


Abstract
Next-generation sequencing has great potential for application in bacterial transcriptomics. However, unlike eukaryotes, bacteria have no clear mechanism to select mRNAs over rRNAs; therefore, rRNA removal is a critical step in sequencing-based transcriptomics. Duplex-specific nuclease (DSN) is an enzyme that, at high temperatures, degrades duplex DNA in preference to single-stranded DNA. DSN treatment has been successfully used to normalize the relative transcript abundance in mRNA-enriched cDNA libraries from eukaryotic organisms. In this study, we demonstrate the utility of this method to remove rRNA from prokaryotic total RNA. We evaluated the efficacy of DSN to remove rRNA by comparing it with the conventional subtractive hybridization (Hyb) method. Illumina deep sequencing was performed to obtain transcriptomes from Escherichia coli grown under four growth conditions. The results clearly showed that our DSN treatment was more efficient at removing rRNA than the Hyb method was, while preserving the original relative abundance of mRNA species in bacterial cells. Therefore, we propose that, for bacterial mRNA-seq experiments, DSN treatment should be preferred to Hyb-based methods.
INTRODUCTION
RNA-seq is a novel method for elucidating the transcriptome of cells. This method uses high-throughput next-generation sequencing technology and has revolutionized the way in which gene expression profiles are examined (1). The RNA molecules present in prokaryotic cells are mostly rRNA species, whereas mRNA constitutes only 1–5% of total RNA. Therefore, efficient enrichment of mRNA is a critical step for successful mRNA-seq experiments. Because mRNA molecules extracted from bacterial cells mostly lack poly-A tails, the methods developed so far have focused on removing non-mRNAs rather than selecting mRNAs. Several techniques (2–6) have been applied to deplete rRNA from the total bacterial RNA population, but the efficiency and robustness of these methods have not been objectively compared. Recently, He et al. (7) compared the two most popular rRNA removal methods, namely subtractive hybridization (Hyb) and exonuclease digestion, using Illumina-based RNA-seq of synthetic microbial metatranscriptomes. Their results suggested that the Hyb method introduced less bias in the relative proportion of the mRNA population compared to exonuclease digestion. However, no study has been conducted to evaluate these rRNA removal methods in mRNA-seq based on pure cultured strains.
Zhulidov et al. (8) introduced a simple cDNA normalization method based on duplex-specific nuclease (DSN) aimed at enhancing the detection of rare transcripts in eukaryotic cDNA libraries by decreasing the prevalence of highly abundant transcripts. This DSN method includes the denaturation of cDNA, its subsequent reassociation and enzymatic degradation of the double-stranded (ds) DNA fraction using DSN isolated from the Kamchatka crab (9). Because the Hyb rate for each transcript is proportional to the square of its concentration (10), abundant transcripts form ds DNA more effectively during the reassociation step and are subjected to DSN-mediated degradation. DSN has a strong preference for cleaving dsDNA, and there is no significant cleavage of single-stranded (ss) DNA under the directed working conditions of the enzyme (9).
The DSN method has been successfully applied to normalize transcripts in cDNA libraries from various eukaryotes (11). The treatment was usually performed on cDNA libraries enriched with mRNAs using either mRNA-specific poly(A) tail selection in the RNA state (12) or an oligo(dT) primer approach for reverse transcription from total RNA (13,14). However, the application of the DSN method for the purpose of rRNA removal from total RNA has not been reported in prokaryotic or eukaryotic transcriptome studies. Here, the use of DSN normalization as an rRNA removal method was evaluated and compared to the conventional subtractive Hyb method. Illumina deep sequencing of the transcriptomes of Escherichia coli grown under four conditions demonstrated that the DSN method is suitable for rRNA removal while preserving the original relative abundance of each mRNA transcript.
MATERIALS AND METHODS
Bacterial cultures
Escherichia coli K-12/MG1655 was grown in LB medium (Difco) at 37°C with continuous shaking. The freshly grown cells were inoculated into two culture flasks containing LB medium and incubated under anaerobic or aerobic conditions. At exponential phase (OD0.6), the aerobic culture was evenly distributed into three sterile culture flasks under aseptic conditions. Three aliquots were then subjected to three different conditions as follows. The first aliquot was subjected to instant RNA extraction at exponential phase, and the second was extracted at stationary phase (OD2.0). The third aliquot was subjected to heat shock stress by incubating the culture at 42°C for 30 min. The anaerobic cells were also grown to exponential phase (OD0.6), and the cells were subjected to instant RNA extraction.
RNA extraction, rRNA removal and sequencing library construction
Total RNA was independently extracted from the four culture conditions (aerobic exponential, aerobic stationary, aerobic heat shock and anaerobic exponential) using the hot phenol method with additional purification using an RNeasy Mini kit (Qiagen) following the manufacturer's instructions. The quantity and quality of the RNA were evaluated before and after the rRNA removal processes using RNA electropherograms (Agilent 2100 Bioanalyzer) and the RNA integrity number (RIN) (15). Total RNAs from cultures treated under the four conditions were aliquoted into three portions. The first aliquot of total RNA (200 ng) was used to generate a sequencing library using an mRNA-seq library prep kit (Illumina) without any other treatment. The RNA was directly subjected to fragmentation without the mRNA purification step (poly-A selection). The second aliquot was subjected to a subtractive Hyb-based rRNA removal process using the MICROBExpress Bacterial mRNA Enrichment Kit (Ambion). The resultant RNA (100 ng) was used for sequencing library construction using the mRNA-seq library prep kit, omitting the poly-A selection step. The last aliquot (200 ng) was used to generate a sequencing library using an mRNA-seq library prep kit with some modifications. The RNA was directly subjected to fragmentation without the mRNA purification step (poly-A selection). The first- and second-strand cDNA was synthesized from the fragmented RNA using random hexamer primers. End repair, A-tailing, adaptor ligation, cDNA template purification and enrichment of the purified cDNA templates using PCR were then performed. The resulting sample libraries were subjected to DSN treatment using the Trimmer-Direct cDNA Normalization Kit (Evrogen) as follows. The sample library mixed with Hyb buffer was denatured at 98°C for 2 min and incubated at 68°C for 5 h. DSN buffer and 2 μl of the DSN enzyme were added to the mixture and incubated at 68°C for 25 min followed by the addition of stop solution. After purification of the DSN-treated library using SPRI beads, the library was enriched by PCR using PE1.0 and PE2.0 primers. The library construction was completed by final purification of the PCR product using SPRI beads. Because the commercial Hyb and DSN kits already employed highly optimized reagents and conditions for E. coli RNA, we adopted the procedures suggested by the respective manufacturers. The summary of the experimental procedures of the control, DSN and Hyb methods is given in Supplementary Figure S1.
Sequencing and alignment of the transcriptome
RNA deep sequencing was performed using two runs of the Illumina Genome Analyzer IIx to generate single-ended 36-bp reads. The genome sequence and functional annotation information of this strain were obtained from the NCBI database (accession number NC_000913.2). Quality-filtered reads were aligned to the reference genome sequence using CLC Genomics Workbench 4.0 (CLC bio). Mapping was based on the minimal length of 32 bp with an allowance of up to two mismatches. The relative transcript abundance was measured in reads per kilobase of exon per million mapped sequence reads (16) (RPKM) using the following formula:
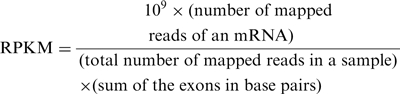 |
Determination of detection threshold
The library-size normalization was performed by dividing the raw read count of each mRNA by the number of total mapped reads in each Illumina lane and then multiplying by the average total mapped read numbers of four control samples. The detection threshold was determined by calculating the number of reads of an mRNA that was significantly different from zero read considering the experimental errors. For this calculation, duplicate control RNA data were analyzed under the assumption that controls 1 and 2 should have an identical true expression level, but measurement errors may cause different observed expression levels. The sample standard deviation was used to calculate the confidence interval that distinguished the observed expression level of an mRNA from an undetected mRNA. The detailed calculations are as follows.
It was assumed that xi1(xi2) was an observed expression level for control 1 (control 2) and that μi was an unknown true expression level for both xi1 and xi2. If xi1 was 0, it was removed from the analysis, and it was assumed that xi1 was larger than 0. For the measurement error, the following log-normal distributions for xi1 and xi2 were assumed, respectively:
 |
where xi1 and xi2 are independent. The variance in xi1 and xi2 depends on μi because the proportion of the measurement error got smaller for higher μi, and results in Supplementary Figure S3a also confirmed that the inverse of their variance was proportional to the true expression level. Because the mean parameters for xi1 and xi2 were assumed to be equal, the following could be obtained:
As a result, the confidence interval for xi1 given μi could be empirically calculated using 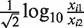 . To minimize the dependence of the variance on μi, only the observed intensities for which
. To minimize the dependence of the variance on μi, only the observed intensities for which 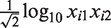 was <1 were considered. If the variance was denoted as
was <1 were considered. If the variance was denoted as 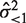 , the normality of the observed expression level resulted in the confidence interval
, the normality of the observed expression level resulted in the confidence interval 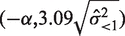 at a 0.001 significance level.
at a 0.001 significance level.
Statistical analyses
Statistical analyses were performed using the RPKM values of mRNAs detected from all experimental conditions after detection threshold filtering using library size-normalized data. The statistical significance (P-value) of differences in rRNA removal efficiency was obtained using the likelihood ratio test based on a generalized linear model based on Poisson regression. The robustness of mRNA relative abundance conservation was analyzed using general linear regression and Lowess nonlinear regression models. Hierarchical clustering was performed using the unweighted pair group method with arithmetic mean (UPGMA) clustering algorithm using Pearson's product-moment correlation coefficient. All statistical analyses were performed using the R package, version 2.11.0 (www.r-project.org).
RESULTS
Illumina deep sequencing
Four RNA samples prepared from E. coli K12/MG1655 cells grown under four conditions (aerobic exponential, aerobic stationary, aerobic heat shock and anaerobic exponential) were aliquoted and subjected to three different protocols. The first aliquot (control) was processed without any rRNA removal treatment; the second aliquot was treated using DSN normalization (DSN; Trimmer-Direct cDNA Normalization Kit, Evrogen); and the third aliquot was treated using subtractive Hyb (MICROBExpress Bacterial mRNA Enrichment kit, Ambion). The RNA quality measured using RNA electropherograms showed that the extracted total RNA was of good quality, with an average RNA integrity number (RIN) of 9.2 (Supplementary Figure S2). The disappearance of rRNA peaks after the rRNA removal process using Hyb was also visualized in the electropherograms.
DNA sequencing was performed using two eight-lane flow cells of the Illumina Genome Analyzer IIx to generate single-ended 36-bp reads. The first run contained eight lanes of untreated control samples, which consisted of duplicate lanes of each condition to allow an increased number of mRNA reads for samples. The second run contained four lanes of DSN-treated samples and four Hyb-treated samples. The number of quality-filtered reads for each sample ranged from 32 to 39 million, and >99.0% of the reads (average 99.4%) were mapped to the reference genome sequence, indicating good sequencing quality and negligible contamination (Table 1). The Illumina reads of duplicate control sample lanes were combined for further analyses.
Table 1.
Sequencing and alignment statistics
| Description | Run ID | Total reads | Mapped reads | mRNA | rRNA | tRNA | misc_RNA | intergenic | Unmapped reads |
|---|---|---|---|---|---|---|---|---|---|
| Control | |||||||||
| Exponential | Ex-C (Ex-C1, Ex-C2) | 73 501 029 | 73 146 186 | 2 752 700 | 69 216 309 | 10 917 | 116 628 | 1 049 632 | 354 843 |
| Stationary | St-C (St-C1, St-C2) | 65 848 252 | 65 554 024 | 2 482 828 | 62 197 077 | 1389 | 195 905 | 676 825 | 294 228 |
| Heat shock | He-C (He-C1, He-C2) | 76 958 662 | 76 537 899 | 2 904 834 | 71 652 945 | 6490 | 183 421 | 1 790 209 | 420 763 |
| Anaerobic | An-C (An-C1, An-C2) | 73 860 611 | 73 469 539 | 2 120 611 | 69 437 582 | 1435 | 274 246 | 1 635 665 | 391 072 |
| DSN | |||||||||
| Exponential | Ex-D | 32 461 464 | 32 346 699 | 18 516 330 | 9 593 620 | 55 732 | 425 762 | 3 755 255 | 114 765 |
| Stationary | St-D | 34 429 138 | 34 274 833 | 24 816 321 | 4 566 206 | 11 204 | 747 676 | 4 133 426 | 154 305 |
| Heat shock | He-D | 33 583 557 | 33 470 900 | 21 926 911 | 7 125 455 | 38 092 | 604 116 | 3 776 326 | 112 657 |
| Anaerobic | An-D | 32 309 941 | 32 179 277 | 16 188 510 | 12 459 843 | 7358 | 727 996 | 2 795 570 | 130 664 |
| Hyb | |||||||||
| Exponential | Ex-H | 39 013 862 | 38 765 857 | 9 461 600 | 25 217 660 | 29 688 | 253 429 | 3 803 480 | 248 005 |
| Stationary | St-H | 35 007 172 | 34 706 767 | 7 897 907 | 22 624 587 | 5341 | 406 012 | 3 772 920 | 300 405 |
| Heat shock | He-H | 39 419 310 | 39 030 251 | 12 374 014 | 23 788 881 | 32 205 | 620 168 | 2 214 983 | 389 059 |
| Anaerobic | An-H | 37 470 516 | 37 106 501 | 5 635 773 | 29 788 740 | 7606 | 519 716 | 1 154 666 | 364 015 |
The statistics of untreated control samples were obtained from data combined from two lanes.
The sequence coverage is defined as the proportion of mRNAs that have one or more mapped reads with respect to all annotated genes (4493 genes in the reference E. coli genome). The average coverage of the control samples was 98.6%, which is not significantly different from those treated with DSN (99.1%) or Hyb (99.2%), suggesting that the sequencing depths in this study were sufficient (Supplementary Table S1). The resultant expression profile of E. coli is shown in Supplementary Table S2, and the top 10 highly expressed genes in each growth and rRNA treatment conditions are summarized in Supplementary Table S3.
The composition of major RNA types, namely rRNA, tRNA, mRNA and other RNA (miscRNA), was similar to that of prokaryotes (7).
rRNA removal efficiency
Illumina deep sequencing of the total RNA in E. coli revealed that rRNA was indeed the major component in all four growth conditions, ranging from 93.1% to 94.5%, whereas a substantially smaller proportion (2.9–3.8%) was identified as mRNA (Table 1). After the rRNA removal treatments, the proportion of rRNA in the cDNA libraries was reduced to 13.3–38.6% using DSN and 60.4–79.5% using Hyb (Figure 1). The mapped mRNAs increased 17.3-fold using DSN and 6.5-fold using Hyb compared to the untreated controls. The difference in the efficiency of rRNA removal treatments was statistically significant (P = 0.00007). The rRNA removal efficiency of DSN was 2.5 times higher than Hyb. However, the ratio of unmapped reads was higher in samples that underwent Hyb (0.86%) than those treated with DSN (0.39%) or than the control (0.50%). The efficiency of rRNA removal in DSN method increased in the order of rRNA size (23 S > 16 S > 5 S), but the ratio of 5 S:16 S:23 S was pretty conserved in DSN (0.1:35:65) compared to control (0.02:32:68), while the ratio was significantly shifted in Hyb (0.4:9:91).
Figure 1.

rRNA removal efficiency of DSN and Hyb methods. The proportion of Illumina reads assigned to mRNA or rRNA indicates that DSN is more effective at rRNA removal than Hyb.
RNA-seq detection threshold
To improve the accuracy of the RNA-seq analysis, the mRNA detection threshold was determined to exclude Illumina reads in which the expression level was severely affected by measurement errors. It was assumed that the biological duplicates, namely controls 1 and 2 under the same conditions, had unknown identical means and that the measurement errors caused the differences in the Illumina read counts. To determine the detection threshold value, read count 1 was used as the one-tailed upper limit of minimum read at the 0.001 significance level. Because the variance of the measurement decreased with the level of the unknown true expression level (Supplementary Figure S3a), the area in which the variance showed normal distribution was determined first (Supplementary Figure S3b) to minimize the dependence of the variance on expression values. For the selected area, the variance was estimated, and the detection threshold of an mRNA was determined to be six Illumina reads in our library size-normalized data at the 0.001 significance level. Consequently, 94.58% of mRNAs that had more than six mapped reads in all samples that passed the filtering. The 234 failed ORFs (5.42%) were omitted from further statistical analyses. By removing the insignificant low read-count genes, the linearity of regression between the two duplicate controls was improved (Supplementary Figure S3c and d).
Robustness of mRNA relative abundance
The correlation of mRNA expression patterns between control samples and the two rRNA removal treatments is summarized in Figure 2a and Supplementary Figure S4. The average slope of the linear regression line between the controls and DSN-treated samples was 0.99 (r = 0.99, Pearson's correlation coefficient), whereas the corresponding value between the controls and Hyb-treated samples was significantly lower (0.75 on average, r = 0.93). Similarly, the Lowess fit of DSN-treated samples converged to linear regression, whereas Hyb-treated samples showed a departure from linearity, with a skewed shape in the area of the low to middle ‘expressers’. Hierarchical clustering analysis of all samples using the Pearson's product-moment correlation coefficient indicated that the mRNA expression profiles of the untreated controls were more similar to that of DSN-treated samples than that of Hyb-treated samples (Figure 2b).
Figure 2.


Comparison of robustness of mRNA relative abundance using DSN and Hyb. (a) Correlation of relative gene expression between the control and rRNA removal treatments. The exponential phase libraries (Ex) are shown as an example. Left, control (Ex-C) versus DSN (Ex-D); right, control (Ex-C) versus Hyb (Ex-H). The points in the plot indicate the RPKM value of each individual mRNA transcript. Red indicates the linear regression line, and blue indicates non-linear regression (Lowess) fit. Green is a straight line with a slope of 1. (b) Hierarchical clustering of all samples tested in this study using UPGMA based on Pearson's correlation. The regression plot and dendrogram represent the superior conservation of mRNA relative abundance in DSN treatment compared to Hyb treatment.
Robustness of mRNA profiles
The fold-change in expression levels of each mRNA was calculated by dividing the RPKM of stationary (St), heat shock (He) or anaerobic (An) samples by the RPKM of the exponential (Ex) sample and these fold-change values were represented as St/Ex, He/Ex or An/Ex, respectively. The fold difference between the control and the two rRNA removal treatments was calculated by subtracting the log-scale fold value of the corresponding control sample from that of DSN- or Hyb-treated sample. The regression analyses (Supplementary Figure S5) and boxplots (Figure 3) indicated a smaller fold difference in samples treated with DSN than Hyb. The slopes of the linear regression lines of DSN- and Hyb-treated samples were 0.96 (r = 0.98) and 0.74 (r = 0.90), respectively. The Lowess fit analysis of Hyb-treated samples also showed a departure from linearity, whereas the fit of DSN-treated samples converged to linear regression. The boxplots (Figure 3) demonstrated no fold differences in DSN-treated samples, with average values close to 0, whereas the corresponding values of Hyb-treated samples ranged between 0.03 and 0.10, implying significant expression level differences caused by the Hyb treatment.
Figure 3.

Boxplots showing fold difference profiles. The fold-change of each mRNA was calculated by dividing the RPKM of St, He or An samples by the RPKM of the Ex sample and is represented as St/Ex, He/Ex or An/Ex, respectively. The fold difference profile caused by rRNA removal treatment was calculated by subtracting the log-scale fold-change of the corresponding control sample from the DSN- or Hyb-treated samples. The average and standard deviation are shown at the top of the plots. No fold difference was observed using DSN, with the average values close to 0, whereas the corresponding values of Hyb ranged between 0.10 and 0.03, implying a significant expression level difference caused by the Hyb treatment.
The transcripts for which relative abundance was severely biased using Hyb treatment were identified using a threshold of 0.5 at Δlog (fold value) because the majority of transcripts of DSN-treated samples were within the threshold (Figure 3). A total of 127 ORFs were identified from Hyb-treated samples (Supplementary Table S4), and their functional categories are provided in Supplementary Figure S6. No correlation between the functional categories of the transcripts and bias was detected. No selective loss or gain of mRNAs depending on GC content was either observed in this data set.
DISCUSSION
Given the prevalence of rRNA in the total transcriptome of prokaryotes, it is essential to enrich mRNA prior to sequencing-based genome-wide gene expression studies. However, it is equally important to preserve the overall gene expression patterns while enriching the mRNA. In this study, we evaluated two commercially available methods for performing such a task using E. coli, the most widely used model bacterium.
In our study, both the Hyb and DSN methods removed a substantial proportion of rRNA species, with DSN being 2.5-fold more efficient than Hyb. Our results suggest that researchers can reduce the sequencing cost of RNA-seq by 2.5-fold by using DSN method, compared to the commercial Hyb kit. The rRNA removal efficiency of Hyb method obtained in this study was comparable with the previous studies. It has been known that the rRNA removal efficiency of Hyb method varies widely for community RNA samples (17,18), as well as for single-species analyses (19). For example, the amount of rRNA remained after the commercial Hyb treatment ranged from 43.6% to 98.6% depending on microbial species (7). This is because the Hyb method is based on Hyb between rRNA and oligonucleotides that target conserved regions of bacterial rRNA; therefore, the removal efficiency is largely dependent on the selected oligonucleotide sequences. In contrast, the DSN method does not depend on particular rRNA sequences; therefore, in theory, it can be used for any organism, including archaea. However, the variation of rRNA removal efficiency between samples was also observed in DSN treatment, but the reason is unclear at this stage.
The regression analyses demonstrated the lower robustness of the Hyb method compared to DSN, especially in the low- to middle-expression range (approximate RPKM < 300). Many transcripts in this range seemed to be expressed at higher levels than those in the untreated controls. The depletion of high expressers may result in the relative overrepresentation of lower expressers. A number of severely biased transcripts were found in samples that underwent Hyb treatment, although no correlation between the functional categories of the transcripts and bias was found. Undesired binding between mRNAs and the capture oligonucleotides may cause this phenomenon. Indeed, in the case of ribosomal proteins (rps, rpl and rpm) that have obvious homology with rRNAs, the average RPKM value was reduced to 76% of the untreated control after Hyb treatment, whereas it was conserved at 105% in DSN-treated samples. An increased proportion of unmapped reads after Hyb treatment compared to the control or DSN treatment was also noted, although the reason for this is unclear.
Because of the function of the DSN enzyme, which preferentially degrades highly abundant transcripts over transcripts of low abundance, we expected that abundant transcripts could be affected by DSN treatment. Because the absolute levels of gene expression in bacteria vary over as much as six orders of magnitude (20–22), the concentration of highly abundant mRNA classes could be affected by DSN degradation. However, as far as we surveyed, even the most abundant mRNA typically comprise 1–7% of mRNAs (6,16,23), and as much as 10% in severe cases (24). Thus, the amount of rRNA in a cell significantly outnumbers even the most abundant mRNA transcripts. In fact, the 10 most expressed genes in each condition showed a little bit declined expression level in DSN (7.9% on average) compared to untreated control as shown in Supplementary Table S3, while the amount of decline was much more severe in Hyb (20.3%). Moreover, the log scale regression analysis in this study proved that the amount of decline observed in the abundant mRNAs did not hamper the overall robustness.
The typical ratio of rRNA:non-rRNA revealed by RNA-seq is 95–99% in a wide taxonomic range of pure cultured bacteria and archaea (7). Thus, our DSN method is, at least, applicable to a broad range of prokaryotes in laboratory culturing condition. In addition, it has been known that the ratio of rRNA compared to mRNA is higher in resting cells than actively growing cells (25,26). Thus, we think that the overwhelming abundance of rRNA compared to non-rRNA would be the case for even slowly growing cells or metabolically inactive cells, though experimental evidence will be required for DSN applicability for these conditions in future. Because the rRNA ratios reported in a metatranscriptomic study are also as high as 74–97% (6), it is fair to say that DSN method would be applicable to mixed population samples.
Several rRNA removal methods are known, but all of these methods have some limitations. The methods based on Hyb between rRNA and DNA targeting rRNA may generate bias depending on the taxonomy of bacteria. These methods include the RNase H digestion method based on reverse transcription with rRNA-specific primers (4) and the Hyb method evaluated in this study. Though recently developed subtractive Hyb method has solved the bias by generating customized oligonucleotide probes targeting sample specific rRNAs (6), it is not still free from non-specific binding of rRNA probe to mRNAs. The size selection method using gel electrophoresis (5) has an apparent limitation because some mRNAs can co-migrate with rRNA and are subsequently omitted. The poly(A) tail addition methods (3,27), which use preferential poly(A) adenylation of mRNA in crude RNA, also have the possible limitation of uneven poly(A) adenylation efficiency among mRNAs, which may generate expression-level bias. Because poly(A) tail addition method have not been tested in pure cultures using RNA-seq, further evaluation is required. In the case of 5′-phosphate-dependent exonuclease digestion of rRNA with 5′ monophosphate, it has already been demonstrated that this method compromises the relative proportion of the mRNA population more severely than the Hyb method (7). This change in the relative proportion may occur because exonuclease can also eliminate 5′-monophosphorylated mRNA species, which are produced during mRNA processing by endoribonucleases (28,29) and RppH (30).
The only drawback of DSN over the Hyb method is that it requires more experimental steps and, therefore, a longer time to prepare cDNA libraries for deep sequencing. However, because the rRNA removal step is performed after cDNA library construction, the possibility of unintentional RNA degradation is greatly reduced compared to other methods involving pre-treatments. In addition, the amount of total input RNA used for deep sequencing library construction was much less in the DSN method (200 ng) than in the conventional mRNA-seq accompanying poly(A) selection (1–10 μg). Considering the loss of RNAs during the mRNA enrichment step using other pre-treatments, the amount of total RNA to be extracted is even smaller using the DSN method.
Another advantage of the DSN method is that it works well even with partially degraded total RNA, whereas the Hyb method requires intact rRNA for the successful binding of the oligonucleotides to targeted conserved sites. Indeed, a well-supported positive correlation between RIN values, which represent the degree of rRNA integrity, and rRNA removal efficiency (r = 0.88) was observed in our Hyb experiments, as well as in a previous report (7); however, this type of correlation was not observed in our DNS treatments (r = 0.27). Although the number of samples (four total RNAs) and the range of RNA degradation (RIN value 8.8–10.0) were not strong enough for statistical analyses, the lower importance of the RNA fragmentation status for effective DNS treatment compared to the Hyb method was clear.
Because the conventional RNA-seq of eukaryotic organisms relies on poly(A)+ capture in the first step of sequencing library preparation, the outcome generally contains only information on poly(A)-tailed RNAs. If the DSN method used in this study is applied to eukaryotic RNA, it probably will provide information on both non-poly(A) RNA sequences and poly(A) RNA. Further study of the feasibility of this method for eukaryotic RNA-seq without poly(A) selection is therefore needed.
In this study, we performed deep sequencing of total RNA of E. coli. Although E. coli is a well studied, widely used model organism, its precise genome-wide expression profile has not been documented previously. To our knowledge, this is the first report on the intact genome-wide RNA profile of E. coli without any treatment and selection. This information clearly demonstrates the overall abundance of different RNA species in a bacterial cell.
DSN-based normalization showed a higher efficiency of rRNA removal than the Hyb method, while preserving the relative abundance of mRNA. The thermodynamic principle of this technique allows its application to any kind of eukaryotic or prokaryotic organism. Therefore, DSN-based mRNA enrichment can be readily used in bacterial mRNA-seq experiments.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Priority Research Centers Program (grant number 2010-0094020); National Research Foundation grant (2011-0016498) through the National Research Foundation of Korea funded by the Ministry of Education, Science and Technology, Republic of Korea. Funding for open access charge: National Science Foundation.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Drs Kangseok Lee and You-Hee Cho for their helpful comments and suggestions on this manuscript.
REFERENCES
- 1.Wang Z, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009;10:57–63. doi: 10.1038/nrg2484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pang X, Zhou D, Song Y, Pei D, Wang J, Guo Z, Yang R. Bacterial mRNA purification by magnetic capture-hybridization method. Microbiol. Immunol. 2004;48:91–96. doi: 10.1111/j.1348-0421.2004.tb03493.x. [DOI] [PubMed] [Google Scholar]
- 3.Frias-Lopez J, Shi Y, Tyson GW, Coleman ML, Schuster SC, Chisholm SW, Delong EF. Microbial community gene expression in ocean surface waters. Proc. Natl. Acad. Sci. USA. 2008;105:3805–3810. doi: 10.1073/pnas.0708897105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dunman PM, Murphy E, Haney S, Palacios D, Tucker-Kellogg G, Wu S, Brown EL, Zagursky RJ, Shlaes D, Projan SJ. Transcription profiling-based identification of Staphylococcus aureus genes regulated by the agr and/or sarA loci. J. Bacteriol. 2001;183:7341–7353. doi: 10.1128/JB.183.24.7341-7353.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.McGrath KC, Thomas-Hall SR, Cheng CT, Leo L, Alexa A, Schmidt S, Schenk PM. Isolation and analysis of mRNA from environmental microbial communities. J. Microbiol. Methods. 2008;75:172–176. doi: 10.1016/j.mimet.2008.05.019. [DOI] [PubMed] [Google Scholar]
- 6.Stewart FJ, Ottesen EA, DeLong EF. Development and quantitative analyses of a universal rRNA-subtraction protocol for microbial metatranscriptomics. ISME J. 2010;4:896–907. doi: 10.1038/ismej.2010.18. [DOI] [PubMed] [Google Scholar]
- 7.He S, Wurtzel O, Singh K, Froula JL, Yilmaz S, Tringe SG, Wang Z, Chen F, Lindquist EA, Sorek R, et al. Validation of two ribosomal RNA removal methods for microbial metatranscriptomics. Nat. Methods. 2010;7:807–812. doi: 10.1038/nmeth.1507. [DOI] [PubMed] [Google Scholar]
- 8.Zhulidov PA, Bogdanova EA, Shcheglov AS, Vagner LL, Khaspekov GL, Kozhemyako VB, Matz MV, Meleshkevitch E, Moroz LL, Lukyanov SA, et al. Simple cDNA normalization using kamchatka crab duplex-specific nuclease. Nucleic Acids Res. 2004;32:e37. doi: 10.1093/nar/gnh031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Shagin DA, Rebrikov DV, Kozhemyako VB, Altshuler IM, Shcheglov AS, Zhulidov PA, Bogdanova EA, Staroverov DB, Rasskazov VA, Lukyanov S. A novel method for SNP detection using a new duplex-specific nuclease from crab hepatopancreas. Genome Res. 2002;12:1935–1942. doi: 10.1101/gr.547002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Young BD, Anderson M. In: Nucleic Acids Hybridisation, a Practical Approach. Hames BD, Higgins SJ, editors. Oxford, Washington DC: IRL Press; 1985. pp. 47–71. [Google Scholar]
- 11.Bogdanova EA, Shagin DA, Lukyanov SA. Normalization of full-length enriched cDNA. Mol. Biosyst. 2008;4:205–212. doi: 10.1039/b715110c. [DOI] [PubMed] [Google Scholar]
- 12.Simon A, Glockner G, Felder M, Melkonian M, Becker B. EST analysis of the scaly green flagellate Mesostigma viride (Streptophyta): implications for the evolution of green plants (Viridiplantae) BMC Plant Biol. 2006;6:2. doi: 10.1186/1471-2229-6-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhu YY, Machleder EM, Chenchik A, Li R, Siebert PD. Reverse transcriptase template switching: a SMART approach for full-length cDNA library construction. Biotechniques. 2001;30:892–897. doi: 10.2144/01304pf02. [DOI] [PubMed] [Google Scholar]
- 14.Danley PD, Mullen SP, Liu F, Nene V, Quackenbush J, Shaw KL. A cricket Gene Index: a genomic resource for studying neurobiology, speciation, and molecular evolution. BMC Genomics. 2007;8:109. doi: 10.1186/1471-2164-8-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schroeder A, Mueller O, Stocker S, Salowsky R, Leiber M, Gassmann M, Lightfoot S, Menzel W, Granzow M, Ragg T. The RIN: an RNA integrity number for assigning integrity values to RNA measurements. BMC Mol. Biol. 2006;7:3. doi: 10.1186/1471-2199-7-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mortazavi A, Williams BA, McCue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 17.Poretsky RS, Bano N, Buchan A, LeCleir G, Kleikemper J, Pickering M, Pate WM, Moran MA, Hollibaugh JT. Analysis of microbial gene transcripts in environmental samples. Appl. Environ. Microbiol. 2005;71:4121–4126. doi: 10.1128/AEM.71.7.4121-4126.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hewson I, Poretsky RS, Dyhrman ST, Zielinski B, White AE, Tripp HJ, Montoya JP, Zehr JP. Microbial community gene expression within colonies of the diazotroph, Trichodesmium, from the Southwest Pacific Ocean. ISME J. 2009;3:1286–1300. doi: 10.1038/ismej.2009.75. [DOI] [PubMed] [Google Scholar]
- 19.Yoder-Himes DR, Chain PS, Zhu Y, Wurtzel O, Rubin EM, Tiedje JM, Sorek R. Mapping the Burkholderia cenocepacia niche response via high-throughput sequencing. Proc. Natl. Acad. Sci. USA. 2009;106:3976–3981. doi: 10.1073/pnas.0813403106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Taniguchi Y, Choi PJ, Li GW, Chen H, Babu M, Hearn J, Emili A, Xie XS. Quantifying E. coli proteome and transcriptome with single-molecule sensitivity in single cells. Science. 2010;329:533–538. doi: 10.1126/science.1188308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wilhelm BT, Marguerat S, Watt S, Schubert F, Wood V, Goodhead I, Penkett CJ, Rogers J, Bahler J. Dynamic repertoire of a eukaryotic transcriptome surveyed at single-nucleotide resolution. Nature. 2008;453:1239–1243. doi: 10.1038/nature07002. [DOI] [PubMed] [Google Scholar]
- 22.Zaslaver A, Bren A, Ronen M, Itzkovitz S, Kikoin I, Shavit S, Liebermeister W, Surette MG, Alon U. A comprehensive library of fluorescent transcriptional reporters for Escherichia coli. Nat. Methods. 2006;3:623–628. doi: 10.1038/nmeth895. [DOI] [PubMed] [Google Scholar]
- 23.Gilbert JA, Field D, Huang Y, Edwards R, Li W, Gilna P, Joint I. Detection of large numbers of novel sequences in the metatranscriptomes of complex marine microbial communities. PLoS One. 2008;3:e3042. doi: 10.1371/journal.pone.0003042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kim JB, Porreca GJ, Song L, Greenway SC, Gorham JM, Church GM, Seidman CE, Seidman JG. Polony multiplex analysis of gene expression (PMAGE) in mouse hypertrophic cardiomyopathy. Science. 2007;316:1481–1484. doi: 10.1126/science.1137325. [DOI] [PubMed] [Google Scholar]
- 25.Johnson LF, Abelson HT, Green H, Penman S. Changes in RNA in relation to growth of fibroblast .1. Amounts of messenger-RNA, ribosomal-RNA, and tertiary RNA in resting and growing cells. Cell. 1974;1:95–100. [Google Scholar]
- 26.ter Kuile BH, Bonilla Y. Influence of growth conditions on RNA levels in relation to activity of core metabolic enzymes in the parasitic protists Trypanosoma brucei and Trichomonas vaginalis. Microbiology. 1999;145:755–765. doi: 10.1099/13500872-145-3-755. [DOI] [PubMed] [Google Scholar]
- 27.Wendisch VF, Zimmer DP, Khodursky A, Peter B, Cozzarelli N, Kustu S. Isolation of Escherichia coli mRNA and comparison of expression using mRNA and total RNA on DNA microarrays. Anal. Biochem. 2001;290:205–213. doi: 10.1006/abio.2000.4982. [DOI] [PubMed] [Google Scholar]
- 28.Matsunaga J, Dyer M, Simons EL, Simons RW. Expression and regulation of the rnc and pdxJ operons of Escherichia coli. Mol. Microbiol. 1996;22:977–989. doi: 10.1046/j.1365-2958.1996.01529.x. [DOI] [PubMed] [Google Scholar]
- 29.Sim SH, Yeom JH, Shin C, Song WS, Shin E, Kim HM, Cha CJ, Han SH, Ha NC, Kim SW, et al. Escherichia coli ribonuclease III activity is downregulated by osmotic stress: consequences for the degradation of bdm mRNA in biofilm formation. Mol. Microbiol. 2010;75:413–425. doi: 10.1111/j.1365-2958.2009.06986.x. [DOI] [PubMed] [Google Scholar]
- 30.Deana A, Celesnik H, Belasco JG. The bacterial enzyme RppH triggers messenger RNA degradation by 5′ pyrophosphate removal. Nature. 2008;451:355–358. doi: 10.1038/nature06475. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.