

Abstract
Increased androgen receptor (AR) transcriptional activity mediated by coactivator proteins may drive castration resistant prostate cancer (CRPC) growth. Vav3, a Rho GTPase guanine nucleotide exchange factor (GEF), is over-expressed in human prostate cancers particularly in models of CRPC progression. Vav3 coactivates AR in a Vav3 pleckstrin homology (PH) domain-dependent but GEF-independent manner. Ectopic expression of Vav3 in androgen-dependent human prostate cancer cells conferred robust castration resistant xenograft tumor growth. Vav3 but not a Vav3 PH mutant greatly stimulated interaction between the AR amino and carboxyl termini (N-C interaction), which is required for maximal receptor transcriptional activity. Vav3 was distributed between the cytoplasm and nucleus with nuclear localization dependent upon the Vav3 PH domain. Membrane targeting of Vav3 abolished Vav3 potentiation of AR activity; whereas, nuclear targeting of a Vav3 PH mutant rescued AR coactivation suggesting that nuclear localization is a key function of the Vav3 PH domain. A nuclear role for Vav3 was further demonstrated by sequential chromatin immunoprecipitation assays, which revealed that Vav3 and AR were recruited to the same transcriptional complexes of an AR target gene enhancer. These data demonstrate the importance of Vav3 in CRPC and define a novel nuclear function of Vav3 in regulating AR activity.
Keywords: Vav3, pleckstrin homology domain, coactivator, androgen receptor, castration resistant prostate cancer, guanine nucleotide exchange factor, tumor xenografts
Introduction
Because the androgen receptor (AR) plays a central role in prostate cancer progression, androgen deprivation therapy is used to manage this malignancy. While most patients respond initially, progression invariably occurs and this recurrent stage is termed castration resistant prostate cancer (CRPC). Despite therapeutic depletion of androgen levels, the AR continues to drive growth of CRPC (Balk, 2002). One mechanism to account for AR action in CRPC is through upregulation of coregulatory proteins that enhance AR activity even when ligand is present at low concentrations. We and others previously showed that levels of Vav3, a guanine nucleotide exchange factor (GEF) for Rho GTPases, are increased during progression of androgen dependent prostate cancer cells to CRPC (Banach-Petrosky et al., 2007; Dong et al., 2006; Lyons and Burnstein, 2006; Lyons et al., 2008). Further, Vav3 serves as a novel AR coactivator in prostate cancer cells even at subnanomolar androgen concentrations, which are often present in CRPC (Lyons and Burnstein, 2006; Mohler et al., 2004; Montgomery et al., 2008). Immunohistochemical analysis of human primary prostate cancer specimens revealed Vav3 overexpression in a significant proportion of samples but not in benign prostatic tissues (Dong et al., 2006). Consistent with a role in prostate cancer development, targeting a constitutively active Vav3 mutant to prostatic epithelium of transgenic mice resulted in adenocarcinoma (Liu et al., 2008).
Vav3 belongs to a small subfamily of diffuse B cell lymphoma (DBL) GEF proteins that also includes the closely related Vav1 and Vav2 (Movilla and Bustelo, 1999). Vav1 expression is typically restricted to cells of hematopoietic lineage while Vav2 and Vav3 are more broadly expressed (Bustelo, 2001). Vav proteins catalyze the exchange of GDP to GTP on Rho family GTPases and this activity is stimulated by growth factor receptor phosphorylation of critical tyrosine residues on the Vav amino terminus (Zeng et al., 2000). Like all DBL family GEFs, Vav proteins contain a highly conserved DBL homology (DH) domain that interacts directly with Rho GTPases and is linked in tandem to a PH domain. Vav3 is best recognized for its cytoplasmic actions whereby Vav3 transmits extracellular growth factor signals to Rho GTPases, including RhoA, RhoG and Rac1 (Lopez-Lago et al., 2000; Movilla and Bustelo, 1999; Zeng et al., 2000). Rho GTPases then interact with downstream effectors to mediate many cellular responses including actin cytoskeleton reorganization, gene expression and cell cycle regulation (Jaffe and Hall, 2005).
Surprisingly, we found that Vav3 potentiation of hormone-inducible AR transcriptional activity is independent of Vav3 GEF activity but requires the pleckstrin homology (PH) domain (Lyons and Burnstein, 2006). In the present study, we established a critical role for Vav3 in conferring castration-resistant tumor xenograft growth and sought to define the molecular mechanism by which Vav3 enhances AR activity. Vav3, but not a Vav3 PH domain mutant, potently enhanced AR N-C interaction, a requirement for maximal AR chromatin binding and transcriptional activity. While many well-characterized steroid receptor coactivator proteins interact with receptors via LxxLL motifs (Heery et al., 1997) neither a conserved LxxLL or a related motif found in Vav3 were required for AR coactivation. We found that the PH domain of Vav3 conferred partial nuclear localization of Vav3 and that Vav3 potentiation of AR transcriptional activity was dependent on Vav3 nuclear localization. Vav3 was co-recruited with AR to the enhancer of the AR target gene prostate specific antigen (PSA). Thus, this study establishes the importance of Vav3 in CRPC growth and defines a novel nuclear function for Vav3 via its PH domain.
Results
Vav3 expression confers castration-resistant prostate cancer tumorigenesis
Because Vav3 is up-regulated following progression of prostate cancer cells and tumors to castration resistance (Banach-Petrosky et al., 2007; Dong et al., 2006; Lyons and Burnstein, 2006), we examined the effect of ectopic expression of wild type Vav3 in VCaP cells, a human prostate cancer cell line that is strictly dependent on androgen for in vitro and in vivo growth (Korenchuk et al., 2001). VCaP cells stably expressing either Vav3 or GFP (control) were inoculated into the hind flanks of immunocompromised SCID mice and mice were castrated when tumors reached approximately 300 mm3. Following castration, xenograft tumors derived from the VCaP/Vav3 cells grew rapidly whereas VCaP/GFP tumors exhibited a lag in growth that is characteristic of VCaP and other androgen-dependent cell lines in response to castration (Fig. 1A). Differences in tumor growth may be due to Vav3 promotion of VCaP cell survival as we observed in androgen-depleted media (Supplementary Fig. 1). Immunoblots from lysates of excised tumors showed that Vav3 expression was retained in these late-stage tumors (Fig. 1A). PSA is secreted from human but not mouse prostate epithelial cells and elevated circulating levels are indicative of tumor growth and AR activity. In line with accelerated tumor growth following castration, mice bearing VCaP/Vav3 tumors had increased circulating PSA levels compared to mice with VCaP/GFP tumors (Fig. 1B).
Figure 1. Vav3 confers castration resistant growth of tumor xenografts.
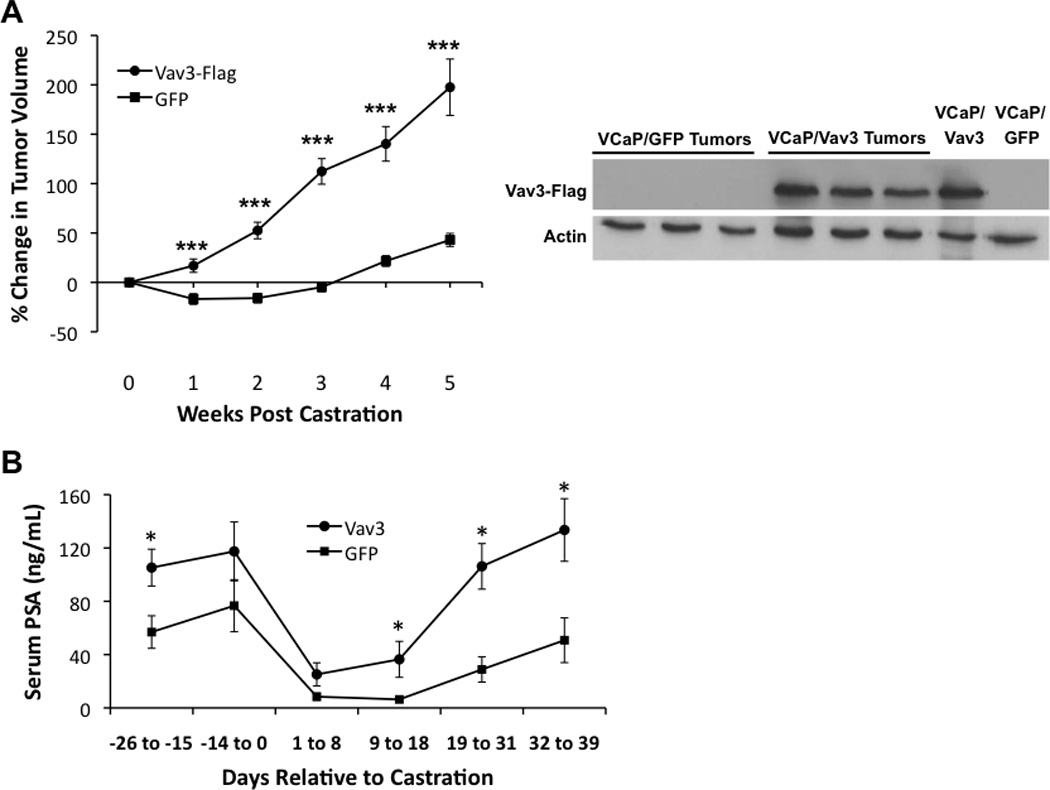
A Left panel: VCaP cells stably expressing Vav3 or GFP (control) were inoculated bilaterally into the hind flanks of immunocompromised SCID mice. Mice were castrated when tumors reached approx. 300 mm3. Tumor volumes (n=12 for VCaP/GFP; n=13–16 for VCaP/Vav3 tumors) were estimated weekly using calipers. Data are plotted as percent change in tumor volume (± SEM) relative to tumor size at time of castration. Significance was determined using a two-tailed Student’s t test (***< 0.001). Right panel: Tumors were excised from euthanized animals five weeks after castration. Vav3-FLAG levels were determined from tumor lysates and from cultured VCaP/Vav3 and VCaP/GFP cells. B. PSA levels were determined from serum samples. Data are plotted as concentration of serum PSA (ng/ml ± SEM) relative to day of castration (n=7–11 for VCaP/GFP; n=6–13 for VCaP/Vav3 tumor-bearing mice). Significance was determined using a two-tailed Student’s t test (*p<0.05, ** p<0.01; ***p<0.005).
To determine whether Vav3 effects on prostate cancer cell proliferation occurred preferentially in AR-expressing cells, ALVA-31 cells, which express Vav3 but not AR were used. AR or vector was stably introduced into ALVA-31 cells and Vav3 activity inhibited using a dominant negative Vav3 mutant, Vav3.1 (Trenkle et al., 2000) (Supplementary Fig. 2A). Inhibition of Vav3 activity caused decreased cell proliferation to a much greater extent in ALVA-31 cells that expressed AR compared to the AR-negative controls (Supplementary Fig. 2B). Together these findings show that Vav3 expression was sufficient for CRPC and that modulation of cell proliferation by Vav3 was most pronounced in AR expressing cells. These results prompted us to examine the mechanism of Vav3 coactivation of AR.
All three Vav family members coactivate AR but not through conserved Vav LxxLL type motifs
Since Vav1 and Vav2 share substantial sequence identity with Vav3, we examined the possibility that other Vav family members coactivate AR. Reporter gene assays using the promoter/enhancer regions of the AR target gene PSA showed that like Vav3, Vav1 and Vav2 potentiated AR activity (Fig. 2A).
Figure 2. Vav family members enhance AR transcriptional activity but a conserved LxxLL and related motif are not required.
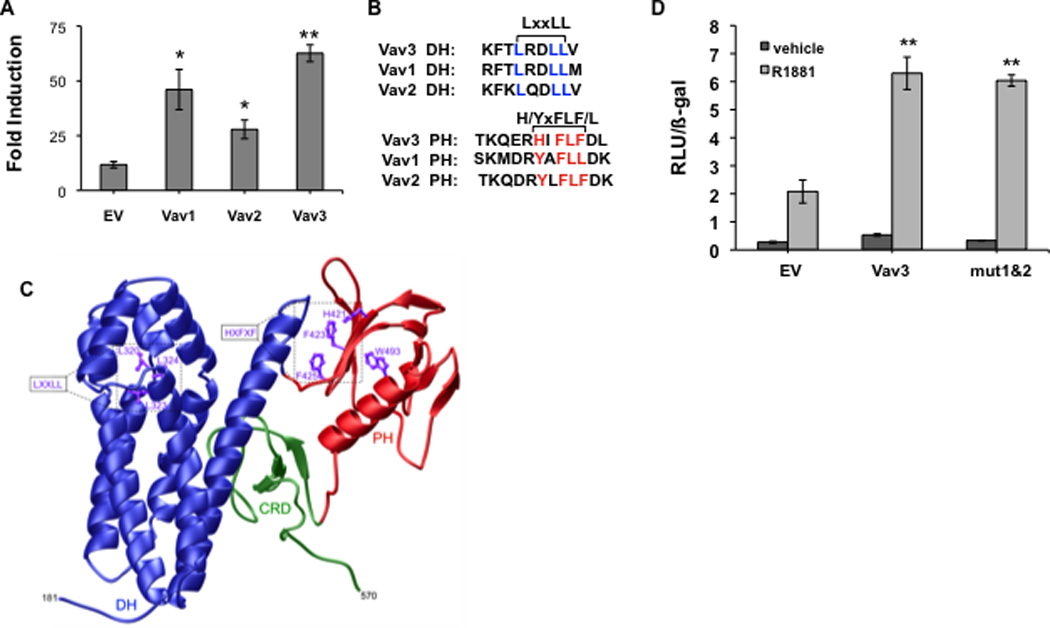
A. PC3 cells transfected with an AR cDNA, PSA-luciferase reporter, a beta-galactosidase expression vector (for normalization) and either Vav1, Vav2, Vav3 or control empty vector (EV) were treated with vehicle or the synthetic androgen R1881 (5 nM) for 48h. Luciferase activity was determined and normalized to beta-galactosidase activity. Data from 3 independent experiments performed in triplicate are plotted as fold induction (relative luciferase units in hormone-treated divided by vehicle-treated samples) ± SEM. B. Sequence alignment of Vav family proteins was performed using AlignX (Vector NTI). DH and PH domains contain a conserved leucine rich motif (LxxLL) in the DH domain and a related aromatic region (HxFxF) in the PH domain. C. Three-dimensional structural model of the DH-PH-CRD contiguous module of human Vav3 is shown. The dbl homology (DH), pleckstrin homology (PH) and cysteine-rich (CRD) domains are respectively colored blue, red and green. The sidechain moieties of conserved residues within the LxxLL and HxFxF motifs are also denoted and colored purple for clarity. The numerals indicate the amino acid boundaries of the DH-PH-CRD module (residues 181–570) within human Vav3. D. PC3 cells transfected with AR, PSA-luciferase, a beta-galactosidase expression vector, wild type Vav3 or Vav3 double mutant (mut 1&2) harboring LxxLL to AxxAA mutations in Vav3 DH domain and HxFLF to AxVAA mutations in Vav3 PH domain or an equal amount of the empty vector (EV) were treated with vehicle or R1881 for 48 hours. Data from a representative experiment (of two independent experiments) performed in triplicate are plotted as relative luciferase units (RLU ± SD) normalized for beta-galactosidase activity. Significance was determined by two-tailed Student’s t test compared with EV transfected cells (A) or EV transfected cells with R1881 (5 nM) treatment (D) (*p<0.05, ** p<0.01).
Many nuclear receptor coactivators contain LxxLL nuclear receptor binding motifs. These motifs interact directly with AF2 regions located in receptor ligand binding domains (Heery et al., 1997). All three Vav family proteins contain an LxxLL sequence in the DH (GEF) domain as well as another conserved hydrophobic region H/YxFLF/L in the PH domain (Fig. 2B). A three dimensional structural model of Vav3 was generated based on the crystal structure of Vav1 (Rapley et al., 2008). This model revealed that the HxFLF forms a potential hydrophobic interface that could be used for protein-protein interactions (Fig. 2C). The other conserved motif (LxxLL) appeared to be less accessible for protein-protein interactions based on this model (Fig. 2C). We tested the possible involvement of both motifs in Vav3 coactivation of AR. A Vav3 double mutant (mut 1&2) containing both LxxLL to AxxAA mutation in the Vav3 DH domain and HxFLF to AxVAA mutation in the Vav3 PH domain was made. This Vav3 double mutant (LxxLL/HxFLF to AxxAA/AxVAA) potentiated AR transcriptional activity similar to wild type Vav3 (Fig. 2D). Single mutants in each of these motifs also retained the capacity to coactivate AR (data not shown). These data suggest that AR coactivation by Vav3 does not require the classical LxxLL nuclear receptor AF2 binding motif in the Vav3 DH domain or the HxFLF sequence in the Vav3 PH domain.
Vav3 enhances androgen-induced AR N-C interaction in a PH domain dependent manner
Androgen inducible AR N-C interaction is essential for optimal AR chromatin binding and transcriptional activity (He et al., 1999; He et al., 2002). To examine whether Vav3 and Vav3 mutants (Fig. 3A) affect AR N-C interaction, mammalian two hybrid assays were conducted. This assay detects interaction between the amino and carboxyl termini of AR fusion proteins using a Gal4 binding site-driven reporter gene as the read out. For these experiments, PC3 human prostate cancer cells were transfected with the Gal4-Tata-Luc reporter, Gal4DBD-ARLBD [the AR carboxyl terminus ligand binding domain (LBD) spanning amino acids 614–919 fused to the Gal4 DNA binding domain (DBD)], and VP16AD-ARTAD [the activation domain (AD) of VP16 fused to AR amino acids 1–565 (TAD)]. In control experiments, omission of either the construct encoding the GAL4DBD-ARLBD or the VP16AD-ARTAD resulted in only negligible background levels of luciferase (data not shown). As previously demonstrated, androgen stimulated luciferase activity in cells expressing both AR fusion proteins indicative of AR N-C interaction (He et al., 2002). Both wild type Vav3 and a Vav3 GEF mutant (Vav3 ISO) greatly enhanced androgen-induced AR N-C interaction. In contrast, the Vav3 PH mutant (W493L) failed to promote AR N-C interaction (Fig. 3B). Western blot analysis showed similar levels of wild type Vav3 and Vav3 mutant expression (Fig. 3C). The Vav3 W493L mutant was previously characterized as lacking PH function, while retaining the ability to bind and activate Rho GTPases, to induce F actin reorganization and cause morphological changes similar to the wild type protein (Movilla and Bustelo, 1999). These findings suggest that this mutation while disrupting PH function, does not significantly alter Vav3 GEF activity. Therefore, our findings indicate that the PH domain but not GEF activity was required for Vav3 enhancement of AR N-C interaction, which supports our previous findings that the Vav3 PH domain, but not GEF activity is essential for coactivation of AR by Vav3 (Lyons and Burnstein, 2006).
Figure 3. Vav3 increases androgen-induced AR N-C interaction in a PH dependent but GEF independent manner.
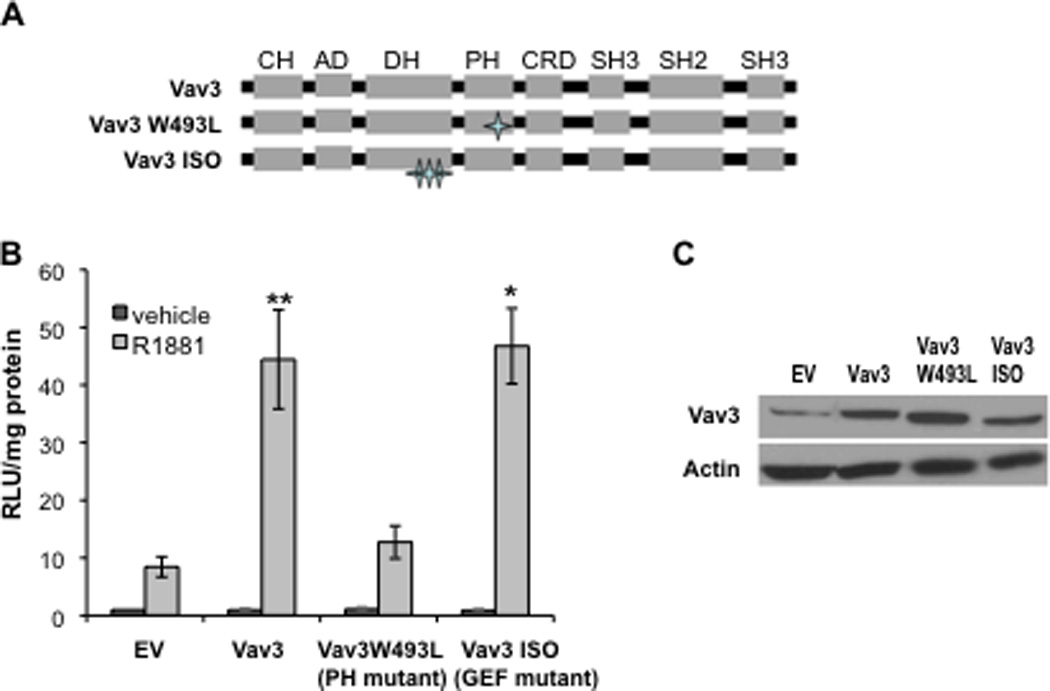
A. Schematic shows Vav3 domain organization and the Vav3 PH and GEF mutants. B. Mammalian two hybrid assays were conducted in which PC3 cells transfected with plasmids encoding: VP16AD-ARTAD [the VP16 activation domain (AD) fused to AR amino acids 1–565], Gal4DBD-ARLBD [the Gal4 DNA binding domain (DBD) fused to the AR ligand binding domain (LBD) 628–919], Gal4-Tata-Luc, and empty vector (EV), Vav3, Vav3 PH mutant W493L or Vav3 GEF mutant (ISO) were treated with either vehicle or R1881 for 24h. Samples were then assayed for luciferase activity and normalized to protein concentration. Data plotted as RLU relative to EV with vehicle treatment (± SEM) are averaged from 3–8 independent experiments. Significance was determined by two-tailed Student’s t test comparing EV transfected cells with R1881 (1 nM) treatment (**p<0.01,*p<0.05). C. Western blot of parallel cultures transfected with indicated plasmids shows expression of Vav3 and Vav3 mutants.
Vav3 PH domain alone does not enhance AR activity but acts as a dominant negative form of Vav3
To determine whether the Vav3 PH domain itself is sufficient to increase AR transcriptional activity, a fusion protein was made with GFP linked to the Vav3 PH domain (GFP-Vav3 PH). GFP-Vav3 PH failed to increase androgen-inducible AR transcriptional activity. However, the GFP-Vav3 PH construct interfered with AR coactivation by full length Vav3, whereas the GFP control did not diminish AR_coactivation, suggesting the inhibitory effect is specific to the Vav3 PH domain (Fig. 4A). GFP-Vav3 PH and full length Vav3 were expressed at comparable levels (Fig. 4B). These results indicate that while the Vav3 PH domain itself was not sufficient for AR coactivation, this domain interfered with full length Vav3 and therefore participates in AR coactivation by Vav3.
Figure 4. Vav3 PH domain alone is not sufficient to enhance AR transcriptional activity but interferes with coactivation of AR by wild type Vav3.
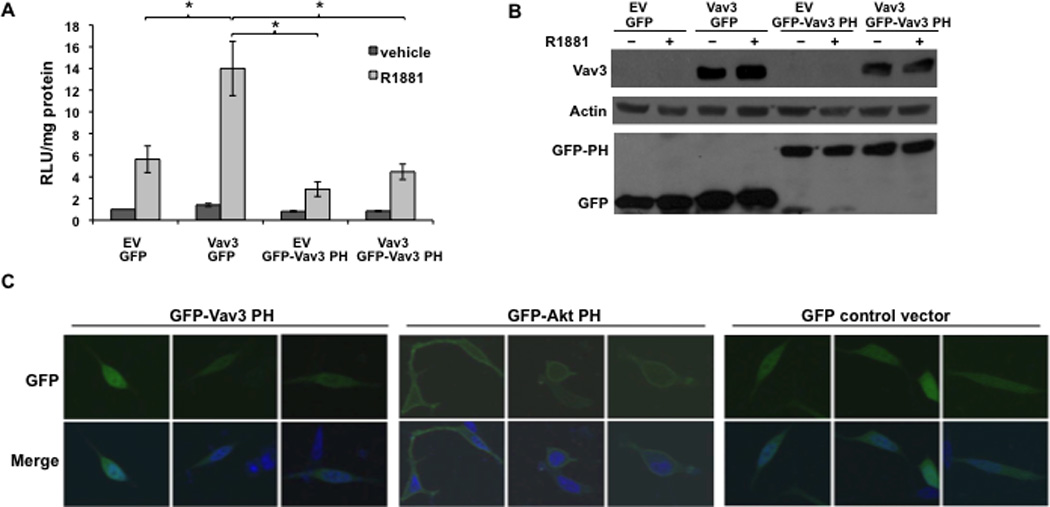
A. PC3 cells transfected with AR, PSA-luc and the indicated plasmids encoding: Vav3 (or EV), GFP fused to the Vav3 PH domain (GFP-Vav3 PH) or GFP, Vav3 plus GFP-Vav3 PH or Vav3 plus GFP were treated with vehicle or R1881 (5 nM). Samples were assayed for luciferase activity and normalized to protein concentration. RLU relative to EV with vehicle treatment from 3 independent experiments performed in triplicate are averaged and plotted (RLU/protein ± SEM). Significance was determined by two-tailed Student’s t test (*p<0.05). B. Parallel cultures transfected with the indicated plasmids were subjected to western blot analysis to assess relative expression levels of Vav3, GFP-Vav3 PH and GFP. C. LNCaP cells transfected with GFP-Vav3 PH domain, GFP-Akt PH domain or GFP control vector were fixed and imaged by confocal microscopy. Representative cells from two independent experiments are shown. Lower images are merged with DAPI staining in blue.
Some PH domains mediate membrane association via high affinity binding to membrane phospholipids (Varnai et al., 2005). We addressed the possibility that the Vav3 PH domain targets Vav3 to the plasma membrane by examining the localization of GFP-Vav3 PH. Since the Akt PH domain is essential for Akt localization to the plasma membrane (Franke et al., 1997; Varnai et al., 2005), GFP fused to the Akt PH domain (GFP-Akt PH) was used as a control. The majority of cells transfected with GFP-Akt PH exhibited GFP localization at the plasma membrane, whereas none of the cells expressing GFP-Vav3 PH exhibited membrane fluorescence. Instead, the GFP-Vav3 PH fusion protein was distributed diffusely in the cytoplasm and nucleus (Fig. 4C). These results suggest that the Vav3 PH domain itself is not sufficient to cause membrane localization.
Membrane targeting of Vav3 disrupts enhancement of AR transcriptional activity
To explore further the possible contribution of Vav3 localization in potentiating AR activity, we targeted Vav3 to the plasma membrane by fusing wild type Vav3 to the C-terminal 18 amino acids of Kras, which includes the CAAX prenylation signal CVIM (Palmby et al., 2002). Addition of this membrane targeting sequence to Vav3 abolished its ability to coactivate AR (Fig. 5A). Vav3 and Vav3-Kras were expressed at similar levels (Fig. 5B). These results suggest that membrane localization of Vav3 is unlikely to be required for AR coactivation.
Figure 5. Membrane targeting of Vav3 eliminates Vav3 coactivation of AR.
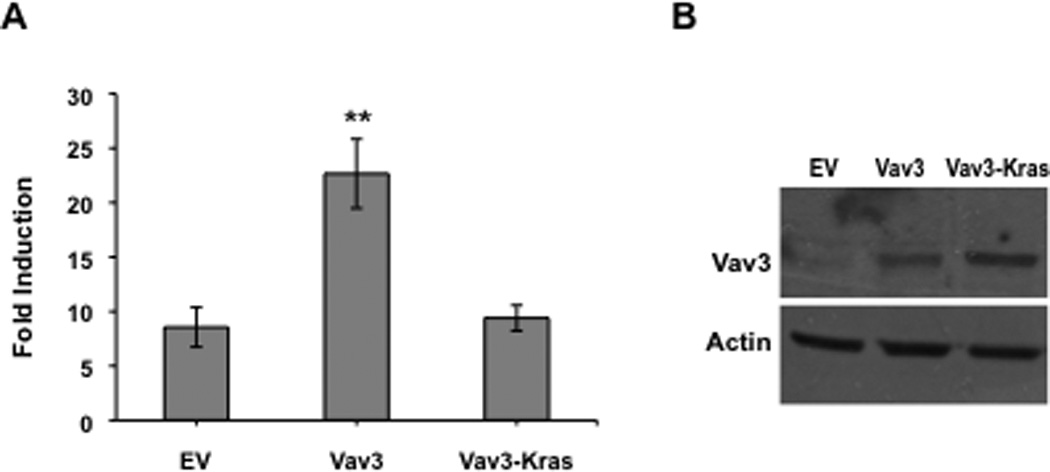
A. PC3 cells were transfected with AR, PSA-luc, Vav3 or a Vav3-Kras fusion proteins containing Kras C-terminal membrane targeting sequence CAAX and treated with either vehicle or R1881 (5 nM). Samples were then assayed for luciferase activity and normalized to protein concentration. Data plotted as fold induction (± SEM) represent the average of 5 independent experiments performed in triplicate. Significance was determined by two-tailed Student’s t test (**p<0.01). B. Parallel cell cultures were subjected to western blot assay using anti-Vav3 and anti-actin antibodies.
Vav3 PH domain confers Vav3 partial nuclear localization
To understand the requirement for the PH domain in Vav3 coactivation of AR, we generated GFP fusions with full length, wild type Vav3 or Vav3W493L (PH domain mutant) and examined subcellular localization. The GFP-Vav3 fusion protein but not GFP-Vav3W493L coactivated AR as expected based on our previous results with the untagged proteins (Lyons and Burnstein, 2006) (data not shown). Cells transfected with GFP-Vav3 exhibited fluorescence in both nuclei and cytoplasm but not in the plasma membrane (Fig. 6A & B) as was also observed by Liu et al (Liu et al., 2010). In contrast, GFP-Vav3W493L was predominately cytoplasmic (Fig. 6 A & B). These findings suggest that the Vav3 PH domain confers partial Vav3 nuclear localization.
Figure 6. Vav3 PH domain confers partial Vav3 nuclear localization.
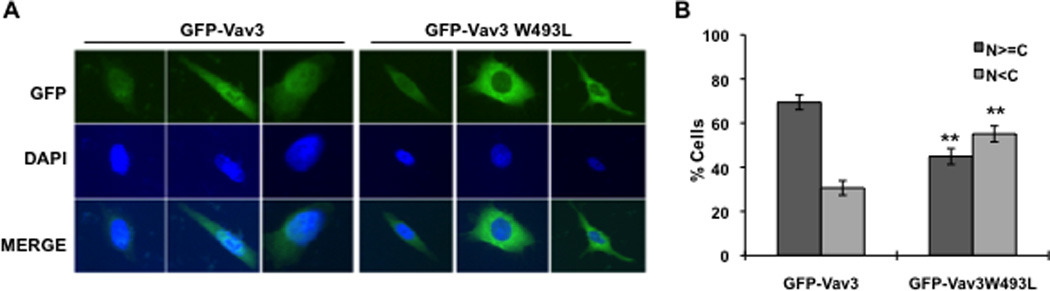
A. PC3 cells were transfected with GFP alone, GFP-Vav3 or GFP-Vav3W493L (PH mutant). 48 hour later, cells were fixed and GFP-Vav3 localization was assessed by fluorescence microscopy. Lower images are merged with DAPI staining in blue. B. Approximately 100 cells from each indicated transfection were scored based on relative nuclear vs. cytoplasmic GFP localization. N≥C indicates greater nuclear or comparable nuclear and cytoplasmic localization; N<C indicates greater cytoplasmic than nuclear localization. Data (summary of 4 independent experiments) are plotted as percentage of total cells counted. Significance was determined by two-tailed Student’s t test compared with GFP-Vav3 (**p<0.01).
Nuclear targeting of a Vav3 PH mutant rescues AR coactivation
To determine whether the reduced nuclear localization of Vav3 W493L was responsible for the impaired ability of this PH mutant to coactivate AR, we constructed Vav3 and Vav3 W493L that were targeted to the nucleus by linking three tandem copies of the SV40 T antigen NLS in frame to the Vav3 C-terminus. Control constructs consisted of Vav3 and Vav3W493L linked only to the myc tag. Prostate cancer cells expressing these Vav3 constructs were subjected to western blotting and were imaged to evaluate expression of the Vav3 fusion proteins and to confirm that addition of the three NLS copies resulted in nuclear localization of Vav3 W493L (Fig. 7 A & B). As shown by reporter assays, nuclear targeting of the Vav3 PH mutant W493L rescued AR coactivation in reporter assays (Fig. 7C). Thus, a critical aspect of Vav3 PH domain function in AR coactivation may be to ensure partial nuclear localization of Vav3.
Figure 7. Nuclear targeting of Vav3 PH domain mutant (W493L) rescues AR coactivation.
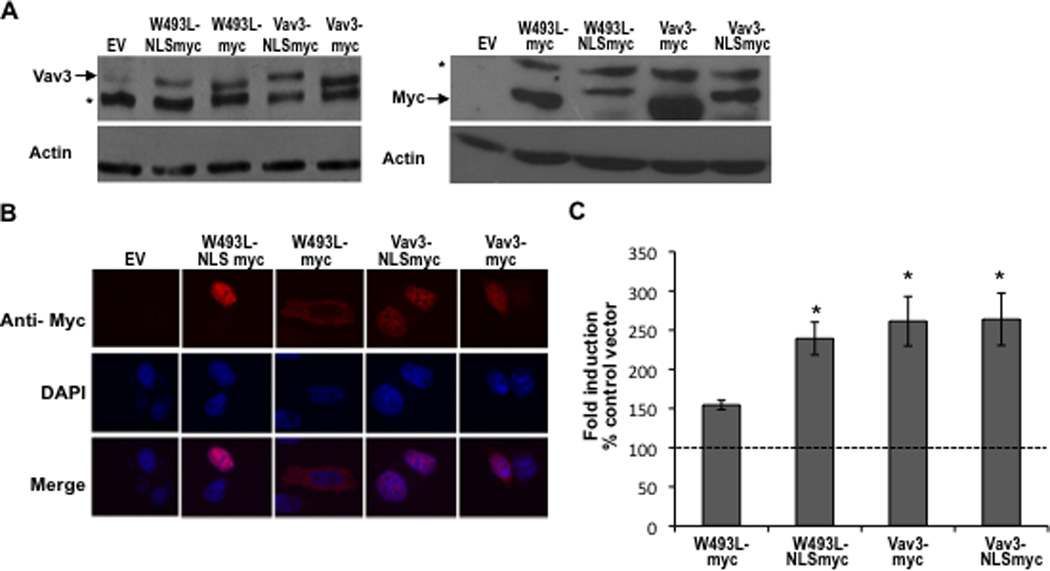
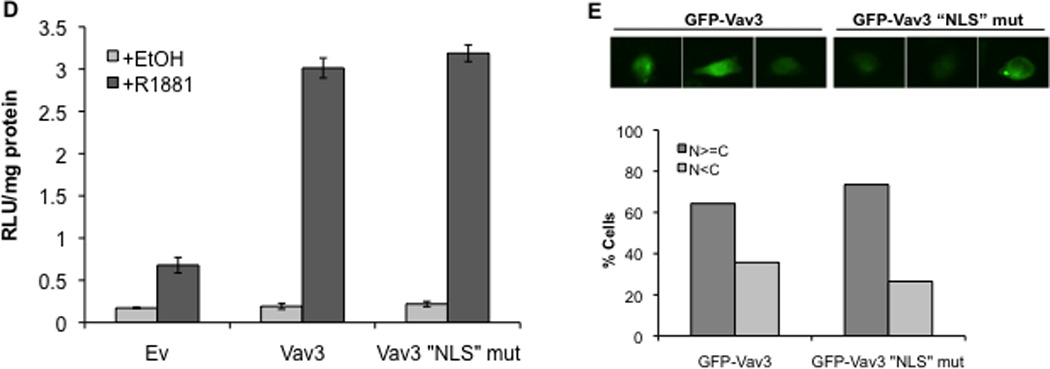
A. Cell lysates from PC3 cells transfected with indicated plasmids (NLS indicates fusion with three copies of the SV40 T antigen nuclear localization sequence) were probed with anti-Vav3 (left) or anti-myc (right) antibodies. Actin was used as the loading control. A representative western blot of each is shown. Asterisks indicate non-specific bands. B. PC3 cells transfected with the indicated plasmids were stained with anti-myc (red) and DAPI (blue) and imaged by confocal microscopy. Lower images are merged with DAPI staining in blue. C. PC3 cells transfected with indicated plasmids, AR, PSA-luc and a beta-galactosidase expression vector were treated with R1881 (1 nM) or vehicle and analyzed by luciferase assay. Data (normalized to beta-galactosidase) are plotted as fold induction relative to control vector (± SEM) and represent the means from 4 independent experiments performed in triplicate. Significance was determined by two-tailed Student’s t test compared to W493L-myc (*p<0.05). D. PC3 cells transfected with AR, PSA-luc, and Vav3, Vav3 NLS mutant (Vav3NLSMUT), or empty vector (Ev) were treated with either vehicle or R1881 (1 nM) for 48 h. Cell lysate were then assayed for luciferase activity. Data from a representative experiment (of three independent experiments) performed in triplicate are plotted as relative luciferase units (mean ± SD) normalized to protein. E. PC3 cells transfected with either GFP-Vav3 or GFP-Vav3“NLS”mutant were imaged by confocal microscopy. Images of 50 cells were scored as in 6B.
The Vav1 PH domain contains a functional nuclear localization sequence (NLS) (KTRELKKK), which is similar to a motif in the Vav3 PH domain (KTKDLKKK). To determine whether this sequence was required for Vav3 coactivation of AR, the putative Vav3 NLS was mutated to ETEDLEEE. Surprisingly, this Vav3 “NLS” mutant retained the capacity to enhance AR activity suggesting that this region is not involved in Vav3 nuclear localization (Fig. 7D). Consistent with this observation, the Vav3 “NLS” mutant, like wild-type Vav3, was localized to both nuclear and cytoplasmic compartments (7E).
Vav3 and AR are in the same transcriptional complexes on the PSA enhancer
Because Vav3 nuclear localization rescued AR coactivation by the Vav3 PH mutant, we investigated the possibility that Vav3 was associated with AREs of an AR target gene in situ. We used the castration resistant derivative of LNCaP cells, LNCaP-R1, which we previously showed express elevated Vav3 levels compared to parental LNCaP cells (Lyons and Burnstein, 2006). Using chromatin immunoprecipitation (ChIP) assays, we observed that endogenous Vav3, like AR, was recruited to the PSA enhancer in LNCaP-R1 cells after androgen treatment (Fig. 8A). Importantly, Vav3 was not found associated with the intervening region of the PSA promoter/enhancer, which lacks AREs (Fig. 8A). These experiments show that native Vav3 was recruited specifically to the ARE-containing enhancer of the PSA gene in response to hormone treatment.
Figure 8. Vav3 and AR are recruited to the same transcriptional complexes in the PSA enhancer.
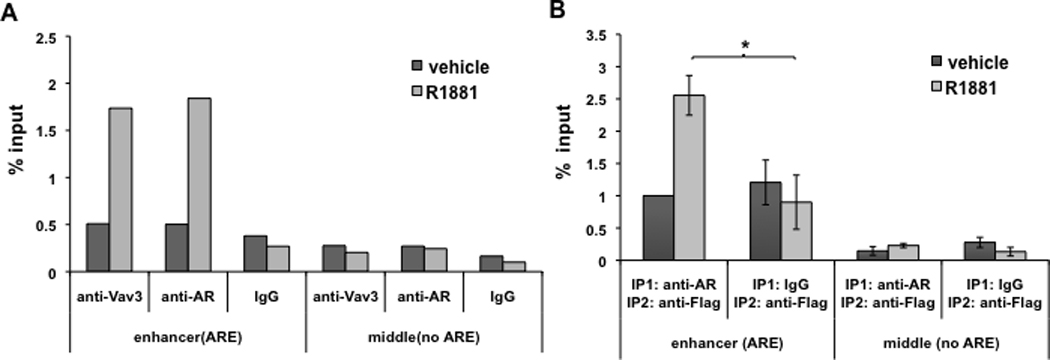
A. LNCaP-R1 cells with endogenous Vav3 expression were maintained in hormone-depleted medium for 72h and then were treated with vehicle or 1nM R1881 for 16h. Chromatin immunoprecipitation (ChIP) was conducted using anti-AR (rabbit), anti-Vav3 (rabbit) antibody, or corresponding rabbit IgG control. Vav3 recruitment to the PSA distal enhancer was determined by real-time PCR. Data plotted as “% input” are the average of two independent experiments. B. LNCaP/Vav3-Flag cells were subjected to sequential ChIP (re-ChIP) assay with anti-AR antibody or IgG control followed by anti-Flag antibody. Vav3 co-recruitment with AR to the PSA distal enhancer was determined by real-time PCR. Data plotted as “Fold ± SEM” relative to vehicle control is the average of three independent experiments. Significance was determined using a two-tailed Student’s t test (*p<0.05).
Since Vav3 and AR were recruited to the PSA enhancer in androgen-treated cells, these proteins might exist in the same transcriptional complexes. To address this possibility, we used sequential chromatin immunoprecipitations in LNCaP cells stably expressing Vav3-Flag. Using anti-AR antibodies or IgG (control) followed by anti-Flag antibodies, Vav3-Flag was detected at the PSA enhancer (Fig. 8B) in the AR immunoprecipitates but not in the control (IgG) immunoprecipitates after androgen treatment. As expected, no AR or Vav3 recruitment was observed in the PSA promoter region lacking AREs. These results support the contention that Vav3 and AR are co-recruited to AREs in chromatin.
Discussion
Vav3 is up-regulated following androgen deprivation and progression to CRPC in human cell-based and tumor xenograft models (Dong et al., 2006; Lyons and Burnstein, 2006; Lyons et al., 2008; Marques et al., 2010; Schmidt et al., 2009). Vav3 expression also increases following progression to CRPC in tumors of the Nkx3.1:Pten mutant mouse model of prostate cancer (Banach-Petrosky et al., 2007; Ouyang et al., 2008). Further, androgen deprivation therapy in men prior to surgical resection of the prostate appears to increase Vav3 gene expression (Holzbeierlein et al., 2004). While Vav3 levels increase with disease progression and Vav3 potently enhances AR activity, its role in CRPC development is unclear. To test this, we examined the effect of Vav3 expression on CPRC progression using an “androgen dependent” human prostate cancer cell xenograft model. While the rate of tumor growth in intact mice was very similar between the Vav3 and GFP-expressing VCaP cells (data not shown), tumors expressing Vav3 did not exhibit the characteristic lag in growth following castration that was exhibited by the control androgen dependent xenografts expressing GFP. Instead, the Vav3 expressing tumors progressed rapidly following castration. These results support the contention that Vav3 is sufficient for castration-resistant tumor growth. Because of the importance of Vav3 in CPRC, we investigated the mechanisms by which Vav3 enhances AR activity and the role of the PH domain in this process.
Our studies indicate that all three Vav family members enhance AR activity. Given the high degree of homology between the three Vav proteins, this is not surprising. Although it is unclear whether Vav1 and Vav2 are involved in prostate cancer development or progression, both proteins appear to have roles in certain other cancers. For example, Vav1 which is normally expressed in cells of hematopoietic lineage, is expressed in a majority of primary neuroblastoma tumors (Hornstein et al., 2003). Additionally, a study which examined 95 pancreatic ductal carcinoma specimens, reported Vav1 expression in over half of the samples (Fujikawa et al., 2003). Vav1 expression was correlated with a reduced survival rate as compared to Vav1-negative tumors (Fernandez-Zapico et al., 2005). Studies of Vav2 have shown its phosphorylation and activation downstream of prolactin signaling and have suggested a role in breast cancer survival and progression (Miller et al., 2005). Vav2 has been reported to be required for angiogenesis and tumor progression in in vivo models of lung carcinoma and melanoma (Brantley-Sieders et al., 2009). Additionally, Vav2 overexpression has been identified in ovarian and head and neck squamous cell carcinoma cell lines (Bourguignon et al., 2001; Patel et al., 2007). These findings and others indicate an emerging role for the Vav proteins in oncogenesis.
Vav3 was first identified as a DBL family Rho GTPase GEF containing a typical tandem arrangement of a DH (GEF) and PH domain. The function of the PH domain of Vav3 however is less well defined as, unlike other DBL GEFs, mutation of the Vav3 PH domain does not disrupt GEF activity (Movilla and Bustelo, 1999). Our previous work supported a unique requirement of the Vav3 PH domain in androgen-mediated enhancement of AR transcriptional activity that is independent of Vav3 GEF activity (Lyons and Burnstein, 2006). PH domains typically share 10–30% sequence identity but have a well-conserved structure (Lemmon, 2004; Lemmon, 2007; Lemmon et al., 2002). While PH domains confer membrane localization of some proteins by virtue of their ability to bind avidly to inositol phospholipids, this characteristic is not universally shared among PH domain-containing proteins (Lemmon, 1999; Lemmon, 2003; Lemmon and Ferguson, 2000; Lemmon and Ferguson, 2001; Varnai et al., 2005). Recent studies suggest that some PH domains may participate in protein nuclear localization; however, the significance and the mechanisms by which PH domains confer differential subcellular localization are not well understood (Houlard et al., 2002; Maffucci et al., 2003; Varnai et al., 2005). Our results suggest that the Vav3 PH domain does not function as a membrane targeting domain but instead participates in Vav3 nuclear localization, which was essential for Vav3 coactivation of AR. The following evidence presented here supports a novel role of the Vav3 PH domain in nuclear localization and potentiation of AR activity: 1. the Vav3 PH domain was essential for Vav3 stimulation of ligand-mediated AR N-C interaction in mammalian two hybrid assays; 2. the Vav3 PH domain alone was not sufficient to mediate membrane localization; 3. Vav3 demonstrated greater nuclear localization compared to a Vav3 PH mutant, which is also impaired in ligand-dependent AR coactivation; 4. nuclear targeting of a Vav3 PH mutant restored AR coactivation; and 5. plasma membrane targeting of Vav3 abrogated its ability to coactivate AR.
Although many PH domain-containing proteins are localized to the plasma membrane, more recent studies have shown that less than ten percent of all known PH domains bind phosphoinositides with high affinity (Lemmon, 2007; Lemmon, 2008). It is noteworthy that PH domains of certain proteins appear to have roles independent of membrane association. Interestingly, several isolated PH domains localize to the nucleus and a role for PH domains in nuclear-cytoplasmic shuttling is emerging. The PH domain of IRS3 is involved in nuclear localization as well as membrane association (Maffucci et al., 2003). Moreover, the very closely related Vav1 PH domain contains a functional nuclear localization signal (NLS). While a similar NLS-like motif is present in Vav3, extensive mutation of this sequence did not affect AR coactivation. Interestingly, our structural model of Vav3 (based on Vav1) showed that the “NLS” motif is not likely to be surface exposed and therefore would not be predicted to be an efficient nuclear targeting motif (data not shown). Our finding that the Vav3 PH domain acted as a dominant negative, outcompeting full length Vav3 for AR coactivation, suggests that the Vav3 PH domain may be involved in protein-protein interactions that permit Vav3 access to the nucleus. Our results are consistent with a mechanism whereby Vav3 is shuttled between nuclear and cytoplasmic compartments, and that nuclear localization relies on the PH domain. These findings are in agreement with other studies that show a reliance on domain interactions for nuclear/cytoplasmic shuttling. For example, in the closely related Vav1 protein, nuclear localization is achieved via the Vav1 PH domain, whereas cytoplasmic localization is conferred by protein-protein interactions involving the C terminal SH3 domain. Signaling events disrupting interaction with the Vav1 SH3 domain relieve cytoplasmic retention and allow nuclear entry (Houlard et al, 2002). Additionally, the casein kinase 2 interacting protein (CKIP 1) shuttles between the nucleus and cytoplasm with the PH domain responsible for nuclear localization. This nuclear localization is opposed by protein interactions involving the C terminal domain. It is hypothesized that the two domains coordinate to determine the subcellular localization of this protein (Xi et al., 2010). Similar mechanisms may regulate Vav3 subcellular localization and function. Our finding that AR coactivation by the Vav3 PH mutant was rescued by nuclear targeting, taken together with our demonstration that the PH domain alone was not sufficient to confer AR coactivation, suggest that although the PH domain was essential for allowing nuclear localization, additional domains of Vav3 must participate in AR coactivation.
Many nuclear receptor coactivators including the well-characterized p160 proteins (e.g. GRIP1/SRC2) contain LxxLL motifs that serve as interfaces for receptor binding (Savkur and Burris, 2004). Typically, coactivator LxxLL motifs interact with receptor AF2 regions present in the carboxy-termini of nuclear receptors. In contrast, the AR AF2 appears to preferentially interact with AR amino terminal motifs that may effectively compete with coactivators (He et al., 2004). Some AR coactivators interact with the amino terminal AF1, which is the more potent activation region of AR (Tindall and Scardino, 2001). We found that the LxxLL motif found in all Vav DH domains was not required for Vav3 enhancement of AR transcriptional activity. While Liu et al (Liu et al., 2010) showed that the DH-PH domains of Vav3 interact with AR, it is not clear that this interaction is retained for full length Vav3 or whether LxxLL motifs are involved. However, like the steroid receptor coactivator, GRIP1/SRC2 (Shen et al., 2005), Vav3 (but not a Vav3 PH mutant) potently increased AR N-C interactions. These findings suggest that Vav3 potentiates AR activity via a unique mechanism. Enhanced AR activity is associated with Vav3 recruitment to the ARE-containing enhancer region of the PSA gene in androgen-treated cells. Importantly, sequential ChIP assays indicated that Vav3 and AR were present in the same transcriptional complexes.
As AR signaling is critical in prostate cancer progression to castration resistance (Chen et al., 2004), targeting the AR remains a major therapeutic strategy for advanced prostate cancer. Since AR activity is governed to a large extent by coactivators, these proteins may also serve as therapeutic targets in the treatment of prostate cancer (O'Malley and Kumar, 2009). We show here that Vav3 expression promoted rapid progression to castration-resistance in tumor xenografts. Vav3 is up-regulated in primary prostate cancer and may increase with disease progression. Our findings that the Vav3 PH domain is critical for Vav3 ligand-dependent AR coactivation is significant as PH domains are considered plausible drug targets (Mahadevan et al., 2008). These studies highlight the importance of Vav3 enhancement of AR activity in prostate cancer progression and support investigation of this protein as a possible therapeutic target.
Materials and methods
Tissue culture
The human prostate cancer cell lines LNCaP.FGC (ATCC cat. No. CRL 1740; batch F-11701) and PC-3 (ATCC cat. No. CRL 1435; batch F-11154) were obtained from American Type Culture Collection (Rockville MD, USA). ). LNCaP-R1 cells were provided by Drs. Shutsung Liao and John Kokontis (University of Chicago). VCaP cells were provided by Dr. Kenneth Pienta, University of Michigan. Cell culture media (RPMI-1640 and DMEM) were obtained from GIBCO-BRL (Gaithersburg, MD, USA). Fetal bovine serum (FBS) was obtained from Hyclone (Logan, Utah, USA). LNCaP and PC-3 cell lines were cultured in RPMI supplemented with 100 IU/ml penicillin, 100ug/ml streptomycin, 2mM L-glutamine (Gibco-BRL, Gathersburg, MD, USA) and 10% FBS. LNCaP cells stably expressing GFP or Vav3-Flag were generated using pQCXIN retroviral transduction system; selected with geneticin (G418, Sigma-Aldrich, St Louis, MO, USA) and cultured in 10% FBS/RPMI medium supplemented with 0.35ug/ml G418. VCaP cells stably expressing GFP or Vav3-Flag were generated using pQCXIP retroviral transduction system; selected with puromycin (Sigma-Aldrich) and cultured in 10%FBS/DMEM supplemented with 400ng/mL puromycin.
Plasmids
The human androgen receptor (AR) cDNA (pCMVhAR) was provided by Dr. Michael McPhaul. The PSA Luciferase plasmid was kindly provided by Dr. Carlos Perez-Stable (University of Miami). Vectors encoding: Gal4DBD-ARLBD, VP16AD-ARTAD, and Gal4-Tata-Luc were generously provided by Dr. Karen Knudsen (University of Cincinnati). Vav3, Vav3 PH mutant W493L and Vav3 GEF mutant Vav3 ISO were previously described (Lyons and Burnstein, 2006). All PCR based approaches were performed using the Expand Hi Fidelity PCR system (Roche Applied Bioscience, Indianapolis, IN, USA). Different Vav3 mutants were created using the quick-change site-directed mutagenesis kit (Stratagene, La Jolla, CA, USA) according to the manufacturer’s instructions. Mutagenic primers of Vav3 mut1 (LxxLL to AxxAA) and mut2 (HxFLF to AxVAA) were as follows: mut1 F: 5’-GAG CAA ATA ATG GGA AAT TTA CTG CTC GAG ACG CGG CTG TGG TTC CTA TGC AAC G-3’ and the following reverse primers: mut1 R: 5’-CGT TGC ATA GGA ACC ACA GCC GCG TCT CGA GCA GTA AAT TTC CCA TTA TTT GCT C-3’; mut2 F: 5’-GAC AAG CAT ACC AAA CAA GAA AGG GCT ATC GTC GCA GCT GAT TTG GCA GTG ATC GTA TGT AAG-3’, mut2 R: 5’-CTT ACA TAC GAT CAC TGC CAA ATC AGC TGC GAC GAT AGC CCT TTC TTG TTT GGT ATG CTT GTC-3’. GFP fusion of Vav3 and isolated Vav3 PH domain were generated by PCR-based approach using the following primers: GFP-Vav3 F: 5’-CCG AAT TCG GGT ATG GAG CCG TGG AAG CAG TGC GCG-3’, R: 5’-GGG TCG ACG GGA TTT GAA TTT ATT CAT CCT CTT CCA C-3’; GFP-Vav3 PH domain F: 5’-CGG AAT TCC TCT ATA GAG ATT TTG AAC CAA CCA GTT TTG-3’, R: 5’- CCG TCG ACT GGT CTT ATG TTA GAC AAA GCC ATT TCA AAC TG-3’. The PCR fragment was digested with EcoRI and SalI and inserted into the same sites of pAcGFP1-C1 vector. Vav3-NLS myc was constructed using the forward primer: 5’-ACA CCA TGG GCA TGG AGC CGT GGA AGC AGT GC-3’ and the reverse primer: 5’-CGG TCG ACT TCA TCC TCT TCC ACA TAT GTG GAT GG-3’. The PCR-amplified fragment was digested with NcoI and SalI and inserted into the same sites of pCMV/myc/nuc vector (Invitrogen Life Technologies, Grand island, NY, USA) and then subcloned into pIRES2-EGFP vector using the forward primer: 5’-GCG AAT TCG CAT GGA GCC GTG GAA GCA GTG CGC GCA GTG GCT C-3’ and the reverse primer: 5’- GAC CCG CGG CTA TGC GGC CCC ATT CAG ATC CTC TTC TGA GAT-3’. The forward primer contains an EcoRI and reverse primer contains SacII site. The PCR fragment was then inserted into pIRES2-EGFP vector using EcoRI and SacII sites. GFP-Vav3W493L and Vav3 W493L-NLSmyc were created by the quick-change site-directed mutagenesis kit as described before using corresponding templates (Lyons and Burnstein, 2006). For Vav3-Flag in retroviral vector, Vav3 in pIRES2-EGFP was firstly subcloned into PacI modified p3XFLAG-CMV™-14 expression vector (Sigma) using EcoRI and KpnI sites. Then Vav3-Flag was subcloned into the retroviral vectors pQCXIN and pQCXIP using NotI and PacI restriction sites. GFP in the vectors pQCXIN and pQCXIP were cloned between NotI and BamHI restriction sites.
Generation of VCaP-Vav3-Flag and VCaP-GFP Xenografts
All experiments involving animals were conducted in a manner approved by the University of Miami Animal Care and Use Committee (Miami, FL). VCaP cells were infected with retroviral constructs encoding either Vav3-Flag or GFP (control) followed by selection in puromycin. VCaP-Vav3-Flag or VCaP-GFP cells (2×106) were injected subcutaneously with an equal volume of Matrigel (BD Biosciences) into both hind flanks of severe combined immunodeficient (SCID) mice (Harlan). Serum samples were taken every two weeks throughout the duration of the experiment. Tumor volume was measured at least twice weekly with calipers calculated using the formula Length*Width*Height*0.52. Upon reaching a tumor volume of approximately 300mm3, all mice were castrated. Tumors were allowed to grow for either 5 weeks or until tumors reached 1000 mm3 and mice were then euthanized. Tumors were excised and quick frozen. Levels of circulating PSA were quantified from serum samples obtained 4 weeks after tumor cell implantation, every two weeks throughout the duration of the experiment, and on the day of euthanasia utilizing an ELISA (Biocheck Inc) using known quantities of purified PSA as control.
Western Blotting
All transfections were carried out using the cationic lipid reagent Lipofectamine (Invitrogen Life Technologies, Grand island, NY,USA) as described (Lyons and Burnstein, 2006). Tumor and cell lysates were subjected to western blot analysis for Vav3 (anti-Vav3, Millipore, Temecula, CA, USA), GFP fusion proteins, (anti-GFP, Invitrogen), myc fusion proteins (anti-myc, Invitrogen), cleaved PARP (Cell Signaling Technology, Beverly, MA, USA) and visualized by enhanced chemiluminescence (ECL) (Amersham Pharmacia Biotech Piscataway, NJ, USA).
Chromatin Immunoprecipitation (ChIP) and sequential ChIP (re-ChIP)
LNCaP cells were maintained in serum-free, phenol-red free RPMI with 2% charcoal stripped serum for 3 days. After 16h treatment with vehicle or R1881 (1nM), cells were fixed with 1% formaldehyde and lysed before sonication. Immunoprecipitation of soluble chromatin was conducted using anti-Vav3 (Millipore), anti-Flag (Sigma, M2), or IgG control (Santa Cruz Biotechnology, Santa Cruz, CA, USA) and protein A or G sepharose beads (Millipore). After several washes, immunocomplexes were eluted off the beads and heated at 65°C to reverse the crosslinking. DNA fragments were purified with the Qiagen PCR purification kit. The fragments were then analyzed by real-time PCR and every sample was run in triplicate. Real-time PCR was performed using an icycler iQ PCR detection system (Bio-Rad Laboratories, Inc., Hercules, CA, USA) with iQ SyberGreen supermix (Bio-Rad, Hercules, CA). The primers for the PSA distal enhancer were as follows: forward, 5’-CCA AAT CTT GTA GGG TGA C-3’; and reverse 5’-CAA TAT GTT CCT CCA GAG TAG-3’ (Wang et al., 2005). The primers for the middle control region were as follows: forward, 5'-TTG GCT CAG ACA TCC TTG-3'; and reverse, 5'-CAA CGC AAC TTA ACC TAA C-3'. For re-ChIP, samples were subjected to sequential rounds of immunoprecipitation. After primary immunoprecipitation with anti-AR (PG-21) (Millipore) or rabbit IgG control (Santa Cruz), the immunocomplexes were eluted from the beads by incubation in 10mM DTT elution buffer. Diluted elution complexes were then subjected to second immunoprecipiation using anti-Flag M2 antibody (Sigma). The remaining steps were the same as described for ChIP. The results were analyzed as percentage of immunoprecipitated DNA vesus total input (% input) using the equation of 100*2^(CTinput−CTsample).
Immunocytochemistry and Immunofluorescence
Cells transfected with Vav3-NLSmyc or W493L-NLSmyc were fixed with 2% formaldehyde and permeabilized with 0.2% Triton X-100. Following 1 h blocking with 3% BSA in PBS, cells were incubated with anti-myc antibody for 1h (1:200), washed with PBS and then incubated with CY3 mouse secondary antibody (1:150) for 30 minutes. After PBS wash, cells were mounted with Prolong Gold antifade reagent containing DAPI and imaged with control microscopy. As for GFP fusion protein, transfected cells were fixed with 2% formaldehyde, washed with PBS and then mounted.
Mammalian two hybrid interaction assay
PC3 cells were transfected with pVP16-AR-(14–565), Gal4-AR-(628–919), Gal4-TATA-LUC reporter, a beta-galactosidase vector (for normalization) and Vav3, Vav3 mutants or control vector for about 4–5 hours and then treated with vehicle or 1nM R1881 for 24h. 48h after transfection cells were assessed for luciferase activity using the Promega luciferase assay kit.
Molecular modeling
Molecular modeling was employed to obtain three-dimensional structure of the DH-PH-CRD contiguous module (residues 181–570) of human Vav3 using the MODELLER software based on homology modeling (Marti-Renom et al., 2000). Briefly, MODELLER employs molecular dynamics and simulated annealing protocols to optimize the modeled structure through satisfaction of spatial restraints derived from amino acid sequence alignment with a corresponding template in Cartesian space. Here, the crystal structure of the DH-PH-CRD contiguous module of human Vav1 (PDB# 2VRW) was used as a template to obtain atomic model of the corresponding module of human Vav3. For amino acid sequence identity between 30–50% between the template and target, MODELLER can generate atomic models with high accuracy (John and Sali, 2003). Importantly, the DH-PH-CRD modules of human Vav1 and human Vav3 share close to 60% amino acid sequence identity. Due to such high sequence identity between the template and target, the atomic model of the DH-PH-CRD module of human Vav3 can be relied upon with a high degree of confidence. A total of 100 atomic models were calculated and the structure with the lowest energy, as judged by the MODELLER Objective Function, was selected for further analysis. The atomic model was rendered using RIBBONS (Carson, 1991).
Supplementary Material
A. PC3 cells transfected with AR, PSA-luc, beta galactosidase (B-gal) and either Vav3, Vav3.1 and Vav3, or empty vector (Ev) were treated with either vehicle or R1881 (1 nM) for 48 h. Cell lysates were then assayed for luciferase activity. Data from a representative experiment (of three independent experiments) performed in triplicate are plotted as relative luciferase units (mean ± SD) normalized to beta-galactosidase activity. B. ALVA 31 cells stably expressing the AR cDNA (ALVA-AR) or neo vector alone (ALVA-NEO) were transfected with a dominant negative Vav3 construct (Vav3.1) or empty vector. Thirty hours later, media were replaced with 3H thymidine-containing media and cells were cultured for 18 h. Acid-soluble radioactivity was removed by TCA and ethanol washes and incorporated radioactivity was determined by liquid scintillation counting.
VCaP cells stably expressing Vav3 or GFP (control) were depleted of androgen and then replated in media containing either 10% FBS (+) or 10% CSS-FBS (−) and cultured for the indicated times. Lysates were probed with antibody against cleaved PARP or actin.
Acknowledgements
We are grateful to Dr. Zafar Nawaz and his laboratory for advice on ChIP assays. We thank Drs. Adena Rosenblatt, Omar Flores and Carlos Perez-Stable for helpful suggestions. Zhe Ma provided assistance with the mammalian two hybrid assays. A National Institutes of Health grant RO1CA132200 (to K.L.B.) supported this work. L.S.L. was supported by U.S. Department of Defense post-doctoral fellowship (PC060504); S.R. was an American Heart Association Predoctoral Fellow; CDF was supported by T32-HL007188 and A.F. acknowledges funding from R01GM083897.
Footnotes
Conflict of Interest Statement: The authors have nothing to disclose.
References
- Balk SP. Androgen receptor as a target in androgen-independent prostate cancer. Urology. 2002;60:132–138. doi: 10.1016/s0090-4295(02)01593-5. discussion 138-9. [DOI] [PubMed] [Google Scholar]
- Banach-Petrosky W, Jessen WJ, Ouyang X, Gao H, Rao J, Quinn J, et al. Prolonged exposure to reduced levels of androgen accelerates prostate cancer progression in Nkx3.1; Pten mutant mice. Cancer Res. 2007;67:9089–9096. doi: 10.1158/0008-5472.CAN-07-2887. [DOI] [PubMed] [Google Scholar]
- Bourguignon LY, Zhu H, Zhou B, Diedrich F, Singleton PA, Hung MC. Hyaluronan promotes CD44v3-Vav2 interaction with Grb2-p185(HER2) and induces Rac1 and Ras signaling during ovarian tumor cell migration and growth. J Biol Chem. 2001;276:48679–48692. doi: 10.1074/jbc.M106759200. [DOI] [PubMed] [Google Scholar]
- Brantley-Sieders DM, Zhuang G, Vaught D, Freeman T, Hwang Y, Hicks D, et al. Host deficiency in Vav2/3 guanine nucleotide exchange factors impairs tumor growth, survival, and angiogenesis in vivo. Mol Cancer Res. 2009;7:615–623. doi: 10.1158/1541-7786.MCR-08-0401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustelo XR. Vav proteins, adaptors and cell signaling. Oncogene. 2001;20:6372–6381. doi: 10.1038/sj.onc.1204780. [DOI] [PubMed] [Google Scholar]
- Carson M. Journal of Applied Crystallography. 1991;Vol. 24:958–961. [Google Scholar]
- Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, et al. Molecular determinants of resistance to antiandrogen therapy. Nat Med. 2004;10:33–39. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- Dong Z, Liu Y, Lu S, Wang A, Lee K, Wang LH, et al. Vav3 oncogene is overexpressed and regulates cell growth and androgen receptor activity in human prostate cancer. Mol Endocrinol. 2006;20:2315–2325. doi: 10.1210/me.2006-0048. [DOI] [PubMed] [Google Scholar]
- Fernandez-Zapico ME, Gonzalez-Paz NC, Weiss E, Savoy DN, Molina JR, Fonseca R, et al. Ectopic expression of VAV1 reveals an unexpected role in pancreatic cancer tumorigenesis. Cancer Cell. 2005;7:39–49. doi: 10.1016/j.ccr.2004.11.024. [DOI] [PubMed] [Google Scholar]
- Franke TF, Kaplan DR, Cantley LC, Toker A. Direct regulation of the Akt proto-oncogene product by phosphatidylinositol-3,4-bisphosphate. Science. 1997;275:665–668. doi: 10.1126/science.275.5300.665. [DOI] [PubMed] [Google Scholar]
- Fujikawa K, Miletic AV, Alt FW, Faccio R, Brown T, Hoog J, et al. Vav1/2/3-null mice define an essential role for Vav family proteins in lymphocyte development and activation but a differential requirement in MAPK signaling in T and B cells. J Exp Med. 2003;198:1595–1608. doi: 10.1084/jem.20030874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B, Gampe RT, Jr, Kole AJ, Hnat AT, Stanley TB, An G, et al. Structural basis for androgen receptor interdomain and coactivator interactions suggests a transition in nuclear receptor activation function dominance. Mol Cell. 2004;16:425–438. doi: 10.1016/j.molcel.2004.09.036. [DOI] [PubMed] [Google Scholar]
- He B, Kemppainen JA, Voegel JJ, Gronemeyer H, Wilson EM. Activation function 2 in the human androgen receptor ligand binding domain mediates interdomain communication with the NH(2)-terminal domain. J Biol Chem. 1999;274:37219–37225. doi: 10.1074/jbc.274.52.37219. [DOI] [PubMed] [Google Scholar]
- He B, Lee LW, Minges JT, Wilson EM. Dependence of selective gene activation on the androgen receptor NH2- and COOH-terminal interaction. J Biol Chem. 2002;277:25631–25639. doi: 10.1074/jbc.M202809200. [DOI] [PubMed] [Google Scholar]
- Heery DM, Kalkhoven E, Hoare S, Parker MG. A signature motif in transcriptional co-activators mediates binding to nuclear receptors. Nature. 1997;387:733–736. doi: 10.1038/42750. [DOI] [PubMed] [Google Scholar]
- Holzbeierlein J, Lal P, LaTulippe E, Smith A, Satagopan J, Zhang L, et al. Gene expression analysis of human prostate carcinoma during hormonal therapy identifies androgen-responsive genes and mechanisms of therapy resistance. Am J Pathol. 2004;164:217–227. doi: 10.1016/S0002-9440(10)63112-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornstein I, Pikarsky E, Groysman M, Amir G, Peylan-Ramu N, Katzav S. The haematopoietic specific signal transducer Vav1 is expressed in a subset of human neuroblastomas. J Pathol. 2003;199:526–533. doi: 10.1002/path.1314. [DOI] [PubMed] [Google Scholar]
- Houlard M, Arudchandran R, Regnier-Ricard F, Germani A, Gisselbrecht S, Blank U, et al. Vav1 is a component of transcriptionally active complexes. J Exp Med. 2002;195:1115–1127. doi: 10.1084/jem.20011701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe AB, Hall A. Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol. 2005;21:247–269. doi: 10.1146/annurev.cellbio.21.020604.150721. [DOI] [PubMed] [Google Scholar]
- John B, Sali A. Comparative protein structure modeling by iterative alignment, model building and model assessment. Nucleic Acids Res. 2003;31:3982–3992. doi: 10.1093/nar/gkg460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korenchuk S, Lehr JE, L MC, Lee YG, Whitney S, Vessella R, et al. VCaP, a cell-based model system of human prostate cancer. In Vivo. 2001;15:163–168. [PubMed] [Google Scholar]
- Lemmon MA. Structural basis for high-affinity phosphoinositide binding by pleckstrin homology domains. Biochem Soc Trans. 1999;27:617–624. doi: 10.1042/bst0270617. [DOI] [PubMed] [Google Scholar]
- Lemmon MA. Phosphoinositide recognition domains. Traffic. 2003;4:201–213. doi: 10.1034/j.1600-0854.2004.00071.x. [DOI] [PubMed] [Google Scholar]
- Lemmon MA. Pleckstrin homology domains: not just for phosphoinositides. Biochem Soc Trans. 2004;32:707–711. doi: 10.1042/BST0320707. [DOI] [PubMed] [Google Scholar]
- Lemmon MA. Pleckstrin homology (PH) domains and phosphoinositides. Biochem Soc Symp. 2007:81–93. doi: 10.1042/BSS0740081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon MA. Membrane recognition by phospholipid-binding domains. Nat Rev Mol Cell Biol. 2008;9:99–111. doi: 10.1038/nrm2328. [DOI] [PubMed] [Google Scholar]
- Lemmon MA, Ferguson KM. Signal-dependent membrane targeting by pleckstrin homology (PH) domains. Biochem J. 2000;350(Pt 1):1–18. [PMC free article] [PubMed] [Google Scholar]
- Lemmon MA, Ferguson KM. Molecular determinants in pleckstrin homology domains that allow specific recognition of phosphoinositides. Biochem Soc Trans. 2001;29:377–384. doi: 10.1042/bst0290377. [DOI] [PubMed] [Google Scholar]
- Lemmon MA, Ferguson KM, Abrams CS. Pleckstrin homology domains and the cytoskeleton. FEBS Lett. 2002;513:71–76. doi: 10.1016/s0014-5793(01)03243-4. [DOI] [PubMed] [Google Scholar]
- Liu Y, Mo JQ, Hu Q, Boivin G, Levin L, Lu S, et al. Targeted overexpression of vav3 oncogene in prostatic epithelium induces nonbacterial prostatitis and prostate cancer. Cancer Res. 2008;68:6396–6406. doi: 10.1158/0008-5472.CAN-08-0645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Wu X, Dong Z, Lu S. The molecular mechanism of Vav3 oncogene on upregulation of androgen receptor activity in prostate cancer cells. Int J Oncol. 2010;36:623–633. doi: 10.3892/ijo_00000538. [DOI] [PubMed] [Google Scholar]
- Lopez-Lago M, Lee H, Cruz C, Movilla N, Bustelo XR. Tyrosine phosphorylation mediates both activation and downmodulation of the biological activity of Vav. Molecular and cellular biology. 2000;20:1678–1691. doi: 10.1128/mcb.20.5.1678-1691.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons LS, Burnstein KL. Vav3, a Rho GTPase guanine nucleotide exchange factor, increases during progression to androgen independence in prostate cancer cells and potentiates androgen receptor transcriptional activity. Mol Endocrinol. 2006;20:1061–1072. doi: 10.1210/me.2005-0346. [DOI] [PubMed] [Google Scholar]
- Lyons LS, Rao S, Balkan W, Faysal J, Maiorino CA, Burnstein KL. Ligand-Independent Activation of Androgen Receptors by Rho GTPase Signaling in Prostate Cancer. Mol Endocrinol. 2008;22:597–608. doi: 10.1210/me.2007-0158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maffucci T, Razzini G, Ingrosso A, Chen H, Iacobelli S, Sciacchitano S, et al. Role of pleckstrin homology domain in regulating membrane targeting and metabolic function of insulin receptor substrate 3. Mol Endocrinol. 2003;17:1568–1579. doi: 10.1210/me.2001-0211. [DOI] [PubMed] [Google Scholar]
- Mahadevan D, Powis G, Mash EA, George B, Gokhale VM, Zhang S, et al. Discovery of a novel class of AKT pleckstrin homology domain inhibitors. Mol Cancer Ther. 2008;7:2621–2632. doi: 10.1158/1535-7163.MCT-07-2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques RB, Dits NF, Erkens-Schulze S, van Weerden WM, Jenster G. Bypass mechanisms of the androgen receptor pathway in therapy-resistant prostate cancer cell models. PLoS One. 2010;5:e13500. doi: 10.1371/journal.pone.0013500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti-Renom MA, Stuart AC, Fiser A, Sanchez R, Melo F, Sali A. Comparative protein structure modeling of genes and genomes. Annu Rev Biophys Biomol Struct. 2000;29:291–325. doi: 10.1146/annurev.biophys.29.1.291. [DOI] [PubMed] [Google Scholar]
- Miller SL, DeMaria JE, Freier DO, Riegel AM, Clevenger CV. Novel association of Vav2 and Nek3 modulates signaling through the human prolactin receptor. Mol Endocrinol. 2005;19:939–949. doi: 10.1210/me.2004-0443. [DOI] [PubMed] [Google Scholar]
- Mohler JL, Gregory CW, Ford OH, 3rd, Kim D, Weaver CM, Petrusz P, et al. The androgen axis in recurrent prostate cancer. Clin Cancer Res. 2004;10:440–448. doi: 10.1158/1078-0432.ccr-1146-03. [DOI] [PubMed] [Google Scholar]
- Montgomery RB, Mostaghel EA, Vessella R, Hess DL, Kalhorn TF, Higano CS, et al. Maintenance of intratumoral androgens in metastatic prostate cancer: a mechanism for castration-resistant tumor growth. Cancer Res. 2008;68:4447–4454. doi: 10.1158/0008-5472.CAN-08-0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Movilla N, Bustelo XR. Biological and regulatory properties of Vav-3, a new member of the Vav family of oncoproteins. Mol Cell Biol. 1999;19:7870–7885. doi: 10.1128/mcb.19.11.7870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Malley BW, Kumar R. Nuclear receptor coregulators in cancer biology. Cancer Res. 2009;69:8217–8222. doi: 10.1158/0008-5472.CAN-09-2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang X, Jessen WJ, Al-Ahmadie H, Serio AM, Lin Y, Shih WJ, et al. Activator protein-1 transcription factors are associated with progression and recurrence of prostate cancer. Cancer Res. 2008;68:2132–2144. doi: 10.1158/0008-5472.CAN-07-6055. [DOI] [PubMed] [Google Scholar]
- Palmby TR, Abe K, Der CJ. Critical role of the pleckstrin homology and cysteine-rich domains in Vav signaling and transforming activity. Journal of Biological Chemistry. 2002;277:39350–39359. doi: 10.1074/jbc.M202641200. [DOI] [PubMed] [Google Scholar]
- Patel V, Rosenfeldt HM, Lyons R, Servitja JM, Bustelo XR, Siroff M, et al. Persistent activation of Rac1 in squamous carcinomas of the head and neck: evidence for an EGFR/Vav2 signaling axis involved in cell invasion. Carcinogenesis. 2007;28:1145–1152. doi: 10.1093/carcin/bgm008. [DOI] [PubMed] [Google Scholar]
- Rapley J, Tybulewicz VL, Rittinger K. Crucial structural role for the PH and C1 domains of the Vav1 exchange factor. EMBO Rep. 2008;9:655–661. doi: 10.1038/embor.2008.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savkur RS, Burris TP. The coactivator LXXLL nuclear receptor recognition motif. J Pept Res. 2004;63:207–212. doi: 10.1111/j.1399-3011.2004.00126.x. [DOI] [PubMed] [Google Scholar]
- Schmidt LJ, Regan KM, Anderson SK, Sun Z, Ballman KV, Tindall DJ. Effects of the 5 alpha-reductase inhibitor dutasteride on gene expression in prostate cancer xenografts. Prostate. 2009;69:1730–1743. doi: 10.1002/pros.21022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen HC, Buchanan G, Butler LM, Prescott J, Henderson M, Tilley WD, et al. GRIP1 mediates the interaction between the amino- and carboxyl-termini of the androgen receptor. Biological chemistry. 2005;386:69–74. doi: 10.1515/BC.2005.009. [DOI] [PubMed] [Google Scholar]
- Tindall DJ, Scardino PJ. State of research for prostate cancer: Excerpt from the report of the Prostate Cancer Progress Review Group. Urology. 2001;57:28–30. doi: 10.1016/s0090-4295(00)00932-8. [DOI] [PubMed] [Google Scholar]
- Trenkle T, McClelland M, Adlkofer K, Welsh J. Major transcript variants of VAV3, a new member of the VAV family of guanine nucleotide exchange factors. Gene. 2000;245:139–149. doi: 10.1016/s0378-1119(00)00026-3. [DOI] [PubMed] [Google Scholar]
- Varnai P, Bondeva T, Tamas P, Toth B, Buday L, Hunyady L, et al. Selective cellular effects of overexpressed pleckstrin-homology domains that recognize PtdIns(3,4,5)P3 suggest their interaction with protein binding partners. J Cell Sci. 2005;118:4879–4888. doi: 10.1242/jcs.02606. [DOI] [PubMed] [Google Scholar]
- Wang Q, Carroll JS, Brown M. Spatial and temporal recruitment of androgen receptor and its coactivators involves chromosomal looping and polymerase tracking. Molecular cell. 2005;19:631–642. doi: 10.1016/j.molcel.2005.07.018. [DOI] [PubMed] [Google Scholar]
- Xi S, Tie Y, Lu K, Zhang M, Yin X, Chen J, et al. N-terminal PH domain and C-terminal auto-inhibitory region of CKIP-1 coordinate to determine its nucleus-plasma membrane shuttling. FEBS Lett. 2010;584:1223–1230. doi: 10.1016/j.febslet.2010.02.036. [DOI] [PubMed] [Google Scholar]
- Zeng L, Sachdev P, Yan L, Chan JL, Trenkle T, McClelland M, et al. Vav3 mediates receptor protein tyrosine kinase signaling, regulates GTPase activity, modulates cell morphology, and induces cell transformation. Mol Cell Biol. 2000;20:9212–9224. doi: 10.1128/mcb.20.24.9212-9224.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A. PC3 cells transfected with AR, PSA-luc, beta galactosidase (B-gal) and either Vav3, Vav3.1 and Vav3, or empty vector (Ev) were treated with either vehicle or R1881 (1 nM) for 48 h. Cell lysates were then assayed for luciferase activity. Data from a representative experiment (of three independent experiments) performed in triplicate are plotted as relative luciferase units (mean ± SD) normalized to beta-galactosidase activity. B. ALVA 31 cells stably expressing the AR cDNA (ALVA-AR) or neo vector alone (ALVA-NEO) were transfected with a dominant negative Vav3 construct (Vav3.1) or empty vector. Thirty hours later, media were replaced with 3H thymidine-containing media and cells were cultured for 18 h. Acid-soluble radioactivity was removed by TCA and ethanol washes and incorporated radioactivity was determined by liquid scintillation counting.
VCaP cells stably expressing Vav3 or GFP (control) were depleted of androgen and then replated in media containing either 10% FBS (+) or 10% CSS-FBS (−) and cultured for the indicated times. Lysates were probed with antibody against cleaved PARP or actin.