

Abstract
A concise total synthesis of (±)-γ-lycorane is described. The key step in the synthesis is the 6π-electrocyclic ring closure of a divinylpyrroline to give a tetrahydroindole, which is subsequently hydrogenated to give the all-cis indolizidine core.
Keywords: Amaryllidaceae alkaloids, Indolizidine, Electrocyclic ring closure, Divinylpyrroline
The Amaryllidaceae family of natural products1 encompasses a structurally diverse range of compounds with a host of biological activities, and has seen extensive investigation by synthetic chemists. In particular, the lycorine sub-class2 has for decades served as a test-ground for a range of synthetic methodologies. In fact, since its initial synthesis in 1966 one single member of this family, γ-lycorane (1, Figure 1), has been prepared in both racemic3 and enantioenriched4 form by over twenty different research groups using a multitude of synthetic approaches. These approaches can broadly be grouped according to the order in which the remaining two rings are constructed from an initial bicyclic compound. All of the approaches to date conclude with construction of the B ring, often by Pictet-Spengler or Bischler-Napierelski cyclization. This allows the existing syntheses to be classed into three groups, based on the order or ring construction.
Figure 1.
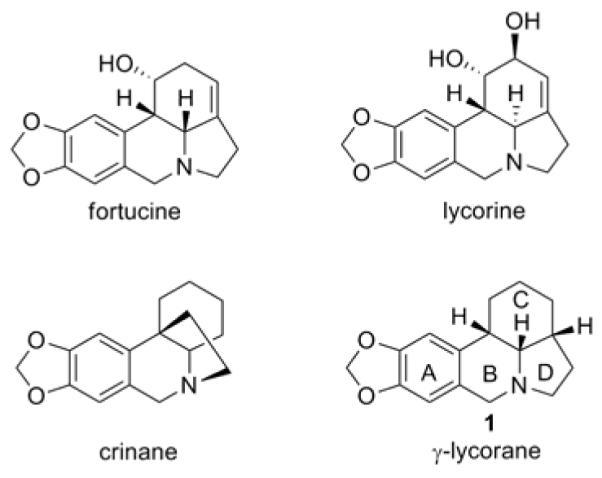
Selected Amaryllidaceae alkaloids
CD → CDA → CDAB approaches
Two examples of the CD → CDA → CDAB approach are shown in Scheme 1. In the first example,3c an intramolecular furan-acrylate cycloaddition is performed on amidofuran 2 to form the CD ring system 3. The A ring is subsequently introduced by acylation, to give aryl amide 4, which is subsequently transformed into the natural product (±)-1.
Scheme 1.

CD → CDA → CDAB and AC→ ACD→ ACDB approaches to γ-lycorane (1)
In the second example,4b Fujioka and Kita prepare the CD ring system by means of an intramolecular haloamination reaction. Condensation of aldehyde 5 with amine 6 gave an aminal that was directly treated with NBS to give bromoaminal 7. A subsequent four step sequence revealed a bromoindolizidinone, to which the piperonyl A ring was appended. A subsequent intramolecular Friedel-Crafts reaction and reduction of the resultant amide gave (−)-γ-lycorane (1).
AC→ ACD→ ACDB Approaches
Gong and coworkers concise synthesis4c began with preparation of an AC ring congener by means of an asymmetric nitroallylation reaction. Treatment of allylic acetate 9 with 3,4-(methylenedioxy)phenylboronic acid in the presence of a rhodium/BINAP catalyst effected allylation to give aryl cyclohexene 10. Conjugate addition of methyl acetate enolate followed by reduction of the nitro substituent with concomitant lactamization gave indolizidinone 12, which was subsequently converted to (+)-γ-lycorane (1).
In a second example of an AC→ACD→ACDB approach, Ojima and Chapsal have reported a recent synthesis,4a drawing heavily on earlier work by Mori,4f in which the A and C rings of γ-lycorane are coupled by means of an allylic alkylation reaction. Dibenzoate 13 was treated with the malonate half-amide 14 in the presence of a monodentate phosphoramidite palladium complex to give the desymmetrized benzoate 15. A subsequent palladium catalyzed tandem allylic amination-intramolecular Heck reaction gave the completed ring system 16.
AD→ ADC→ ADCB approaches
The final class of approaches, beginning with construction of an AD ring congener, has seen little use, with only two reported syntheses. In the first synthesis by the Vollhardt laboratory,3i aldehyde 17 is elaborated to acyl pyrroline 18 (Scheme 2). Treatment with CpCo(CO)2 and irradiation effected closure of the C and B rings to give cyclization product 19. Subsequent demetalation and reduction steps gave (±)-γ-lycorane (1).
Scheme 2.
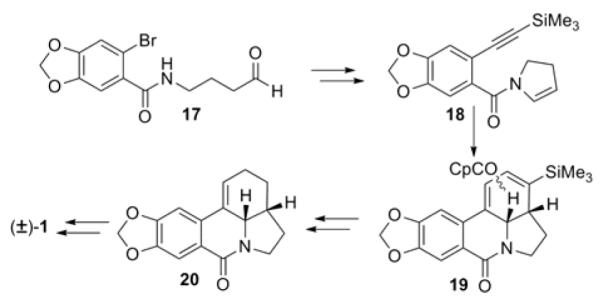
Vollhardt AD→ ADC→ ADCB approach to γ-lycorane (1)
The second example of this strategy, reported by Angle and Boyce,3h begins with Weinreb amide 21, available in six steps from TIPS-pyrrole. Addition of piperonyllithium to this amide and reduction of the resultant ketone gave benzylic alcohol 23 (Scheme 3). Upon treatment with Sn(OTf)2 the alcohol undergoes a cationic cyclization, forming the C ring. Subsequent hydrogenation and closure of the B ring gave (±)-γ-lycorane (1).
Scheme 3.
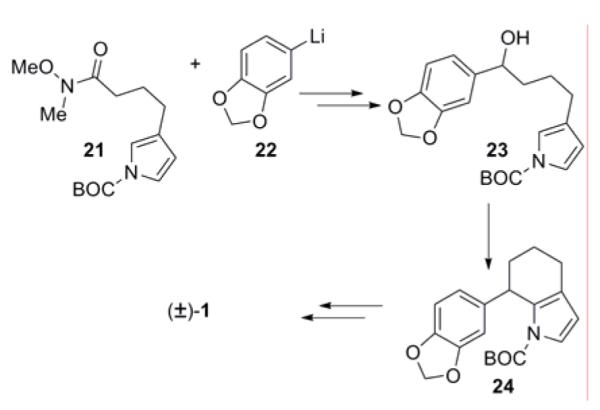
Angle AD→ ADC→ ADCB approach to γ-lycorane (1)
We felt that we could prepare γ-lycorane via the less common AD→ADC→ADBC approach using a modification of methodology recently developed in our laboratory for the synthesis of indoles. This methodology makes use of electrocyclic ring closures of trienecarbamates 25 for the synthesis of highly substituted indole ring systems (Scheme 4).5
Scheme 4.
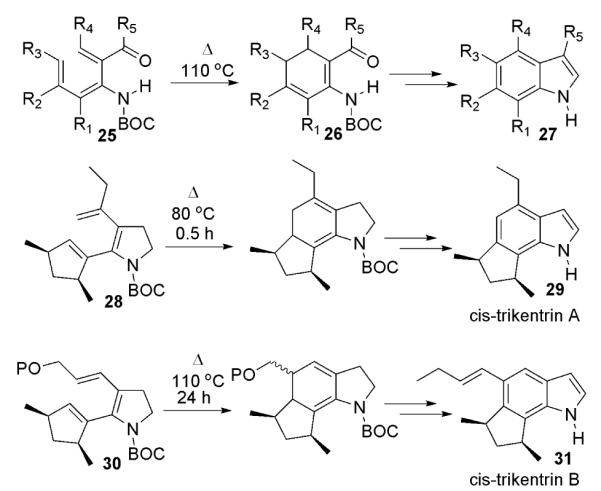
Indole syntheses via trienecarbamate electrocyclizations
The utility of this methodology for the synthesis of natural products has been demonstrated, first through synthesis of the welwistatin A ring system,6 and subsequently via syntheses of (±)-cis-trikentrin A (29) and B (31)7 from divinylpyrroline substrates 28 and 30. In each case, the dihydroaniline intermediates generated from the electrocyclic closures are oxidized to give, ultimately, indole ring systems.
While all of the examples we have reported to date involve aromatization of the dihydroaniline cyclization products, we felt this methodology could be extended to non-aromatic carbocyclic ring systems as well. In particular, γ-lycorane seemed to offer us an ideal initial substrate upon which we could test our methodology.
Our retrosynthetic analysis for the synthesis of γ-lycorane (1) is shown in Scheme 5. We envisaged that the natural product would be formed from indolizidine 32. This amine, in turn, could be derived by asymmetric hydrogenation of diene 33. This diene would result from the 6π-electrocyclic ring closure of divinylpyrroline 34, which would in turn be derived from stannane 35 via Stille coupling with the appropriate vinyl halide 36.
Scheme 5.
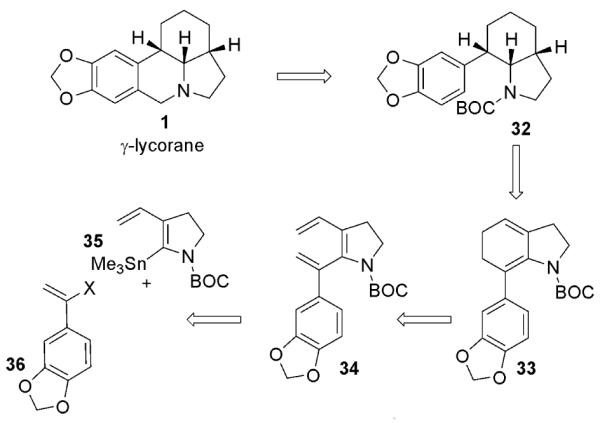
Retrosynthetic analysis for the synthesis of γ-lycorane (1)
To that end, stannane 35 was prepared by Peterson olefination of aldehyde 37 followed by stannylation at C(2) with n-BuLi/trimethyltinchloride5 (Scheme 6). Initial attempts at Stille coupling with the moderately unstable vinyl iodide 36a8 gave poor yields of the desired triene 34.
Scheme 6.
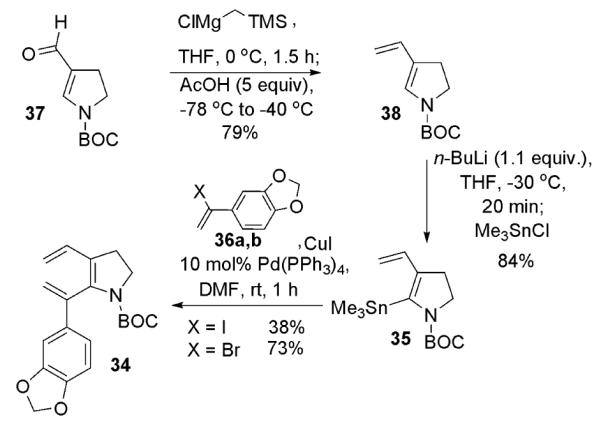
Preparation of trienecarbamate 34
The more stable bromide 36b9 gave the desired triene in acceptable yield however.
With our key substrate in hand, we were pleased to discover that the 6π-electrocyclic ring closure proceeded smoothly in refluxing toluene to give tetrahydroindole 33 in excellent yield (Scheme 7).
Scheme 7.
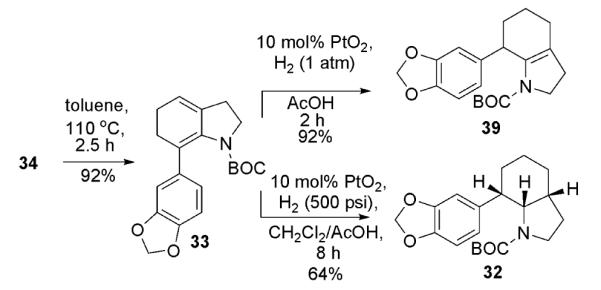
Preparation of the indolizidine substructure of γ-lycorane (1)
Our attention then turned to the hydrogenation of this diene. Under simple heterogeneous palladium catalyzed hydrogenation conditions enecarbamate 39 was formed. While this adduct could be reduced to the desired indolizidine core of γ-lycorane diastereoselectively under more forcing conditions, it was ultimately determined to be more expedient to reduce both double bonds in a single pot using PtO2 in acetic acid, giving indolizidine 32 directly.
With this racemic material in hand we then examined conditions for the corresponding asymmetric hydrogenation of diene 33. Catalytic asymmetric hydrogenation of enamides has been extensively studied, however there remains limited precedent for reduction of tetrasubstituted systems. We therefore entered this investigation with some trepidation.
We began with a brief screening of three Rh-ligand systems, DuPhos10, TangPhos11 and BINAP12. 1H NMR analysis of the crude mixtures revealed that in each of these cases 1,4-hydrogenation to enecarbamate 39 was predominating. Unfortunately, chiral HPLC analysis of the products revealed negligible enantioselectivity.
Use of a monodentate phosphoramide catalyst13 also proved unrewarding, failing to give any identifiable hydrogenation products. Discouraged by the lack of even modest selectivity in this initial screening, we quickly turned our efforts to the synthesis of racemic material.
To that end, deprotection of the carbamate was undertaken to give 40 (Scheme 8), whose spectra correlated well with previously published material.3j Pictet-Spengler ring closure under the conditions reported by Umezawa3n gave γ-lycorane (1).
Scheme 8.
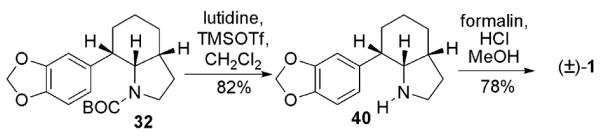
Completion of the γ-lycorane (1) total synthesis
In conclusion, we have completed a concise total synthesis of γ-lycorane in 9 steps from commercially available BOC-2-pyrrolidinone, demonstrating the utility of electrocyclic ring closures of divinylpyrrolines for the synthesis of indolizidine ring systems. Application of this methodology in the synthesis of other ring systems embodied in biologically active natural products is underway in our laboratories.
Acknowledgments
We appreciate the financial support provided by the National Institutes of Health (GM28663).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Martin SF. In: The Alkaloids. Brossi A, editor. Vol. 30. Academic Press; New York: 1987. pp. 251–376. [Google Scholar]
- 2.Lewis JR. Nat Prod. Rep. 1994;11:329. doi: 10.1039/np9941100329. [DOI] [PubMed] [Google Scholar]
- 3.Gao S, Tu YQ, Song Z, Wang A, Fan X, Jiang Y. J. Org. Chem. 2005:6523. doi: 10.1021/jo0507690. [DOI] [PubMed] [Google Scholar]; (b) Yasuhara T, Osafune E, Nishimura K, Yamashita M, Yamada K-I, Muraoka O, Tomioka K. Tetrahedron Lett. 2004;45:3043. [Google Scholar]; (c) Padwa A, Brodney MA, Lynch SM. J. Org. Chem. 2001;66:1716. doi: 10.1021/jo0014109. [DOI] [PubMed] [Google Scholar]; (d) Shao Z, Chen J, Huang R, Wang C, Li L, Zhang H. Synlett. 2003:2228. [Google Scholar]; (e) Cassayre J, Zard SZ. Synlett. 1999:504. [Google Scholar]; (f) Hoang-Cong Z, Quiclet-Sire B, Zard SZ. Tetrahedron Lett. 1999;40:2125. [Google Scholar]; (g) Cossy J, Tresnard L, Pardo DG. Eur. J. Org. Chem. 1999:1925. [Google Scholar]; (h) Angle SR, Boyce JP. Tetrahedron Lett. 1995;36:6185. [Google Scholar]; (i) Grotjahn DB, Vollhardt PC. Synthesis. 1993:579. [Google Scholar]; (j) Pearson WH, Schkeryantz JM. J. Org. Chem. 1992;57:6783. [Google Scholar]; (k) Baeckvall JE, Anderson PG, Stone GB, Gogoll A. J. Org. Chem. 1991;56:2988. [Google Scholar]; (l) Hwang H, Magnus P. J. Chem. Soc., Perkin Trans. 1. 1983:693. [Google Scholar]; (m) Iida H, Yuasa Y, Kibayashi C. J. Org. Chem. 1979;44:1074. [Google Scholar]; (n) Umezawa B, Hoshino O, Sawaki S, Sato S, Numao N. J. Org. Chem. 1977;42:4272. [Google Scholar]; (o) Iida H, Aoyagi S, Kibayashi C. J. Chem. Soc. Perkin Trans. 1. 1975:2502. [Google Scholar]; (p) Hara H, Hoshino O, Umezawa B. Tetrahedron Lett. 1972;13:5031. [Google Scholar]; (q) Ganem B. Tetrahedron Lett. 1971;12:105. doi: 10.1016/j.tetlet.2014.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]; (r) Irie H, Nishutani Y, Sugita M, Uyeo S. J. Chem. Soc., Chem. Commun. 1970:1313. [Google Scholar]; (s) Ueda N, Tokuyam T, Sakan T. Bull. Chem. Soc. Jpn. 1966;39:2012. [Google Scholar]
- 4.Chapsal BD, Ojima I. Org. Lett. 2006;8:1395. doi: 10.1021/ol060181v. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Fujioka H, Murai K, Ohba Y, Hirose H, Kita Y. Chem. Commun. 2006:832. doi: 10.1039/b512161b. [DOI] [PubMed] [Google Scholar]; (c) Dong L, Xu Y-J, Cun L-F, Cui X, Jiang Y-Z, Gong L-Z. Org. Lett. 2005;7:4285. doi: 10.1021/ol051795n. [DOI] [PubMed] [Google Scholar]; (d) Banwell MG, Harvey JE, Hockless DCR. J. Org. Chem. 2000;65:4241. doi: 10.1021/jo991791u. [DOI] [PubMed] [Google Scholar]; (e) Ikeda M, Ohtani S, Sato T, Ishibashi H. Synthesis. 1998:1803. [Google Scholar]; (f) Yoshizki H, Satoh H, Sato S, Nukui S, Shibasaki M, Mori M. J. Org. Chem. 1995;60:2016. [Google Scholar]
- 5.Greshock TJ, Funk RL. J. Am. Chem. Soc. 2006;128:4946. doi: 10.1021/ja060282o. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Greshock TJ, Funk RL. Org. Lett. 2006;8:2643. doi: 10.1021/ol0608799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huntley RJ, Funk RL. Org. Lett. 2006;8:3403. doi: 10.1021/ol061259a. [DOI] [PubMed] [Google Scholar]
- 8 (a).Yuecel B, Arve L, de Meijere A. Tetrahedron. 2005;61:11355. (b) see also: Pross A, Sternhill S. Aust. J. Chem. 1970;23:989.
- 9.Overman LE, Mendelson LT, Jacobsen EJ. J. Am. Chem. Soc. 1983;105:6629. [Google Scholar]
- 10.Burk MJ, Lee JR, Martinez JP. J. Am. Chem. Soc. 1994;116:10847. [Google Scholar]
- 11.Tang W, Zhang X. Angew. Chem. Int. Ed. 2002;41:1612. doi: 10.1002/1521-3773(20020503)41:9<1612::aid-anie1612>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 12.Miyashita A, Yasuda A, Takaya H, Toriumi K, Ito T, Souchi T, Noyori R. J. Am. Chem. Soc. 1980;102:7932. [Google Scholar]
- 13.Van den Berg M, Haak RM, Minnaard AJ, de Vries AHM, de Vries JG, Feringa BL. Adv. Synth Catal. 2002;344:1003. [Google Scholar]
- 14.Sakaitani M, Ohfune Y. J. Org. Chem. 1990;55:870. [Google Scholar]