

Abstract
Holosprosencephaly (HPE) is the most common disorder of the developing forebrain in humans, and is characterized by failed or incomplete cleavage of the cerebral hemispheres and deep brain structures. HPE includes wide phenotypic variability, with a continuum of both brain and craniofacial anomalies.
While “classic” eye findings, including the spectrum of midline anomalies ranging from cyclopia to hypotelorism, as well as chorioretinal coloboma and microphthalmia, have been frequently described in patients with HPE, other subtle eye anomalies may also occur.
In our study we prospectively analyzed a small cohort of 10 patients in whom we identified mutations in SHH, SIX3, ZIC2, or FGF8, the latter of which is a very recently described HPE-associated gene. We found that 9 of 10 patients had at least two ophthalmologic anomalies, including refractive errors, microcornea, microphthalmia, blepharoptosis, exotropia, and uveal coloboma.
These findings contribute to the understanding of the phenotypic variability of the HPE spectrum, and highlight findings in one medically important but often incompletely investigated system.
Keywords: Holoprosencephaly, SHH, SIX3, ZIC2, FGF8, Ocular, Microphthalmia, Coloboma, Strabismus, Microcornea
INTRODUCTION
Holoprosencephaly (HPE) is the most common disorder of the developing forebrain in humans, with a frequency up to 1 in250 concept uses and approximately 1 in10,000 live births [Matsunaga and Shiota, 1977] [Orioli and Castilla, 2007]. HPE is known to be due to chromosomal anomalies (50% of patients), may occur in the context of syndromes such as Smith-Lemli-Opitz syndrome (SLOS), be due to mutations in Sonic Hedgehog (SHH) [Roessler et al., 1996] and other genes that interact with or are a direct part of the SHH-signaling pathway including SIX3 [Wallis et al., 1999], ZIC2 [Brown et al., 1998], TGIF [Gripp et al., 2000], GLI2 [Roessler et al., 2003], PTCH [Ming et al., 2002], NODAL [Roessler et al., 2009c] and others [Arauz et al., 2010; Ribeiro et al., 2010], involve alterations in cholesterol biosynthesis [Haas et al., 2007], or result from exposure to environmental factors (such as ethanol and retinoic acid during embryonic organogenesis) [Lipinski et al., 2010; Roessler and Muenke, 2010].
Holoprosencephaly is characterized by distinct and complex phenotypic variability. The HPE neuroanatomical spectrum, which includes a progressive gradation of structural brain anomalies, consists of failed or incomplete cerebral hemisphere separation. Classically defined types of HPE range from the alobar form of HPE at the most severe end of the spectrum, to the lobar form of HPE, agenesis of the corpus callosum (ACC), and septo-opticor septo-preoptic dysplasia at the less severe end of the spectrum of patients with structural brain anomalies [Hahn and Barnes, 2010; Hahn et al., 2010].
This spectrum of brain malformations is typically accompanied by a craniofacial phenotypic spectrum of characteristic midline deficits, which includes facial features such as cyclopia, hypotelorism, proboscis, absent nose, absent nasal bone, single nostril, pyriform aperture stenosis, choanal stenosis, and cleft lip/palate. These facial features often correlate with the severity of brain anomalies [Demyer et al., 1964], with some exceptions, such as the case of patients with mutations in ZIC2 [Solomon et al., 2010a]. Some types of ophthalmologic anomalies in patients with HPE are relatively easily ascertained on routine physical examination. These include cyclopia, synophthalmia, hypotelorism, and uveal coloboma. However, more subtle ophthalmologic anomalies, may also occur [Schimmenti et al., 2003]. For example, Schimmenti et al. described a family without any structural brain anomalies in whom a mutation (c.1210_1233del, p.Gly404_Gly411del) in the autocatalytic C-terminus of protein encoded by the SHH gene (NM_000193) was associated with chorioretinal coloboma and iristhinning [Schimmenti et al., 2003]. This provides an example of how alterations of the SHH-signaling pathway can result in abnormal morphogenesis causing subtle eye anomalies in patients with HPE or microforms of HPE (a diagnosis of “microform” HPE is often given to mildly-affected individuals with craniofacial malformations consistent with the HPE spectrum, but who do not have characteristic structural brain anomalies).
The evaluation of patients and their families affected by HPE may include the following: dysmorphology examination, brain imaging, cytogenetic studies, and molecular genetics analyses[Hahn and Plawner, 2004; Levey et al., 2010; Pineda-Alvarez et al., 2010]. However, a careful ophthalmologic assessment is not routinely performed after the initial diagnostic period, especially by specialists highly familiar with the condition. As a consequence, subtle ophthalmological anomalies are, in our experience, often overlooked. As these anomalies may have health consequences, we describe a series of patients with the goal of detailing specific ophthalmologic findings.
METHODS
We analyzed 10 patients with previously identified intragenic mutations or deletions affecting HPE-associated genes (SHH, ZIC2, SIX3 or FGF8, the last of which is a newly identified HPE-associated gene) (Table I). These patients were evaluated at the National Institutes of Health Clinical Center (NIH-CC) in a 2-year period by a multi-disciplinary team including an ophthalmologist geneticist (BPB) highly familiar with the disorder.
TABLE I.
Molecular findings in patients with holoprosencephaly
| Affected Gene | Inheritance | Sequence Variant | Domain | Predicted effect | References | Comments | |
|---|---|---|---|---|---|---|---|
| Patients 1 and 2 | SHH | Familial | c.588_602del (p.Gly197_Gly201 del) | C-terminus | In-frame deletion including autocleavage site (Cys 198), resulting in possible alteration of protein processing | Patient 2 is the mother of Patient 1 | |
| Patient 3 | SIX3 | de novo | c.769C>T (p.Arg257Trp) | Homeodomain | Intermediate loss-of-function | [Domené et al., 2008] | - |
| Patient 4 | SHH | de novo | 46, XX, del(7)(q36). arr cgh 7q36.1q36.3(149, 597,103–158,739,800)×1 including SHH | Whole gene | Whole gene deletions have previously been described to cause severe holoprosencephaly; however, incomplete penetrance and variable expressivity are high for this particular locus | [Solomon et al., 2010b] | - |
| Patients 5 and 6 | SHH | Familial | c.1051C>T (p.Gln351*) | C-terminus | Truncation of C-terminus, resulting in possible alteration of protein processing. | [Roessler et al., 2009a] | Patient 6 is the mother of Patient 5 |
| Patient 7 | FGF8 | Familial | c.686C>T (p.Thr229Met) | C-terminus | Hypomorphic mutation related with midline defects, including HPE, and idiopathic hypogonadotropic hypogonadism | [Arauz et al., 2010; Falardeau et al., 2008] | - |
| Patients 8 and 9 | SIX3 | Familial | c.743_745del (p.Thr248_Gln249delinsLys) | Homeodomain | Possible loss-of-function | [Lacbawan et al., 2009] | Patient 9 is the mother of Patient 8 |
| Patient 10 | ZIC2 | de novo | c.612del (p.Tyr205ThrFS* 13) | 5′ to Homeodomain | Predicted null due to absence of the homeodomain | [Roessler et al., 2009b] | - |
Due to logistic considerations, most of the patients invited to the NIH-CC to participate in our study had relatively less severe phenotypes and were stable enough to be evaluated in the outpatient clinics.
RESULTS
General characteristics
Eight of the 10 patients were female, and two were male. Six patients presented with microforms of HPE, two patients had semilobar HPE, and one had lobar HPE. Additionally all patients had documented pathogenic mutations in genes associated with HPE, including SHH in five patients; SIX3 in three patients; and ZIC2 and FGF8, in one patient each, respectively (Table I).
Dysmorphological findings
All the patients evaluated had craniofacial anomalies consistent with HPE. Among these anomalies were: mild hypotelorism (10/10 patients); depressed midface with depressed nasal bridge (6/10), and single central maxillary incisor (2/10). However, additional structural ophthalmological anomalies were not detected in any of the patients with the initial physical examination prior to examination by an ophthalmologic geneticist. The in-depth ophthalmological evaluation detected a spectrum of ocular and extraocular anomalies in 9/10 patients (Table II). The only patient without such findings was a patient with microform HPE due to a mutation in FGF8 [Arauz et al., 2010].
TABLE II.
Clinical findings in patients with holoprosencephaly
| Patient (Age and Gender) | Gene (Sequence Variant) | Physical exam findings | Other Clinical Characteristics | HPE Brain Phenotype | Summary of Ophthalmologic Anomalies |
|---|---|---|---|---|---|
| Patient 1 (34-year-old female) | SHH: (c.588_602del; p.Gly197_Gly201del) | Single central incisor | None | Microform of HPE | Horizontal corneal diameters 10.5 mm OU. RI: −0.75 OD, −1.75 OS, Not requiring correction. |
| Patient 2 (12-year-old male) | SHH: (c.588_602del; p.Gly197_Gly201del) | Mild microcephaly, hypotelorism, mildly depressed nasal bridge, single central incisor | Attention deficit/hyperactivity disorder, hypopituitarism, corrected hypospadias. | Microform of HPE | Large scleral crescent in OS reminiscent of uveal coloboma, nanophthalmus (axial length of the eye, 21.69mm and 22.48mm in OD and OS respectively), foveal hypoplasia in OD, horizontal corneal diameters 11.5mm OU, unilateral blepharoptosis. RI: +1.75 OD and +2.50 OS, not requiring correction. |
| Patient 3 (9-year-old female) | SIX3: (c.769C>T; p.Arg257Trp) | Microcephaly, hypotelorism, depressed nasal bridge, single central incisor. | Non-verbal, seizure disorder, diabetes insipidus, small kidneys. | Lobar HPE, absent septum pellucidum, azygous anterior cerebral artery | Myopia and astigmatism, horizontal corneal diameters: 11mm OU. RI: −4.5 OU, requiring correction. |
| Patient 4 (6-year-old female) | SHH: (46, XX, del(7)(q 36). arr cgh 7q36.1q36.3(149,597,103–158,739,800)×1) | Mild microcephaly, hypotelorism, upslanting palpebral fissures. | Austism spectrum disorder, non-verbal. | Hypoplasia of the left frontal lobe | Hyperopia, astigmatism, 30–40 prism diopters esotropia, horizontal corneal diameters: 11mm OU. RI:+3.5 OU, requiring correction. |
| Patient 5 (2.5-year-old male) | SHH: (c.1051C>T; p.Gln351*) | Hypotelorism, choanal stenosis, depressed nasal bridge, single central incisor, ear tags. | Corrected umbilical hernia | Microform of HPE | Myopia, iris and chorioretinal coloboma OD, microcornea OD, horizontal corneal diameters 10mm OD, 12mm OS, 40 prism diopter right exotropia and, unilateral (left) blepharoptosis. RI: −4.5 OD, −4.25 OI, requiring correction. |
| Patient 6 (25-year-old female) | SHH: (c.1051C>T; p.Gln351*) | Hypotelorism and apparent proptosis, depressed nasal bridge | None | Microform of HPE | Myopia, horizotal corneal diameters, 11.6mm and 11.5 OD and OI respectively, asymmetric cupping of the optic nerve (0.5mm OD vs 0.3mm OS), and thinning of the optic nerves nasally OU, more in OS. RI: −3.25 OD, −3.5 OS, requiring correction. |
| Patient 7 (8-year-old female) | FGF8: (c.686C>T (p.Thr229Met)) | Hypotelorism, narrow nasal bridge, single central incisor. | Light pigmentary pattern | Microform of HPE | Blonde fundus consistent with cutaneous pigmentation. RI: +0.75 and +1.00, not requiring correction. |
| Patient 8 (8-year-old female) | SIX3: (c.743_745del; p.Thr248_Gln249delinsLys) | Microcephaly, hypotelorism, depressed nasal bridge, post-surgically corrected midline cleft lip/palate. | Hypopituitarism, diabetes insipidus | Semilobar HPE | Myopia, central visual impairment, horizontal corneal length 11mm OU, mildly dysplastic optic nerve on OD (cup-to-disk ratio: 0.3), fine multivectorial nystagmus, and 30 prism diopters exotropia. RI: −2.5 OU, requiring correction. |
| Patient 9 (40-year-old female) | SIX3: (c.743_745del; p.Thr248_Gln249delinsLys) | Hypotelorism, and single central incisor | Hypothyroidism | Microform of HPE | Myopia, bilateral punctate cortical cataracts (+1), myopic crescent in OS, 14 prism diopter hypertropia on OS, and apparent bilateral inferior oblique underaction. RI: −8.25 OD, −6.75 OS, requiring correction. |
| Patient 10 (5-year-old female) | ZIC2: (c.612del; p.Tyr205ThrFS* 13) | Microcephaly, hypotelorism, and depressed nasal bridge. | Diabetes insipidus | Semilobar HPE, absence of the corpus callosum. | Hyperopia, hyperopic astigmatism microcornea with horizontal corneal diameters of 10mm OU. RI: +1.50 OU. |
Ocular anomalies
Among the ocular anomalies identified, the most common anomalies were refractive errors. Nine of 10 patients had refractive errors of varying severity, six of which necessitated corrective lenses; one of the six also suffered cortical visual impairment. Six of 10 patients presented with smaller than average horizontal corneal diameters (Fig 1A). Of the six patients with small corneal diameters, two had borderline microcornea, with horizontal corneal diameters of 10 mm [Rufer et al., 2005]. In one of these patients (Patient 5), a borderline horizontal corneal diameter was accompanied by a chorioretinal coloboma. Although, the horizontal corneal diameters appeared to be smaller than average, the number of patients in this cohort is too small to attempt further statistical analyses regarding this finding.
FIGURE 1.
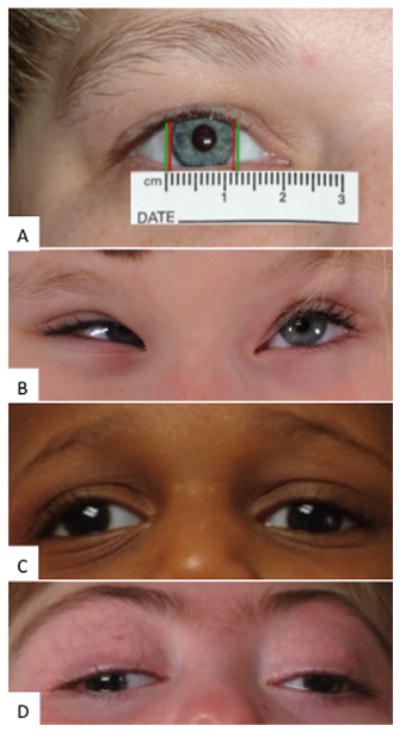
A: Illustration of the horizontal corneal diameter of Patient 1, an adult female patient with a microform of HPE due to a SHH mutation in the C-terminus of the gene; corneal diameters measured ~10mm (distance between red bars), while the average is ~12mm for adult women (distance between green bars). B: Patient 4, a child with a 7q36 deletion including SHH, presented with hypotelorism, upslanting palpebral fissures, and esotropia. C: Patient 5, a child with a microform of HPE due to a SHH mutation in the C-terminus of the gene, presented with hypotelorism, right exotropia and left blepharoptosis. D: Patient 8, a child with semilobar HPE due to a SIX3 mutation, presented with hypotelorism, and right exotropia.
Other ocular anomalies presented with lower frequency. These included: chorioretinal coloboma (1/10, same patient mentioned above), subtle retinal and optic nerve anomalies (3/10), simple microphthalmia, and narrow anterior chamber resulting in an increased risk for the development of closed-angle glaucoma (1/10). One patient with a SIX3 mutation had “pre-senile” cortical cataracts, which while perhaps not medically significant, may indicate a developmental alteration related to the HPE-associated mutation.
Extraocular anomalies
Besides anomalies that involve eye spacing (in our cohort, evident as hypotelorism), other extraocular anomalies were identified. These included strabismus (4/10) (Fig 1): exotropia (2/10), esotropia (1/10), and inferior oblique muscle underaction, in addition, to hypertropia (1/10). The other extraocular manifestation noted was unilateral blepharoptosis, which was present in two unrelated patients in our cohort, who had two different mutations in the C-terminus of SHH.
DISCUSSION
From a developmental standpoint, eye morphogenesis is a complex process, and proper patterning requires extremely well-coordinated temporo-spatial and dosage-dependent interactions between molecules, including PAX6, SHH, PAX2, FGF8, BMP4, and TBX5. Alterations in the signaling of any of these molecules, all of which are also involved in forebrain patterning, have been postulated to produce defects in the antero-posterior and ventral-dorsal development of the eye, resulting in intraocular anomalies [Kobayashi et al., 2010].
PAX6, for instance, is the main inducer of the optic vesicle and is expressed in either side of the diencephalon of the developing embryo. PAX6 expression is repressed by the prechordal plate (presumably due to SHH expression in the ventral midline), maintaining eye-field separation. Variable degrees of loss-of–function within the SHH pathway may explain mild to severe presentation of eye non-separation (ranging from mild hypotelorism to synthophthalmia and cyclopia) [England et al., 2006].
On the other hand, PAX2 expression is induced by SHH activity in the ventral aspect of the optic vesicle, and PAX2 [Kobayashi et al., 2010]has an important role in the closure of the optic fissure along the ventral optic cup and stalk [Carlson, 2009]. Failure of optic fissure closure results in a uveal coloboma, compromising one or multiple structures of the eye, which can include the iris, ciliary body, choroid, retina, and/or optic nerve [Guercio and Martyn, 2007]. This supports the role of SHH in the pathogenesis of this defect in patients reported in this and other studies [Bakrania et al., 2010; Schimmenti et al., 2003], although, coloboma in patients with mutations in PAX2 occurs at a rather low frequency.
Correlating with the concepts above, four of the patients with mutations in the C-terminus of SHH presented with craniofacial microforms of HPE and ocular anomalies, but without structural brain anomalies. Two additional studies have demonstrated the occurrence of mutations in the C-terminus of SHH in patients with isolated ocular anomalies [Bakrania et al., 2010]. Although the functional consequences of mutations in the C-terminus of SHH were initially unknown, a recent study demonstrated that mutations in this region of the gene affect autocatalylic cleavage, altering normal processing to mature SHH (SHH-Np). In this scenario, although the full-length (ie, non-cleaved) SHH has activity, its ability to generate further signaling is impaired [Tokhunts et al., 2010; Traiffort et al., 2004]. These findings may help explain part of the phenotypic variability observed in patients with mutations in SHH [Solomon et al., 2010b], and highlights the role of SHH and the molecules related to its signaling pathway in eye development.
Attempting to correlate the “extraocular anomalies” with the molecular defects in this patient cohort is challenging for a number of reasons, including the small number of patients studied. For instance, strabismus may be seen in patients with structural brain anomalies regardless of the underlying condition, although only 1 of the 4 patients in our cohort who had strabismus had a brain malformation. We observed 2 unrelated proposita with blepharoptosis who had mutations in the C-terminus of SHH; carrier siblings of both patients (not included in this cohort) were observed to have blepharoptosis as well. It is difficult to extrapolate further from this observation, except that patients with SHH mutations bear scrutiny for this and other ophthalmologic disturbances. Nevertheless, from a developmental standpoint, this observation is interesting, as SHH has been described to regulate the patterning of nuclei in the midbrain in mice[Perez-Balaguer et al., 2009]. Some of these nuclei are involved in the control of ocular and palpebral motility.
From the clinical standpoint, many of these ocular anomalies cause complications that may be anticipated and managed through follow-up by a specialized ophthalmologist familiar with HPE. For instance, besides the visual defects, a severe coloboma compromising the retina or optic nerve increases the risk of retinal detachment and choroidal neovascularization. Moreover, patients with microphthalmia are at risk of closed-angle glaucoma due to a smaller anterior chamber [Guercio and Martyn, 2007]. Although patients in this cohort did not present with high degree myopia (defined as a refraction index less than -5D), the natural history of myopia in individuals with HPE-associated mutations is not well characterized, and comparisons with “unaffected” individuals are difficult, hence the incidence of complications are not known. Certain patients, especially those with brain anomalies, present with cortical visual impairment; for these patients, vision does not improve despite adequate correction of the refraction error.
Strabismus appears to be relatively frequent in patients within the HPE phenotypic spectrum; it is of extreme importance to detect heterotropias, since, even slit deviations may result in amblyopia and loss of stereopsis. Patients presenting with documented ocular deviations (and presumably without major brain anomalies) should be treated appropriately, with the goal of establishing binocular vision and orthophoria (proper eye alignment) [Olisky et al., 2007]. In summary, we show that subtle intraocular anomalies such as refraction errors, borderline microcornea, retinal thinning or positioning of the optic nerve, uveal and chorioretinal coloboma, microphthalmia (including nanophthalmia), and extraocular anomalies such as strabismus and blepharoptosis are common in patients within the HPE phenotypic spectrum and with mutations in SHH or genes related to its signaling pathway. Therefore, routine and careful ophthalmologic evaluation of these patients is indicated, as some of these subtle anomalies require intervention and follow-up. Further, these findings may provide insights into potential genotype-phenotype correlations and our understanding of the broad and emerging clinical spectrum of HPE.
Acknowledgments
We would like to thank the patients, and families for their continued support of research investigations into the genetic basis of HPE and its clinical manifestations. This research was supported by the Division of Intramural Research of the National Human Genome Research Institute, the National Institutes of Health.
References
- Arauz RF, Solomon BD, Pineda-Alvarez DE, Gropman AL, Parsons JA, Roessler E, Muenke M. A Hypomorphic Allele in the FGF8 Gene Contributes to Holoprosencephaly and Is Allelic to Gonadotropin-Releasing Hormone Deficiency in Humans. Mol Syndromol. 2010;1:59–66. doi: 10.1159/000302285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakrania P, Ugur Iseri SA, Wyatt AW, Bunyan DJ, Lam WW, Salt A, Ramsay J, Robinson DO, Ragge NK. Sonic hedgehog mutations are an uncommon cause of developmental eye anomalies. Am J Med Genet A. 2010;152:1310–1313. doi: 10.1002/ajmg.a.33239. [DOI] [PubMed] [Google Scholar]
- Brown SA, Warburton D, Brown LY, Yu CY, Roeder ER, Stengel-Rutkowski S, Hennekam RC, Muenke M. Holoprosencephaly due to mutations in ZIC2, a homologue of Drosophila odd-paired. Nat Genet. 1998;20:180–183. doi: 10.1038/2484. [DOI] [PubMed] [Google Scholar]
- Carlson BM. Human embryology and developmental biology. xvi. Philadelphia, PA: Mosby/Elsevier; 2009. p. 541. [Google Scholar]
- Demyer W, Zeman W, Palmer CG. The Face Predicts the Brain: Diagnostic Significance of Median Facial Anomalies for Holoprosencephaly (Arhinencephaly) Pediatrics. 1964;34:256–263. [PubMed] [Google Scholar]
- Domené S, Roessler E, El-Jaick KB, Snir M, Brown JL, Velez JI, Bale S, Lacbawan F, Muenke M, Feldman B. Mutations in the human SIX3 gene in holoprosencephaly are loss of function. Hum Mol Genet. 2008;17:3919–3928. doi: 10.1093/hmg/ddn294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- England SJ, Blanchard GB, Mahadevan L, Adams RJ. A dynamic fate map of the forebrain shows how vertebrate eyes form and explains two causes of cyclopia. Development. 2006;133:4613–4617. doi: 10.1242/dev.02678. [DOI] [PubMed] [Google Scholar]
- Falardeau J, Chung WC, Beenken A, Raivio T, Plummer L, Sidis Y, Jacobson-Dickman EE, Eliseenkova AV, Ma J, Dwyer A, Quinton R, Na S, Hall JE, Huot C, Alois N, Pearce SH, Cole LW, Hughes V, Mohammadi M, Tsai P, Pitteloud N. Decreased FGF8 signaling causes deficiency of gonadotropin-releasing hormone in humans and mice. J Clin Invest. 2008;118(8):2822–2831. doi: 10.1172/JCI34538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gripp KW, Wotton D, Edwards MC, Roessler E, Ades L, Meinecke P, Richieri-Costa A, Zackai EH, Massague J, Muenke M, Elledge SJ. Mutations in TGIF cause holoprosencephaly and link NODAL signalling to human neural axis determination. Nat Genet. 2000;25:205–208. doi: 10.1038/76074. [DOI] [PubMed] [Google Scholar]
- Guercio JR, Martyn LJ. Congenital malformations of the eye and orbit. Otolaryngol Clin North Am. 2007;40:113–140. vii. doi: 10.1016/j.otc.2006.11.013. [DOI] [PubMed] [Google Scholar]
- Haas D, Morgenthaler J, Lacbawan F, Long B, Runz H, Garbade SF, Zschocke J, Kelley RI, Okun JG, Hoffmann GF, Muenke M. Abnormal sterol metabolism in holoprosencephaly: studies in cultured lymphoblasts. J Med Genet. 2007;44(5):298–305. doi: 10.1136/jmg.2006.047258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn JS, Barnes PD. Neuroimaging advances in holoprosencephaly: Refining the spectrum of the midline malformation. Am J Med Genet C Semin Med Genet. 2010;154C:120–132. doi: 10.1002/ajmg.c.30238. [DOI] [PubMed] [Google Scholar]
- Hahn JS, Barnes PD, Clegg NJ, Stashinko EE. Septopreoptic holoprosencephaly: a mild subtype associated with midline craniofacial anomalies. AJNR Am J Neuroradiol. 2010;31:1596–1601. doi: 10.3174/ajnr.A2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn JS, Plawner LL. Evaluation and management of children with holoprosencephaly. Pediatr Neurol. 2004;31:79–88. doi: 10.1016/j.pediatrneurol.2004.03.006. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Yasuda K, Araki M. Coordinated regulation of dorsal bone morphogenetic protein 4 and ventral Sonic hedgehog signaling specifies the dorso-ventral polarity in the optic vesicle and governs ocular morphogenesis through fibroblast growth factor 8 upregulation. Dev Growth Differ. 2010;52:351–363. doi: 10.1111/j.1440-169X.2010.01170.x. [DOI] [PubMed] [Google Scholar]
- Lacbawan F, Solomon BD, Roessler E, El-Jaick K, Domene S, Velez JI, Zhou N, Hadley D, Balog JZ, Long R, Fryer A, Smith W, Omar S, McLean SD, Clarkson K, Lichty A, Clegg NJ, Delgado MR, Levey E, Stashinko E, Potocki L, Vanallen MI, Clayton-Smith J, Donnai D, Bianchi DW, Juliusson PB, Njolstad PR, Brunner HG, Carey JC, Hehr U, Musebeck J, Wieacker PF, Postra A, Hennekam RC, van den Boogaard MJ, van Haeringen A, Paulussen A, Herbergs J, Schrander-Stumpel CT, Janecke AR, Chitayat D, Hahn J, McDonald-McGinn DM, Zackai EH, Dobyns WB, Muenke M. Clinical spectrum of SIX3-associated mutations in holoprosencephaly: correlation between genotype, phenotype and function. J Med Genet. 2009;46:389–398. doi: 10.1136/jmg.2008.063818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levey EB, Stashinko E, Clegg NJ, Delgado MR. Management of children with holoprosencephaly. Am J Med Genet C Semin Med Genet. 2010;154C:183–190. doi: 10.1002/ajmg.c.30254. [DOI] [PubMed] [Google Scholar]
- Lipinski RJ, Godin EA, O’Leary-Moore SK, Parnell SE, Sulik KK. Genesis of teratogen-induced holoprosencephaly in mice. Am J Med Genet C Semin Med Genet. 2010;154C:29–42. doi: 10.1002/ajmg.c.30239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsunaga E, Shiota K. Holoprosencephaly in human embryos: epidemiologic studies of 150 cases. Teratology. 1977;16:261–272. doi: 10.1002/tera.1420160304. [DOI] [PubMed] [Google Scholar]
- Ming JE, Kaupas ME, Roessler E, Brunner HG, Golabi M, Tekin M, Stratton RF, Sujansky E, Bale SJ, Muenke M. Mutations in PATCHED-1, the receptor for SONIC HEDGEHOG, are associated with holoprosencephaly. Hum Genet. 2002;110:297–301. doi: 10.1007/s00439-002-0695-5. [DOI] [PubMed] [Google Scholar]
- Olisky SE, Hug D, Smith LP. Disorders of Eye Movement and Alignment. In: Kliegman RM, editor. Nelson textbook of pediatrics. 18 2007. [Google Scholar]
- Orioli IM, Castilla EE. Clinical epidemiologic study of holoprosencephaly in South America. Am J Med Genet A. 2007;143:3088–3099. doi: 10.1002/ajmg.a.32104. [DOI] [PubMed] [Google Scholar]
- Perez-Balaguer A, Puelles E, Wurst W, Martinez S. Shh dependent and independent maintenance of basal midbrain. Mech Dev. 2009;126(5–6):301–313. doi: 10.1016/j.mod.2009.03.001. [DOI] [PubMed] [Google Scholar]
- Pineda-Alvarez DE, Dubourg C, David V, Roessler E, Muenke M. Current recommendations for the molecular evaluation of newly diagnosed holoprosencephaly patients. Am J Med Genet C Semin Med Genet. 2010;154C:93–101. doi: 10.1002/ajmg.c.30253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro LA, Quiezi RG, Nascimento A, Bertolacini CP, Richieri-Costa A. Holoprosencephaly and holoprosencephaly-like phenotype and GAS1 DNA sequence changes: Report of four Brazilian patients. Am J Med Genet A. 2010;152:1688–1694. doi: 10.1002/ajmg.a.33466. [DOI] [PubMed] [Google Scholar]
- Roessler E, Belloni E, Gaudenz K, Jay P, Berta P, Scherer SW, Tsui LC, Muenke M. Mutations in the human Sonic Hedgehog gene cause holoprosencephaly. Nat Genet. 1996;14:357–360. doi: 10.1038/ng1196-357. [DOI] [PubMed] [Google Scholar]
- Roessler E, Du YZ, Mullor JL, Casas E, Allen WP, Gillessen-Kaesbach G, Roeder ER, Ming JE, Ruiz i Altaba A, Muenke M. Loss-of-function mutations in the human GLI2 gene are associated with pituitary anomalies and holoprosencephaly-like features. Proc Natl Acad Sci U S A. 2003;100:13424–13429. doi: 10.1073/pnas.2235734100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roessler E, El-Jaick KB, Dubourg C, Velez JI, Solomon BD, Pineda-Alvarez DE, Lacbawan F, Zhou N, Ouspenskaia M, Paulussen A, Smeets HJ, Hehr U, Bendavid C, Bale S, Odent S, David V, Muenke M. The mutational spectrum of holoprosencephaly-associated changes within the SHH gene in humans predicts loss-of-function through either key structural alterations of the ligand or its altered synthesis. Hum Mutat. 2009a doi: 10.1002/humu.21090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roessler E, Lacbawan F, Dubourg C, Paulussen A, Herbergs J, Hehr U, Bendavid C, Zhou N, Ouspenskaia M, Bale S, Odent S, David V, Muenke M. The full spectrum of holoprosencephaly-associated mutations within the ZIC2 gene in humans predicts loss-of-function as the predominant disease mechanism. Hum Mutat. 2009b;30:E541–54. doi: 10.1002/humu.20982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roessler E, Muenke M. The molecular genetics of holoprosencephaly. Am J Med Genet C Semin Med Genet. 2010;154C:52–61. doi: 10.1002/ajmg.c.30236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roessler E, Pei W, Ouspenskaia MV, Karkera JD, Velez JI, Banerjee-Basu S, Gibney G, Lupo PJ, Mitchell LE, Towbin JA, Bowers P, Belmont JW, Goldmuntz E, Baxevanis AD, Feldman B, Muenke M. Cumulative ligand activity of NODAL mutations and modifiers are linked to human heart defects and holoprosencephaly. Mol Genet Metab. 2009c;98(1–2):225–34. doi: 10.1016/j.ymgme.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rufer F, Schroder A, Erb C. White-to-white corneal diameter: normal values in healthy humans obtained with the Orbscan II topography system. Cornea. 2005;24:259–261. doi: 10.1097/01.ico.0000148312.01805.53. [DOI] [PubMed] [Google Scholar]
- Schimmenti LA, de la Cruz J, Lewis RA, Karkera JD, Manligas GS, Roessler E, Muenke M. Novel mutation in sonic hedgehog in non-syndromic colobomatous microphthalmia. Am J Med Genet A. 2003;116:215–221. doi: 10.1002/ajmg.a.10884. [DOI] [PubMed] [Google Scholar]
- Solomon BD, Lacbawan F, Mercier S, Clegg NJ, Delgado MR, Rosenbaum K, Dubourg C, David V, Olney AH, Wehner LE, Hehr U, Bale S, Paulussen A, Smeets HJ, Hardisty E, Tylki-Szymanska A, Pronicka E, Clemens M, McPherson E, Hennekam RC, Hahn J, Stashinko E, Levey E, Wieczorek D, Roeder E, Schell-Apacik CC, Booth CW, Thomas RL, Kenwrick S, Cummings DA, Bous SM, Keaton A, Balog JZ, Hadley D, Zhou N, Long R, Velez JI, Pineda-Alvarez DE, Odent S, Roessler E, Muenke M. Mutations in ZIC2 in human holoprosencephaly: description of a Novel ZIC2 specific phenotype and comprehensive analysis of 157 individuals. J Med Genet. 2010a;47:513–524. doi: 10.1136/jmg.2009.073049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon BD, Mercier S, Velez JI, Pineda-Alvarez DE, Wyllie A, Zhou N, Dubourg C, David V, Odent S, Roessler E, Muenke M. Analysis of genotype-phenotype correlations in human holoprosencephaly. Am J Med Genet C Semin Med Genet. 2010b;154C:133–141. doi: 10.1002/ajmg.c.30240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokhunts R, Singh S, Chu T, D’Angelo G, Baubet V, Goetz JA, Huang Z, Yuan Z, Ascano M, Zavros Y, Therond PP, Kunes S, Dahmane N, Robbins DJ. The full-length unprocessed hedgehog protein is an active signaling molecule. J Biol Chem. 2010;285:2562–2568. doi: 10.1074/jbc.M109.078626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traiffort E, Dubourg C, Faure H, Rognan D, Odent S, Durou MR, David V, Ruat M. Functional characterization of sonic hedgehog mutations associated with holoprosencephaly. J Biol Chem. 2004;279:42889–42897. doi: 10.1074/jbc.M405161200. [DOI] [PubMed] [Google Scholar]
- Wallis DE, Roessler E, Hehr U, Nanni L, Wiltshire T, Richieri-Costa A, Gillessen-Kaesbach G, Zackai EH, Rommens J, Muenke M. Mutations in the homeodomain of the human SIX3 gene cause holoprosencephaly. Nat Genet. 1999;22:196–198. doi: 10.1038/9718. [DOI] [PubMed] [Google Scholar]