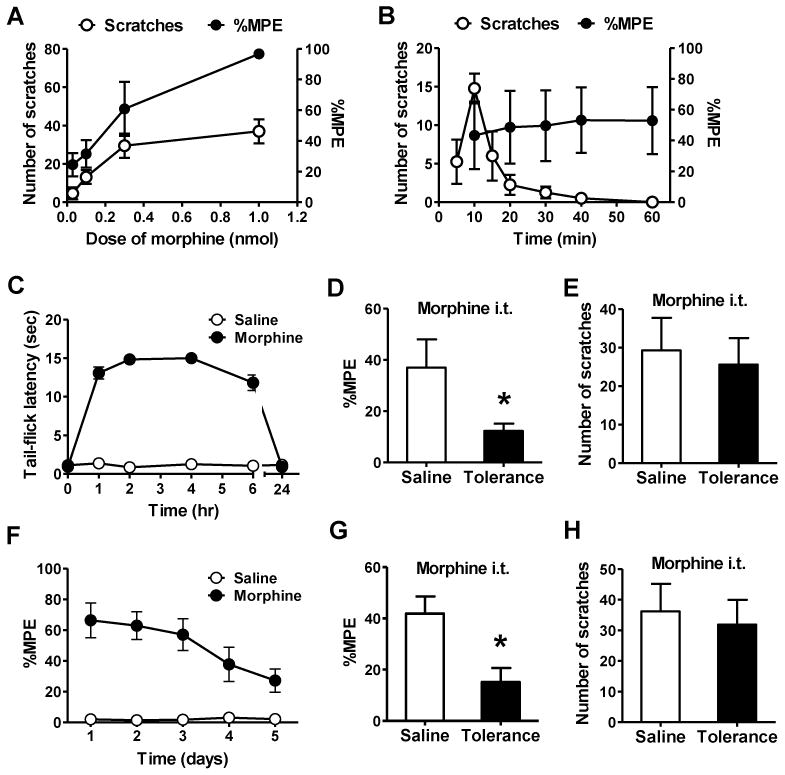
(A) Dose effect of i.t. morphine on MIS and MIA in 30 min.
(B) Time course of morphine on MIS and MIA.
(C) Induction of acute MIA tolerance with morphine (100 mg/kg, s.c.) or saline. Mice had returned to the basal nociceptive latencies 24 hr after injection.
(D) Acute MIA tolerance was tested with i.t. morphine 24 hr after s.c. morphine treatment.
(E) I.t. morphine induced comparable scratches in acute MIA tolerant and control mice.
(F) Induction of chronic MIA tolerance by daily injection of morphine (10 mg/kg, s.c.) and MIA was examined daily.
(G) After 5 days of systemic morphine injection, i.t. morphine also showed antinociceptive tolerance.
(H) I.t. morphine induced comparable scratches in chronic MIA tolerant and control mice. In all experiments, the dose of i.t. morphine is 0.3 nmol. n = 6~8 per group. MIA results are expressed as % maximum possible effect (MPE). *p < 0.05. Error bars represent standard error of the mean.
MOR1D is an Itch-Specific Receptor
The finding that MIS is separable from MIA prompted us to study its molecular basis. Mice lacking the Oprm gene displayed loss of MIA (Loh et al., 1998; Matthes et al., 1996; Sora et al., 1997). MIS was nearly abolished in mice lacking the coding exons 2 and 3 of the Oprm gene (Loh et al., 1998), whereas gastrin-releasing peptide-induced scratching (GIS) was not affected (Figure 2A). Consistent with previous studies (Ballantyne et al., 1988; Ko et al., 2004), MIS was also abolished by naloxone, a non-specific MOR antagonist (Figure 2B). In contrast, neither naloxone (Figure S1A) nor beta-FNA (Figure S1B) impacted GIS. The mouse Oprm gene consists of 16 coding exons, comprising dozens of spliced isoforms that primarily differ at the C-terminus (Pan, 2005; Pasternak). For example, MOR1 consists of exon 1~4, while MOR1D of exons 1~3 and 8~9 (Figure 2C). The multiplicity of the Oprm isoform system has been suggested to underlie the heterogeneity and variability of analgesic and scratching effects exerted by different agonists (Andoh et al., 2008; Pasternak, 2004; Ravindranathan et al., 2009). We postulated that different isoforms are responsible for MIS and MIA respectively. To test this, we performed exon-specific siRNA knockdown experiments in the spinal cord of mice, followed by an examination of MIS. Knockdown of either exon 1 contained by the majority of MOR isoforms including MOR1, or exon 9 contained by isoforms 1C, 1D and 1E significantly attenuated MIS (Figure 2D). However, siRNA knockdown of exon 4 contained by MOR1 or exon 7 contained by 1C and 1E failed to reduce MIS significantly (Figure 2D). Interestingly, knockdown of exon 1 or 4 markedly attenuated MIA, whereas knockdown of exon 7 or 9 had no effect on MIA (Figure 2E). Quantitative RT-PCR tests confirmed that spinal MOR1 mRNA significantly reduced after exon 1 or exon 4 siRNA (Figure 2F and 2G), and that spinal MOR1D mRNA significantly reduced after exon 1 or exon 9 siRNA (Figure 2F and 2H). In contrast, neither MOR1 nor MOR1D expression in DRG neurons was compromised by siRNA treatments (Figures S1C and S1D). The knockdown of MOR1D protein in the spinal cord by exon 9 siRNA was verified by Western blot and confirmed that MOR1 and GRPR protein levels were not affected (Figure 2I and 2J). To further exclude the possibility that exon 9 siRNA treatment might affect GRPR function, we examined i.t. GIS, and found no significant reduction of GIS after MOR isoform knockdown (Figure S1E). These results indicate that exon 9 is critical for MIS but not for MIA, whereas exon 4 is critical for MIA but not for MIS. Thus, spinal MOR1D has emerged as a MIS-specific isoform, whereas MOR1 possesses a MIA-specific function.
Figure 2. Identification of MIA- and MIS-Specific MOR isoforms.

(A) MIS was severely impaired in MOR KO mice, whereas GIS in MOR KO mice was comparable to that in wild-type littermate control mice.
(B) MIS was significantly reduced by naloxone (3 mg/kg, s.c.).
(C) Schematic representation of partial alternative Oprm splicing in the mouse. Clear rectangles represent the targeting exons by siRNA.
(D) MIS was significantly reduced by MOR siRNA targeting at exon 1 (MOR1, 1C, 1D, and 1E) and exon 9 (MOR1C, 1D, and 1E), but not by siRNA targeting at exon 4 (MOR1) or exon 7 (MOR1C and 1E). *p < 0.05. Sequence of siRNAs are included in supplementary file.
(E) MOR siRNA targeting at exon 1 and exon 4, but not exon 7 or exon 9 significantly reduced morphine analgesic effect.
(F) Representative gel images showing decreased spinal MOR1 mRNA level after exon 1 and exon 4 specific siRNA treatments and decreased spinal MOR1D mRNA level after exon 1 and exon 9 specific siRNA treatments. 18S RNA, an internal control, was comparable among all groups.
(G) Exon 1 and exon 4 specific siRNA significantly knocked down MOR1 mRNA in spinal cord as detected by q-RT-PCR.
(H) Spinal MOR1D mRNA level was significantly reduced by siRNA specific to MOR exon 1 and exon 9 as detected by qRT-PCR.
(I and J) Western blot (I) and quantified data (J) showed MOR exon 9 siRNA treatment specifically reduced protein level of MOR1D but not that of MOR1 or GRPR in the spinal cord. In all experiments, n = 5~8 per group. *p < 0.05. Error bars represent standard error of the mean. Also see Figure S1
Co-Localization of GRPR and MOR1D in the Dorsal Horn of the Spinal Cord
To determine the expression pattern of MOR1D, we generated an antibody specific against the unique MOR1D C-terminus using a strategy previously described (Abbadie et al., 2000). MOR1D and MOR1 antibodies specifically recognized human embryonic kidney 293 (HEK 293) cells transfected with either MOR1D or MOR1 (Figure S2A), with no cross activity observed between the two antibodies. These data validate the specificity of the MOR1D antibody. Immunostaining using MOR1D antibody indicates that MOR1D is expressed mainly in lamina I of the spinal cord (Figure 3A and Figure S2B); no staining was observed in the spinal cord of MOR KO mouse (Figure S2B). In contrast, MOR1 staining is largely restricted to lamina II with a few in lamina I (Figure 3B). Importantly, no co-localization of MOR1 and MOR1D was detected (Figure 3C). We next examined whether MOR1D expression overlaps with GRPR expression. Double-staining of MOR1D and GRPR revealed that the expression of the two receptors overlaps in lamina I cells (Figures 3D–3F). In twenty-five sections across the lumbar spinal cord, approximately 31% of GRPR+ cells co-stained with MOR1D, and ~65% of MOR1D+ cells co-stained with GRPR. No overlapping expression between GRPR and MOR1 was observed (Figures 3G–3I). This result suggests that GRPR signaling pathway may play a role in MIS.
Figure 3. Co-Expression of GRPR and MOR1D in Lamina I of the Spinal Cord.

(A–C) Double immunostaining of GRPR (red, lamina I) and MOR1D (green, lamina II) revealed no co-localization of GRPR and MOR1 in the dorsal spinal cord.
(D–F) Double immunostaining of GRPR (red) and MOR1D (green) in lamina I of the spinal cord. Arrows indicate co-expression (yellow) and arrowheads indicate singular expression. Cells co-expressing GRPR (11/33) and MOR1D (11/17), which represent ~31% of GRPR-positive cells and ~65% of MOR1D-positive cells respectively, were found in 25 lumbar spinal cord sections.
(G–I) Double immunostaining revealed no co-localization of GRPR (red, lamina I) and MOR1 (green, lamina II) in the dorsal spinal cord.
Scale bar, 50 μm. Also see Figure S2
Abolition of Opioid-Induced Scratching by the Blockade of the GRPR Function in the Spinal Cord
To examine whether GRPR is important for mediating opioid-induced itch, we compared MIS between GRPR KO and wild-type mice. Strikingly, MIS was nearly abolished in GRPR KO mice (Figure 4A). In contrast, no significant difference in MIA was observed between the groups (Figure 4B). The abolition of MIS in GRPR KO mice was recapitulated when MOR agonists (DAMGO or fentanyl) was intrathecally injected (Figures 4C and 4E). Analgesic effects of DAMGO or fentanyl did not differ between GRPR KO and their littermate controls (Figures 4D and 4F). Consistently, i.t. injection of the GRPR antagonist significantly inhibited MIS (Figure 4G), whereas MIA remained unchanged (Figure 4H and Figure S3A). These findings demonstrate that GRPR is required for MIS, but not for anti-nociceptive transmission. Importantly, the GRPR antagonist itself has no significant effect on acute pain as tested by tail immersion assay (Figure S3B) and von Frey (Figure S3C). Therefore, GRPR is essential for mediating opioid-induced itch, but not for opioid-mediated anti-nociception.
Figure 4. GRPR is Important for Opioid-Induced Scratching Behavior.

(A) MIS was nearly abolished in GRPR KO mice compared with wild-type littermate mice.
(B) MIA was comparable between GRPR KO and wild-type littermates.
(C) Scratching behavior induced by i.t. DAMGO was significantly reduced in GRPR KO mice.
(D) Analgesic effect of i.t. DAMGO was comparable between GRPR KO and wild-type littermates.
(E and F) Scratching behavior induced by i.t. fentanyl was significantly reduced in GRPR KO mice (E), while the analgesic effect of fentanyl was not affected (F).
(G) MIS was significantly inhibited by co-injection with the GRPR antagonist (0.1, 1 nmol).
(H) MIA was not significantly affected by co-injection of the GRPR antagonist.
In all experiments, the dose of i.t. morphine is 0.3 nmol. n = 6~9 per group. *p < 0.05. Error bars represent standard error of the mean. See also Figure S3.
Heterodimerization and Co-Internalization of MOR1D and GRPR
The co-expression of GRPR and MOR1D, along with their requirement for MIS, prompted us to ask whether GRPR and MOR1D may physically interact through receptor heterodimerization, a mechanism commonly employed by GPCRs to increase their diverse pharmacological and physiological properties (Bouvier, 2001; Milligan, 2009). Co-immunoprecipitation (co-IP) was performed using extracts of HEK 293 cell stably expressing Myc-tagged GRPR, together with HA-tagged MOR1D or HA-tagged MOR1. Anti-HA antibody precipitated a band corresponding in size to Myc-GRPR in cells co-expressing Myc-GRPR and HA-MOR1D (Figure 5A, L4). Conversely, precipitation with anti-Myc antibody identified a band corresponding to HA-MOR1D in cells co-expressing GRPR and MOR1D (Figure 5B, L4). This physical interaction is specific to MOR1D because HA-MOR1 and Myc-GRPR did not precipitate each other in cells co-expressing GRPR and MOR1 (Figure 5A and 5B, L3). To examine the physical interaction of MOR1D and GRPR in vivo, we performed co-IP experiments using the spinal cord membrane preparation. GRPR was co-precipitated with MOR1D by anti-MOR1D antibody (Figure 5C, L3), but not by MOR1 antibody (Figure 5C, L4) or a non specific rabbit IgG (Figure 5C, L2). Together, these results indicate that physical interactions between GRPR and MOR1D exist both in vitro and in vivo.
Figure 5. Co-Immunoprecipitation and Co-Internalization of GRPR and MOR1D.

(A) Myc-GRPR (43 kDa) was detected in membrane fraction of HA-MOR1/Myc-GRPR cells (L1) and HA-MOR1D/Myc-GRPR cells (L2). Anti-HA antibody co-precipitated Myc-GRPR from HA-MOR1D/Myc-GRPR cells (L4), but not from HA-MOR1/Myc-GRPR cells (L3).
(B) Expression of HA-MOR1 (44 kDa) in HA-MOR1/Myc-GRPR cells (L1) and expression of HA-MOR1D (44 kDa) in HA-MOR1D/Myc-GRPR cells (L2) were revealed by anti-HA immunoblotting. An HA-MOR1D band (44 kDa) was precipitated by anti-Myc antibody from HA-MOR1D/Myc-GRPR cells (L4). Anti-Myc antibody failed to precipitate HA-MOR1 from HA-MOR1/Myc-GRPR cells (L3). IP: immunoprecipitaion, IB: immunoblotting, kDa: kilodaltons.
(C) GRPR, MOR1D and MOR1 were detected in the membrane extract of dorsal horn (L1). GRPR was co-precipitated by anti-MOR1D (L3) but not by anti-MOR1 (L4) or an non specific rabbit IgG (L2).
(D and E) Immunostaining (D) and ELISA (E) revealed internalization of HA-MOR1D but not HA-MOR1 or Myc-GRPR upon morphine treatment, while GRP induced internalization of GRPR but not MOR1D or MOR1.
(F and G) Immunostaining (F) and ELISA (G) revealed that Myc-GRPR was co-internalized with MOR1D, but not with MOR1 upon morphine stimulation. GRP only induced internalization of GRPP but not MOR1D or MOR1.
(H) Naloxone dose-dependently blocked morphine-induced internalization of Myc-GRPR and HA-MOR1D.
(I) The GRPR antagonist blocked morphine-induced internalization of Myc-GRPR, but not HA-MOR1D.
Data are expressed as mean and standard error of three independent experiments. Error bars represent standard error of the mean. *p < 0.05. Also see Figure S4.
To test whether MOR1D may cross-activate GRPR and undergo co-internalization with GRPR in response to morphine, we first examined internalization of GRPR, MOR1D and MOR1 in stably expressing HEK 293 cells various combinations of Myc-GRPR, HA-MOR1D and HA-MOR1 after morphine stimulation. Morphine failed to induce GRPR internalization in cells expressing GRPR alone (Figure 5D and 5E) or in cells co-expressing MOR1 and GRPR (Figure 5F and 5G). In contrast, GRPR internalization was significantly enhanced in HEK 293 cells co-expressing MOR1D and GRPR (Figure 5F and 5G). Consistent with a previous study (Whistler et al., 1999), no internalization of HA-MOR1 by morphine was found (Figure 5D–5G). However, cells expressing MOR1D (Figure 5D and 5E) or MOR1D and GRPR (Figure 5F and 5G) showed significant MOR1D internalization in response to morphine. In addition, GRPR internalization in response to DAMGO occurred only in MOR1D/GRPR cells, but not in MOR1/GRPR cells (Figure S4). These results suggest that the co-existence of GRPR and MOR1D is a prerequisite for morphine-mediated GRPR internalization.
Next we assessed whether naloxone would affect morphine-induced MOR1D-GRPR internalization. Naloxone inhibited morphine-induced GRPR or MOR1D internalization in a dose-dependent manner and at a dose of 10 μM could nearly abolish MOR1D-GRPR internalization (Figure 5H). Interestingly, the GRPR antagonist inhibited morphine-induced internalization of GRPR but not MOR1D (Figure 5I). Consistently, GRP was able to internalize GRPR, regardless of whether GRPR was co-expressed with MOR1D or MOR1 (Figure 5D–G). However, neither MOR1D nor MOR1 internalized upon GRP stimulation, regardless of whether they were co-expressed with GRPR (Figure 5D–G). Taken together, these results indicate that despite the co-expression of MOR1D and GRPR, they cannot be reciprocally activated; only MOR1D is able to cross-activate GRPR in response to morphine, not vice versa.
Cross-Activation of the GRPR Signaling Transduction Pathway by MOR1D upon Morphine Stimulation
GRPR can activate multiple signaling pathways including the phospholipase C (PLC)/inositol 1,4, 5-trisphosphate (IP3)/Ca2+ signaling pathway in response to GRPR agonists in a number of heterologous cell lines (Jensen et al., 2008; Kroog et al., 1995). To ascertain whether GRPR-dependent calcium responses might be induced by morphine, we examined Ca2+ signals in HEK 293 cells stably expressing various combinations of MOR1, MOR1D and GRPR. Both morphine and GRP induced calcium spikes in cells co-expressing MOR1D and GRPR (Figure 6A), suggesting an activation of GRPR by morphine or GRP. Morphine- or GRP-induced calcium signals were not affected in calcium free extracellular buffer, indicating the endoplasmic reticulum origin of calcium (Figure S5A). However, morphine failed to evoke Ca2+ spikes in cells co-expressing MOR1 and GRPR or in cells containing only GRPR; neither morphine nor GRP generated a calcium response in cells expressing MOR1D alone (Figure 6A).
Figure 6. Cross Activation of the GRPR Signal Transduction Pathway by MOR1D in Response to Morphine.

The responses of HEK 293 cells expressing vary receptors to morphine or GRP, tested using calcium imaging.
(A) HEK 293 cells co-expressing MOR1D and GRPR showed calcium response to both morphine and GRP. Cells co-expressing MOR1 and GRPR were unable to respond to morphine, whereas they responded to GRP.
(B) In cells co-expressing MOR1D and GRPR the GRPR antagonist completely blocked morphine- or GRP-induced Ca2+ increase, while naloxone blocked morphine- and reduced GRP-induced Ca2+ response.
(C) Both PLC inhibitor U73122 and IP3 receptor antagonist 2-APB blocked the calcium response to morphine and GRP in cells co-expressing MOR1D and GRPR. U73343, an inactive structural analog of U73122 had no such effect.
(D) Quantified data comparing peak intracellular calcium concentration. Naloxone significantly reduced GRP-induced [Ca2+]i increase in cells co-expressing MOR1D and GRPR.
(E and F) GRPR+ cells in superficial dorsal horn were ablated by bombesin-saporin. The superficial dorsal horn was dissected for qRT-PCR. Gel image (E) and quantitative analysis
(F) showed that PLCβ3 mRNA was lost in bombesin-saporin-treated group. PLCβ1 and IP3R3 mRNA were significantly decreased by bombesin-saporin treatment.
(G) Two days after the last injection of PLCβ siRNA (1.25 μg, i.t.), MIS was significantly reduced.
(H) MIA was not significantly affected by PLCβ siRNA.
(I) PLCβ mRNA level in the superficial dorsal horn was significantly reduced by i.t. injection of PLCβ siRNA.
(J) Two days after i.t. IP3R3 siRNA, MIS was significantly reduced.
(K) MIA was not affected by IP3R3 siRNA.
(L) IP3R3 mRNA level in the superficial dorsal horn was significantly reduced by i.t. injection of IP3R3 siRNA.
In all experiments, n = 6~7 per group. *p < 0.05. Error bars represent standard error of the mean. Also see Figure S5.
To ascertain whether morphine-induced calcium spike is a consequence of a cross-activation of GRPR, we pretreated cells co-expressing MOR1D and GRPR with the GRPR antagonist or naloxone. Morphine-induced calcium spikes were blocked by the GRPR antagonist and naloxone (Figure 6B). GRP-induced calcium spikes were completely blocked by the GRPR antagonist and significantly reduced by naloxone (Figure 6B and 6D). Both morphine- and GRP-evoked Ca2+ spikes were blocked by U73122 (a selective PLC inhibitor that prevents IP3 liberation) or 2-APB (an IP3 receptor (IP3R) antagonist), while U73343 (an inactive structural analog control for U73122) had no effect on calcium response to morphine or GRP (Figure 6C). These data suggest that morphine cross-activates GRPR through MOR1D, leading to activation of the PLC/IP3/Ca2+ signaling pathway.
Co-Expression of PLCβ isoforms, IP3R3 and GRPR in the Spinal Cord
In order for PLC and IP3R signaling molecules to act downstream of GRPR, they must be co-expressed in GRPR+ cells. To circumvent the difficulties of double staining each individual PLC and IP3R isoform with GRPR, we took advantage of mice whose GRPR neurons+ can be ablated specifically in the spinal cord by bombesin-saporin treatment (Sun et al., 2009), and used qRT-PCR to compare the mRNA change of individual isoforms in the superficial dorsal horn between mice treated with bombesin-saporin and with blank-saporin. As confirmed by the significant decrease of GRPR mRNA (Figure S5B), there was a complete loss of PLCβ3 expression and a significant decrease of PLCβ1, IP3R type 3 (IP3R3), and MOR1D mRNA in bombesin-saporin-treated tissues as compared to the control (Figures 6E, 6F and S5B). These results reveal co-expression of PLCβ1/3, IP3R3, MOR1D and GRPR in GRPR+ cells.
Inhibition of PLC/IP3 Signaling Significantly Attenuates MIS but not MIA
To determine the physiological relevance of morphine-induced signaling transduction in vivo, spinal siRNA knockdown approach was employed to investigate whether PLC/IP3 signaling is important for MIS. Consistently, siRNA knockdown of either PLCβ1/3 or IP3R3 in mice significantly compromised MIS (Figures 6G and 6J). In contrast, the same treatments did not alter MIA (Figures 6H and 6K). The efficiency and selectivity of siRNA were determined by qRT-PCR; spinal PLCβ and IP3R3 mRNA levels significantly decreased by approximately 62% and 33%, respectively (Figures 6I and 6L). No significant knockdown of PLCβ or IP3R3 mRNA in DRG neurons was observed (Figures S5C and S5D). The reduction of PLCβ3 and IP3R3 protein levels in spinal cord was further confirmed by Western blot (Figures S5E and S5F). Interestingly, i.t. injection of either U73122 or 2-APB significantly attenuated MIS but had no impact on MIA (Figure S5G–J), suggesting an existence of MIS-specific PLC/IP3 signaling in vivo.
MOR1D C-Terminus is Critical for MIS and MOR1D-GRPR Heterodimeric Interaction
The difference between MOR1 and MOR1D isoforms lies in a motif consisting of seven amino acids (RNEEPSS) in MOR1D C-terminus (Figure 7A). This motif is likely to be essential for MOR1D and GRPR physical interaction. To test this, a Tat-fusion peptide (Tat-MOR1DCT) containing a Tat (YGRKKRRQRRR), a trans-activating domain of HIV protein that can permeate the cell membrane (Schwarze et al., 1999), and the RNEEPSS motif was synthesized (Figure 7A) and intrathecally injected. Introduction of Tat-MOR1DCT causes competition with MOR1D for physical contacts with GRPR in vivo. Remarkably, i.t. injection of Tat-MOR1DCT specifically blocked MIS (Figure 7B), while leaving GIS (Figure 7B) and MIA (Figure 7C) unperturbed. Subsequent co-IP analysis revealed that Tat-MOR1DCT significantly reduced the amount of GRPR precipitated by the MOR1D antibody (Figure 7D and 7E). These results demonstrate that MOR1D C-terminus is critical for MOR1D-GRPR dimerization and for MIS.
Figure 7. MOR1D C-Terminus is Critical for MIS and MOR1D/GRPR Dimerization.

(A) Sequence comparison of MOR1D and MOR1 reveals a unique motif in MOR1D C-terminus. Synthesized peptide Tat-MOR1DCT contains a Tat domain from human immunodeficiency virus-type 1 and the motif from MOR1DCT. Control peptide contains Tat domain and scrambled sequence of MOR1DCT.
(B) Tat-MOR1DCT blocked MIS without affecting GIS.
(C) Tat-MOR1DCT had no effect on MIA.
(D and E) Co-IP by anti-MOR1D (D) and quantified O.D. ratio of GRPR and MOR1D (E) showing Tat-MOR1DCT decreased GRPR/MOR1D interaction in the lumbar spinal cord.
In all experiments, n = 6~8 per group. *p < 0.05. Error bars represent standard error of the mean.
DISCUSSION
In this study, we present molecular, cellular, biochemical and behavioral data that demonstrate uncoupling of opioid-induced itch and opioid-induced anti-nociception in the spinal cord. Functionally, morphine tolerance tests show no correlation between MIA and MIS. At the molecular level, we identify MOR1D as an isoform critical for mediating MIS but not MIA, whereas the MOR1 isoform is required for MIA but not MIS. At the cellular level, we show that MOR1D expression is largely restricted to lamina I and overlaps with GRPR, whereas MOR1 is mainly located in lamina II in the spinal cord. MOR1D is the first MOR isoform that does not possess the cardinal function of an opioid receptor. These data argue against the prevailing view that opioid induces itch as result of pain inhibition, and reveal opioid-induced itch to be an active process, independently initiated by MOR1D-mediated activation of GRPR. Coupled with the finding that MIA remains unaffected in GRPR KO mice, the present studies further support the notion that GRPR is an itch-specific receptor (Sun and Chen, 2007) and that GRPR-expressing neurons represent a labeled line for itch in the spinal cord (Sun et al., 2009).
Unidirectional Cross Activation of GRPR by MOR1D through Heterodimeric Interactions
GRP is an itch-specific peptide that is presumably released from primary afferents to activate spinal GRPR in response to pruritic stimuli (Sun and Chen, 2007). Spinal morphine may promote presynaptic release of GRP to activate central GRPR signaling. Several observations, however, suggest that GRP is dispensable for morphine-induced activation of GRPR. First, MOR antagonist naloxone abolished MIS but did not change GIS. Consistently, GIS is normal in MOR KO mice. Second, in HEK 293 cells expressing both MOR1D and GRPR, GRP failed to cause MOR1D internalization. These results indicate that the activation of GRPR in response to morphine is mediated via a postsynaptic mechanism. Indeed, MOR1D and GRPR dimers are detected by co-IP in heterologous cells, and MOR1D and GRPR can also be co-immunoprecipitated from spinal cord membrane preparation. Thus, spinal opiates produce itch through MOR1D and GRPR heterodimerization. Importantly, in vivo interference with Tat-MOR1DCT markedly reduces co-IP of GRPR and MOR1D and blunts MIS. Taken together, these data demonstrate the importance of physical interactions between MOR1D and GRPR in MIS.
Calcium imaging studies illustrate that neither GRPR nor MOR1D alone are able to elicit a calcium response to morphine. Strikingly, a blockade of PLCβ and IP3R abolished morphine-induced calcium signaling in cells co-expressing MOR1D and GRPR. These results are in accordance with previous observations that the ability of the Gi-coupled receptors to evoke calcium signaling often depends on a concomitant activation of the Gq-coupled receptors (Samways and Henderson, 2006). Distinguished from previous studies, the present study provides behavioral relevance for the PLCβ/IP3-dependent Ca2+ signaling evoked by morphine. Interestingly, PLCβ3 in DRG neurons has been shown to be required for MIA (Xie et al., 1999) as well as for histaminergic itch (Han et al., 2006). The fact that spinal opioid-induced itch is histamine-independent (Ko et al., 2004), along with our finding that no change of PLCβ and IP3R occurs in DRG neurons by siRNA knockdown, indicates that the canonical PLCβ/IP3R3/Ca2+ signal transduction pathway in the spinal cord is itch-specific, and is different from its function in DRG neurons.
GPCR heterodimerization synergistically modulates respective receptor activity, resulting in either enhanced or inhibited ligand binding properties, or conferring novel function not originally possessed by the singular receptors (George et al., 2000; Jordan and Devi, 1999; Lopez and Salome, 2009). In contrast to the reciprocal regulation of each receptor by respective agonists that commonly occur with GPCR heterodimerization, which allows for coincidental detection, our results uncover a unidirectional signaling model for GPCR crosstalk: while morphine-encoded itch information is transmitted from MOR1D to GRPR, GRP-encoded itch signaling cannot be reversely relayed to MOR1D by GRPR. Interestingly, the observation that a MOR1D-GRPR co-immunoprecipitated band from spinal cord membrane preparation is detected in the absence of morphine stimulation indicates a constitutive presence of MOR1D-GRPR heterodimeric assembly in vivo. How can GRPR be activated and internalized by morphine via MOR1D, whereas MOR1D cannot be internalized by GRP? One can envision the possibility that MOR1D and GRPR heterodimers may exist in a relatively unstable and dynamic equilibrium state that can be either strengthened/activated upon morphine stimulation, resulting in co-internalization, or weakened in response to GRP, leading to a dissociation of heterodimers so that only GRPR internalizes. This is reminiscent of agonist-dependent dimerization and internalization of the δ opioid receptor (Cvejic and Devi, 1997), and may also explain why the GRPR antagonist blocks morphine-mediated GRPR but not MOR1D internalization. Such a unidirectional signaling may ensure that opioid-encoded itch information is correctly relayed to the GRPR- signaling machinery, and avoids accidental engagement of MOR1D that may result in inappropriate signaling, such as a condition when GRPR is activated by GRP released from primary afferents. This one-way communication mechanism allows for added versatility to the physiological significance for GPCR heterodimerization, and enables opioid receptors to carry out an unorthodox function.
Why has such a mechanism evolved to permit cross activation of itch signaling by opioids ? One plausible explanation is that opioid-induced pruritus may serve as the body’s warning sign for opiate overdose or for internal metabolism disorders. For example, patients with cholestasis often suffer from terrible pruritus, which has been attributable to enhanced endogenous opioidergic signaling that is centrally mediated because opiate antagonists could ameliorate cholestasic itch, along with several other systemic itch conditions (Bergasa, 2005; Jones and Bergasa, 1990; Metze et al., 1999).
Our study cannot exclude the possibility that MOR1D may additionally regulate GRPR signaling through the Gi-coupled intracellular cross-talk. In this regard, MIS provides a reliable, unique and unparalleled behavioral paradigm for facilitating further dissection of detailed intracellular signaling mechanisms of MOR1D and GRPR interactions and for understanding the corresponding physiological relevance.
Uncoupling of Itch and Pain: Therapeutic Implications
The identification of itch-specific MOR1D may shed light into the physiological and therapeutic relevance of the multiplicity of the MOR system. Although opioid antagonists may be used clinically to ameliorate spinal opioid-induced itch, their undiscriminating actions on both MOR1D and MOR1 might hinder opioid analgesia (Szarvas et al., 2003). Our finding, which uncouples MIS and MIA, enables us to envisage new therapeutic strategies. Disruption of GPCR heterodimerization with the use of pharmacological compounds or antibodies may allow highly specific targeting approach (Agnati et al., 2003; Hipser et al.; Waldhoer et al., 2005), and the unique C terminus of MOR1D may be one of the best therapeutic targets. This heterodimeric-specific approach would not perturb the normal function of GRPR or MOR1D in other tissues where they are singularly expressed and where their physiological function may be important. Likewise, if MOR1D-GRPR signaling were involved in cholestatic itch, a specific blockade may overcome side effects such as withdrawal-like symptoms often associated with the use of opioid antagonists in cholestsic itch (Bergasa, 2005). The present study implies that the physiological significance of multiple MOR isoforms may go beyond their normal anti-nociception paradigm that has been primarily restricted to the heterogeneity of opioid analgesia and patients (Pasternak, 2010). Although the dissociation between centrally mediated MIA and non-neural tissue-mediated side effects of opioids has been reported (Ling et al., 1989; Manara et al., 1986), it is much more difficult to separate MIA from side effects originating centrally. In this regard, an interesting question arises as to whether MOR1D may mediate other types of opiate side effects since it is expressed in other brain areas such as the nucleus of the solitary tract, in which no co-localization with MOR1 has been found (Abbadie et al., 2000). The uncoupling of MIA and MIS underscores the necessity of elucidating the function of individual MOR isoforms, which may promise novel pain therapy without debilitating side effects.
EXPERIMENTAL PROCEDURES
Animals
Generation and genotyping of GRPR KO and MOR KO were described previously (Hampton et al., 1998; Loh et al., 1998). All the experiments were performed in accordance with the guidelines of the National Institutes of Health and were approved by the Animal Studies Committee at Washington University School of Medicine.
Drugs and Reagents
Morphine, DAMGO, fentanyl, GRP, naloxone, bombesin-saporin (Advanced Targeting), the GRPR antagonist (D-Phe-6-Bn(6-13)OMe), U73122, U73343, 2-APB, siRNA (Sigma), Tat-MOR1DCT and sequence-scrambled control peptide were administered intrathecally.
Behavior
Scratching behavior and tail immersion assay were performed as previously described (Sun and Chen, 2007). Morphine antinociceptive tolerance was induced as described (Fairbanks and Wilcox, 1999) (Zhao et al., 2007).
Preparation and I.T. Injection of siRNA
Selective siRNA duplexes for mouse Oprm exons, PLCβ1/3 and IP3R3 were intrathecally injected daily for 3 consecutive days. Behavior testing and tissue harvest were carried out at 48 hr after the last injection.
Laser Capture Microdissection (LCM)
LCM were performed as previously described (van Baarlen et al., 2009), and laminae I&II of the spinal cord were dissected using the Pix-Cell II with HS caps (Arcturus).
Quantitative RT-PCR (qRT-PCR)
RNA was isolated from the LCM sample caps using the PicoPure RNA isolation kit (Arcturus). qRT-PCR amplification was performed using an Mx3000 QPCR system (Stratagene). All samples were run in triplicate.
Generation of MOR1D antibody and Immunohistochemistry
Rabbit anti-MOR1D antibody was generated as described (Abbadie et al., 2000). Double staining was performed using standard protocols.
Cell Culture and Transfections
To generate lines co-expressing Myc-tagged GRPR and HA-tagged MOR1D or MOR1 receptors, the cells were subjected to G418/hygromycin double selection. Clones expressing Myc-GRPR, HA-MOR1, HA-MOR1D, HA-MOR1/Myc-GRPR and HA-MOR1D/Myc-GRPR were examined using quantitative Western blot analysis to ensure that clones co-express GRPR and MOR (or MOR1D) in 1:1 ratio.
Co-Immunoprecipitation and Western Blot Analysis
HEK 293 cells expressing MOR1D/GRPR or MOR1/GRPR were exposed to the cross-linking agent dithiobis-(succinimidylpropionate) (Pierce) and subsequently lysed as described (Koch et al., 2001). The receptor proteins were incubated with HA antibody (BD bioscience), or c-Myc antibody (Covance). The complex was precipitated, deglycosylated and separated on SDS gels (Invitrogen). Proteins were incubated with c-Myc antibody or HA antibody first, and then with goat horseradish peroxidase-linked secondary antibodies (Santa Cruz). Immunoblots were developed with the enhanced chemiluminescence reagents (Amersham).
Internalization Assays
The receptor internalization assay was performed as described previously (Pfeiffer et al., 2002).
Calcium Imaging
The cells were loaded with Fura 2-acetomethoxy ester (Molecular Probes) for ratiometric studies. Cells were imaged at 340 and 380 nm excitation to detect intracellular free calcium. Each experiment was done in triplicate, and at least 50 cells were analyzed each time.
Statistical Analysis
Statistical comparisons were performed with two-way analysis of variance (ANOVA) or Student’s t-test. All data were expressed as the mean ± standard error of the mean (s.e.m.) and error bars represent s.e.m. P < 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We thank J. Yin, S. Weng, YC. Pan and ZQ. Zhao for technical help and comments, CJ. Coscia, YX. Pan and GW. Pasternak for MOR and MOR1D plasmids, E. Wada and J. Battey for GRPR antibodies, P. Sternweis for PLCβ3 antibody, E. Liman for IP3R3 antibody, D. Coy for a GRPR antagonist, BW. Li for qRT-PCR help, and W. Huh for LCM. We also thank M. Bruchas and A. Munanairi for comments. J. Jeffry has been supported by a NIDA training grant. The work was supported by a NIAMS grant (R01AR056318) to ZF.C.
Footnotes
Supplemental Information includes Extended Experimental Procedures and five figures.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbadie C, Pan Y, Drake CT, Pasternak GW. Comparative immunohistochemical distributions of carboxy terminus epitopes from the mu-opioid receptor splice variants MOR-1D, MOR-1 and MOR-1C in the mouse and rat CNS. Neuroscience. 2000;100:141–153. doi: 10.1016/s0306-4522(00)00248-7. [DOI] [PubMed] [Google Scholar]
- Agnati LF, Ferre S, Lluis C, Franco R, Fuxe K. Molecular mechanisms and therapeutical implications of intramembrane receptor/receptor interactions among heptahelical receptors with examples from the striatopallidal GABA neurons. Pharmacol Rev. 2003;55:509–550. doi: 10.1124/pr.55.3.2. [DOI] [PubMed] [Google Scholar]
- Alvarez VA, Arttamangkul S, Dang V, Salem A, Whistler JL, Von Zastrow M, Grandy DK, Williams JT. mu-Opioid receptors: Ligand-dependent activation of potassium conductance, desensitization, and internalization. J Neurosci. 2002;22:5769–5776. doi: 10.1523/JNEUROSCI.22-13-05769.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andoh T, Yageta Y, Konno M, Yamaguchi-Miyamoto T, Takahata H, Nojima H, Nemoto H, Kuraishi Y. Evidence for separate involvement of different mu-opioid receptor subtypes in itch and analgesia induced by supraspinal action of opioids. J Pharmacol Sci. 2008;106:667–670. doi: 10.1254/jphs.08004sc. [DOI] [PubMed] [Google Scholar]
- Ballantyne JC, Loach AB, Carr DB. Itching after epidural and spinal opiates. Pain. 1988;33:149–160. doi: 10.1016/0304-3959(88)90085-1. [DOI] [PubMed] [Google Scholar]
- Bergasa NV. The pruritus of cholestasis. J Hepatol. 2005;43:1078–1088. doi: 10.1016/j.jhep.2005.09.004. [DOI] [PubMed] [Google Scholar]
- Bouvier M. Oligomerization of G-protein-coupled transmitter receptors. Nat Rev Neurosci. 2001;2:274–286. doi: 10.1038/35067575. [DOI] [PubMed] [Google Scholar]
- Carstens E. Responses of rat spinal dorsal horn neurons to intracutaneous microinjection of histamine, capsaicin, and other irritants. J Neurophysiol. 1997;77:2499–2514. doi: 10.1152/jn.1997.77.5.2499. [DOI] [PubMed] [Google Scholar]
- Carstens EE, Carstens MI, Simons CT, Jinks SL. Dorsal horn neurons expressing NK-1 receptors mediate scratching in rats. Neuroreport. 2010;21:303–308. doi: 10.1097/WNR.0b013e328337310a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaney MA. Side effects of intrathecal and epidural opioids. Can J Anaesth. 1995;42:891–903. doi: 10.1007/BF03011037. [DOI] [PubMed] [Google Scholar]
- Cvejic S, Devi LA. Dimerization of the delta opioid receptor: implication for a role in receptor internalization. J Biol Chem. 1997;272:26959–26964. doi: 10.1074/jbc.272.43.26959. [DOI] [PubMed] [Google Scholar]
- Davidson S, Giesler GJ. The multiple pathways for itch and their interactions with pain. Trends Neurosci. 2010;33:550–558. doi: 10.1016/j.tins.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson S, Zhang X, Yoon CH, Khasabov SG, Simone DA, Giesler GJ., Jr The itch-producing agents histamine and cowhage activate separate populations of primate spinothalamic tract neurons. J Neurosci. 2007;27:10007–10014. doi: 10.1523/JNEUROSCI.2862-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fairbanks CA, Wilcox GL. Spinal antinociceptive synergism between morphine and clonidine persists in mice made acutely or chronically tolerant to morphine. J Pharmacol Exp Ther. 1999;288:1107–1116. [PubMed] [Google Scholar]
- George SR, Fan T, Xie Z, Tse R, Tam V, Varghese G, O'Dowd BF. Oligomerization of mu- and delta-opioid receptors. Generation of novel functional properties. J Biol Chem. 2000;275:26128–26135. doi: 10.1074/jbc.M000345200. [DOI] [PubMed] [Google Scholar]
- Hales P. Pruritus after epidural morphine. Lancet. 1980;2:204. doi: 10.1016/s0140-6736(80)90090-2. [DOI] [PubMed] [Google Scholar]
- Hampton LL, Ladenheim EE, Akeson M, Way JM, Weber HC, Sutliff VE, Jensen RT, Wine LJ, Arnheiter H, Battey JF. Loss of bombesin-induced feeding suppression in gastrin-releasing peptide receptor-deficient mice. Proc Natl Acad Sci U S A. 1998;95:3188–3192. doi: 10.1073/pnas.95.6.3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han SK, Mancino V, Simon MI. Phospholipase Cbeta 3 mediates the scratching response activated by the histamine H1 receptor on C-fiber nociceptive neurons. Neuron. 2006;52:691–703. doi: 10.1016/j.neuron.2006.09.036. [DOI] [PubMed] [Google Scholar]
- Hipser C, Bushlin I, Gupta A, Gomes I, Devi LA. Role of antibodies in developing drugs that target G-protein-coupled receptor dimers. Mt Sinai J Med. 77:374–380. doi: 10.1002/msj.20199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikoma A, Steinhoff M, Stander S, Yosipovitch G, Schmelz M. The neurobiology of itch. Nat Rev Neurosci. 2006;7:535–547. doi: 10.1038/nrn1950. [DOI] [PubMed] [Google Scholar]
- Jensen RT, Battey JF, Spindel ER, Benya RV. International Union of Pharmacology. LXVIII. Mammalian bombesin receptors: nomenclature, distribution, pharmacology, signaling, and functions in normal and disease states. Pharmacol Rev. 2008;60:1–42. doi: 10.1124/pr.107.07108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones EA, Bergasa NV. The pruritus of cholestasis: from bile acids to opiate agonists. Hepatology. 1990;11:884–887. doi: 10.1002/hep.1840110526. [DOI] [PubMed] [Google Scholar]
- Jordan BA, Devi LA. G-protein-coupled receptor heterodimerization modulates receptor function. Nature. 1999;399:697–700. doi: 10.1038/21441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith DE, Murray SR, Zaki PA, Chu PC, Lissin DV, Kang L, Evans CJ, von Zastrow M. Morphine activates opioid receptors without causing their rapid internalization. J Biol Chem. 1996;271:19021–19024. doi: 10.1074/jbc.271.32.19021. [DOI] [PubMed] [Google Scholar]
- Kieffer BL. Opioids: first lessons from knockout mice. Trends Pharmacol Sci. 1999;20:19–26. doi: 10.1016/s0165-6147(98)01279-6. [DOI] [PubMed] [Google Scholar]
- Ko MC, Naughton NN. An experimental itch model in monkeys: characterization of intrathecal morphine-induced scratching and antinociception. Anesthesiology. 2000;92:795–805. doi: 10.1097/00000542-200003000-00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko MC, Song MS, Edwards T, Lee H, Naughton NN. The role of central mu opioid receptors in opioid-induced itch in primates. J Pharmacol Exp Ther. 2004;310:169–176. doi: 10.1124/jpet.103.061101. [DOI] [PubMed] [Google Scholar]
- Koch T, Schulz S, Pfeiffer M, Klutzny M, Schroder H, Kahl E, Hollt V. C-terminal splice variants of the mouse mu-opioid receptor differ in morphine-induced internalization and receptor resensitization. J Biol Chem. 2001;276:31408–31414. doi: 10.1074/jbc.M100305200. [DOI] [PubMed] [Google Scholar]
- Kroog GS, Jensen RT, Battey JF. Mammalian bombesin receptors. Med Res Rev. 1995;15:389–417. doi: 10.1002/med.2610150502. [DOI] [PubMed] [Google Scholar]
- Kuraishi Y, Yamaguchi T, Miyamoto T. Itch-scratch responses induced by opioids through central mu opioid receptors in mice. J Biomed Sci. 2000;7:248–252. doi: 10.1007/BF02255473. [DOI] [PubMed] [Google Scholar]
- Lagerstrom MC, Rogoz K, Abrahamsen B, Persson E, Reinius B, Nordenankar K, Olund C, Smith C, Mendez JA, Chen ZF, et al. VGLUT2-dependent sensory neurons in the TRPV1 population regulate pain and itch. Neuron. 2010;68:529–542. doi: 10.1016/j.neuron.2010.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law PY, Wong YH, Loh HH. Molecular mechanisms and regulation of opioid receptor signaling. Annu Rev Pharmacol Toxicol. 2000;40:389–430. doi: 10.1146/annurev.pharmtox.40.1.389. [DOI] [PubMed] [Google Scholar]
- Ling GS, Paul D, Simantov R, Pasternak GW. Differential development of acute tolerance to analgesia, respiratory depression, gastrointestinal transit and hormone release in a morphine infusion model. Life Sci. 1989;45:1627–1636. doi: 10.1016/0024-3205(89)90272-5. [DOI] [PubMed] [Google Scholar]
- Liu Y, Abdel Samad O, Zhang L, Duan B, Tong Q, Lopes C, Ji RR, Lowell BB, Ma Q. VGLUT2-dependent glutamate release from nociceptors is required to sense pain and suppress itch. Neuron. 2010;68:543–556. doi: 10.1016/j.neuron.2010.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loh HH, Liu HC, Cavalli A, Yang W, Chen YF, Wei LN. mu Opioid receptor knockout in mice: effects on ligand-induced analgesia and morphine lethality. Brain Res Mol Brain Res. 1998;54:321–326. doi: 10.1016/s0169-328x(97)00353-7. [DOI] [PubMed] [Google Scholar]
- Lopez A, Salome L. Membrane functional organisation and dynamic of mu- opioid receptors. Cell Mol Life Sci. 2009;66:2093–2108. doi: 10.1007/s00018-009-0008-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manara L, Bianchi G, Ferretti P, Tavani A. Inhibition of gastrointestinal transit by morphine in rats results primarily from direct drug action on gut opioid sites. J Pharmacol Exp Ther. 1986;237:945–949. [PubMed] [Google Scholar]
- Matthes HW, Maldonado R, Simonin F, Valverde O, Slowe S, Kitchen I, Befort K, Dierich A, Le Meur M, Dolle P, et al. Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the mu-opioid-receptor gene. Nature. 1996;383:819–823. doi: 10.1038/383819a0. [DOI] [PubMed] [Google Scholar]
- McMahon SB, Koltzenburg M. Itching for an explanation. Trends Neurosci. 1992;15:497–501. doi: 10.1016/0166-2236(92)90102-e. [DOI] [PubMed] [Google Scholar]
- Metze D, Reimann S, Beissert S, Luger T. Efficacy and safety of naltrexone, an oral opiate receptor antagonist, in the treatment of pruritus in internal and dermatological diseases. J Am Acad Dermatol. 1999;41:533–539. [PubMed] [Google Scholar]
- Milligan G. G protein-coupled receptor hetero-dimerization: contribution to pharmacology and function. Br J Pharmacol. 2009;158:5–14. doi: 10.1111/j.1476-5381.2009.00169.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols ML, Allen BJ, Rogers SD, Ghilardi JR, Honore P, Luger NM, Finke MP, Li J, Lappi DA, Simone DA, et al. Transmission of chronic nociception by spinal neurons expressing the substance P receptor. Science. 1999;286:1558–1561. doi: 10.1126/science.286.5444.1558. [DOI] [PubMed] [Google Scholar]
- Pan YX. Diversity and complexity of the mu opioid receptor gene: alternative pre- mRNA splicing and promoters. DNA Cell Biol. 2005;24:736–750. doi: 10.1089/dna.2005.24.736. [DOI] [PubMed] [Google Scholar]
- Pasternak GW. Multiple opiate receptors: deja vu all over again. Neuropharmacology. 2004;47(Suppl 1):312–323. doi: 10.1016/j.neuropharm.2004.07.004. [DOI] [PubMed] [Google Scholar]
- Pasternak GW. Molecular insights into mu opioid pharmacology: From the clinic to the bench. Clin J Pain. 2010;26(Suppl 10):S3–9. doi: 10.1097/AJP.0b013e3181c49d2e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel KN, Dong X. An itch to be scratched. Neuron. 2010;68:334–339. doi: 10.1016/j.neuron.2010.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paus R, Schmelz M, Biro T, Steinhoff M. Frontiers in pruritus research: scratching the brain for more effective itch therapy. J Clin Invest. 2006;116:1174–1186. doi: 10.1172/JCI28553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfeiffer M, Koch T, Schroder H, Laugsch M, Hollt V, Schulz S. Heterodimerization of somatostatin and opioid receptors cross-modulates phosphorylation, internalization, and desensitization. J Biol Chem. 2002;277:19762–19772. doi: 10.1074/jbc.M110373200. [DOI] [PubMed] [Google Scholar]
- Ravindranathan A, Joslyn G, Robertson M, Schuckit MA, Whistler JL, White RL. Functional characterization of human variants of the mu-opioid receptor gene. Proc Natl Acad Sci U S A. 2009;106:10811–10816. doi: 10.1073/pnas.0904509106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross SE, Mardinly AR, McCord AE, Zurawski J, Cohen S, Jung C, Hu L, Mok SI, Shah A, Savner EM, et al. Loss of Inhibitory Interneurons in the Dorsal Spinal Cord and Elevated Itch in Bhlhb5 Mutant Mice. Neuron. 2010;65:886–898. doi: 10.1016/j.neuron.2010.02.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samways DS, Henderson G. Opioid elevation of intracellular free calcium: possible mechanisms and physiological relevance. Cell Signal. 2006;18:151–161. doi: 10.1016/j.cellsig.2005.08.005. [DOI] [PubMed] [Google Scholar]
- Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. In vivo protein transduction: delivery of a biologically active protein into the mouse. Science. 1999;285:1569–1572. doi: 10.1126/science.285.5433.1569. [DOI] [PubMed] [Google Scholar]
- Sora I, Takahashi N, Funada M, Ujike H, Revay RS, Donovan DM, Miner LL, Uhl GR. Opiate receptor knockout mice define mu receptor roles in endogenous nociceptive responses and morphine-induced analgesia. Proc Natl Acad Sci U S A. 1997;94:1544–1549. doi: 10.1073/pnas.94.4.1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun YG, Chen ZF. A gastrin-releasing peptide receptor mediates the itch sensation in the spinal cord. Nature. 2007;448:700–703. doi: 10.1038/nature06029. [DOI] [PubMed] [Google Scholar]
- Sun YG, Zhao ZQ, Meng XL, Yin J, Liu XY, Chen ZF. Cellular basis of itch sensation. Science. 2009;325:1531–1534. doi: 10.1126/science.1174868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szarvas S, Harmon D, Murphy D. Neuraxial opioid-induced pruritus: a review. J Clin Anesth. 2003;15:234–239. doi: 10.1016/s0952-8180(02)00501-9. [DOI] [PubMed] [Google Scholar]
- Trafton JA, Abbadie C, Marek K, Basbaum AI. Postsynaptic signaling via the [mu]-opioid receptor: responses of dorsal horn neurons to exogenous opioids and noxious stimulation. J Neurosci. 2000;20:8578–8584. doi: 10.1523/JNEUROSCI.20-23-08578.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Baarlen P, Troost FJ, van Hemert S, van der Meer C, de Vos WM, de Groot PJ, Hooiveld GJ, Brummer RJ, Kleerebezem M. Differential NF-kappaB pathways induction by Lactobacillus plantarum in the duodenum of healthy humans correlating with immune tolerance. Proc Natl Acad Sci U S A. 2009;106:2371–2376. doi: 10.1073/pnas.0809919106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldhoer M, Fong J, Jones RM, Lunzer MM, Sharma SK, Kostenis E, Portoghese PS, Whistler JL. A heterodimer-selective agonist shows in vivo relevance of G protein-coupled receptor dimers. Proc Natl Acad Sci U S A. 2005;102:9050–9055. doi: 10.1073/pnas.0501112102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whistler JL, Chuang HH, Chu P, Jan LY, von Zastrow M. Functional dissociation of mu opioid receptor signaling and endocytosis: implications for the biology of opiate tolerance and addiction. Neuron. 1999;23:737–746. doi: 10.1016/s0896-6273(01)80032-5. [DOI] [PubMed] [Google Scholar]
- Xie W, Samoriski GM, McLaughlin JP, Romoser VA, Smrcka A, Hinkle PM, Bidlack JM, Gross RA, Jiang H, Wu D. Genetic alteration of phospholipase C beta3 expression modulates behavioral and cellular responses to mu opioids. Proc Natl Acad Sci U S A. 1999;96:10385–10390. doi: 10.1073/pnas.96.18.10385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao ZQ, Gao YJ, Sun YG, Zhao CS, Gereau RWt, Chen ZF. Central serotonergic neurons are differentially required for opioid analgesia but not for morphine tolerance or morphine reward. Proc Natl Acad Sci U S A. 2007;104:14519–14524. doi: 10.1073/pnas.0705740104. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.