
Figure 2.
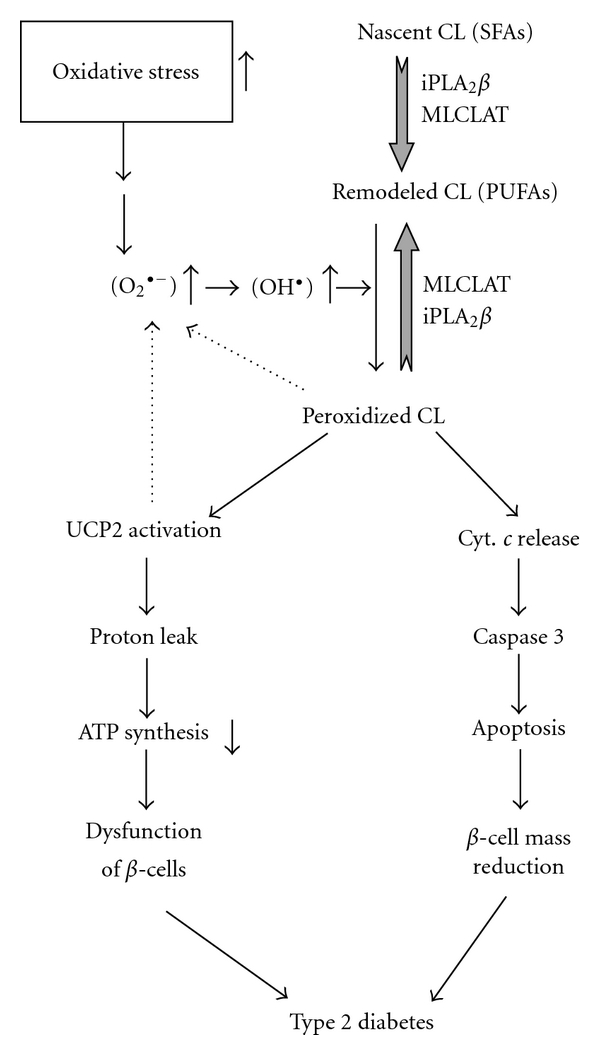
Schematic summary of the proposed role of mitochondrial cardiolipin oxidation in β-cell failure in type 2 diabetes mellitus. Oxidative stress results in increased mitochondrial ROS generation in β-cells. With moderate oxidative stress, ROS oxidize polyunsaturated fatty acid (PUFA) substituents in mitochondrial cardiolipin molecules, which may generate signals that mitigate ROS production via effects on respiratory electron transport chain complexes or on uncoupling protein 2 (UCP2) (dotted arrows). After delivery of the signal from the ROS-PUFA interaction, the oxidized cardiolipin molecule is repaired in a pathway in which iPLA2 β excises the oxidized PUFA residue to yielded monolysocardiolipin (MLCL), which is then reacylated with an unoxidized PUFA substituent by MLCL acyltransferase (MLCLAT) to complete the oxidation and repair cycle. Under conditions of overwhelming oxidative stress imposed by high metabolic loads, the rate of cardiolipin oxidation exceeds the capacity of the repair mechanism and oxidized cardiolipin molecules accumulate and compromise mitochondrial membrane integrity, and this leads to cytochrome c (Cyt. c) release into the cytosol and induction of apoptosis, which eventuates in β-cell failure and the development of T2DM. Circumstances in which the capacity of the repair mechanism is overwhelmed in this way would include reductions in iPLA2 β activity caused by genetic deficiency, pharmacologic inhibition, or yet to be defined regulatory influences on expression. Block arrows denote the iPLA2 β-mediated deacylation; line arrows denote the stimulatory pathway. SFAs: saturated fatty acids.