

Abstract
Charge transfer between metal ions occupying distinct crystallographic sublattices in an ordered material is a strategy to confer visible light absorption on complex oxides to generate potentially catalytically active electron and hole charge carriers. CaCu3Ti4O12 has distinct octahedral Ti4+ and square planar Cu2+ sites and is thus a candidate material for this approach. The sol−gel synthesis of high surface area CaCu3Ti4O12 and investigation of its optical absorption and photocatalytic reactivity with model pollutants are reported. Two gaps of 2.21 and 1.39 eV are observed in the visible region. These absorptions are explained by LSDA+U electronic structure calculations, including electron correlation on the Cu sites, as arising from transitions from a Cu-hybridized O 2p-derived valence band to localized empty states on Cu (attributed to the isolation of CuO4 units within the structure of CaCu3Ti4O12) and to a Ti-based conduction band. The resulting charge carriers produce selective visible light photodegradation of 4-chlorophenol (monitored by mass spectrometry) by Pt-loaded CaCu3Ti4O12 which is attributed to the chemical nature of the photogenerated charge carriers and has a quantum yield comparable with commercial visible light photocatalysts.
Introduction
The potential for inorganic materials to harvest and convert solar energy has attracted great interest in recent years.1−5 In particular, the ability of some metal oxides to photocatalytically break-down various water pollutants into harmless chemicals is an important and viable application of these materials: cheap, nontoxic metal oxides could, in the future, be used to clean polluted wastewater from chemical and textile industries.(6) Photocatalysts have also shown activity toward nondestructive organic transformations, a mild alternative to conventional synthesis with many potential applications.7,8
While a number of metal oxides are already, to some extent, able to photocatalytically break-down various water pollutants, the yields of these reactions remain low, and the vast majority of the reactions occur only upon UV light irradiation. As UV light accounts for only approximately 4% of solar energy at the Earth’s surface,(9) one of the main challenges in developing an economically viable photocatalytic material is to extend the catalytic activity beyond this 4% region. Thus, it is desirable to develop photocatalytic materials that are active under visible light, as this accounts for approximately half of all solar energy.(9)
Visible light photocatalytic activity has previously been achieved through the doping of colored transition metal ions into the structure of TiO2,10−12 a well-known UV-light-active photocatalyst. However, this method often detrimentally affects the catalytic efficiency of the material, compared to the parent compound, by inhibiting charge migration through the metal oxide(13) because of carrier localization produced by the breaking of translational symmetry upon the positionally disordered introduction of dopant species. Thus, if a material is to possess high activity in the visible range, other methods for the absorption of visible light must be employed.
There are numerous examples of UV-active complex metal oxide photocatalysts. These materials chiefly contain one of the closed-shell cations Ti4+, Nb5+, Ta5+, W6+, or Zn2+, whose binary oxides have themselves been studied extensively as UV-active photocatalysts.14,15 The influence of d0 transition metals on the electronic and optical properties of ABO3 perovskite materials has been investigated.(16) In contrast to a simple oxide such as TiO2, there are two ordered metal sublattices for the large A and smaller B cations. Layered structures have received particular attention in recent years, partly owing to the opportunities provided by exfoliation and restacking techniques.17,18 The number of visible-light-active complex metal oxide photocatalysts where positionally disordered doping is not used is far fewer. To afford visible light activity, elements other than the aforementioned d0 and d10 metals must be incorporated. In contrast to doping, incorporation into an extended and ordered lattice facilitates the formation of energy bands that extend throughout the structure, rather than localized defect sites, and thus does not hinder charge migration through the semiconductor particle. The occupancy of both the A and B sites of complex oxides such as perovskites can be tailored in an ordered way, unlike the disorder prevalent in doped materials,(19) and this has been investigated over a range of structure types. The double perovskites A2InNbO6 and A3CoNb2O9 (A = Ca, Sr, Ba), which accommodate two different cations on the ordered B sites,20,21 have shown visible light activity toward H2 and O2 evolution from aqueous solution under sacrificial conditions, with in this case only the BO6 octahedral network expected to be catalytically active.22,23 The substitution of Ca2+ for Pb2+ on the A site of the UV-active Aurivillius phase CaBi4Ti4O15 yields the visible-light-active photocatalyst PbBi4Ti4O15,(24) where both the B and disordered A sublattices, with their closed-shell 6s2 and 3d0 configurations, are involved in the photocatalysis. The work on such materials has focused primarily on H2 generation from water;(14) far less is currently known about organic photo-oxidation pathways in the presence of complex metal oxides.
Charge transfer between occupied and empty states located on different metals is a potential route to introduce charge carriers which does not require UV excitation and is suitable for combination with the distinct metal sublattice strategy. Ce3+ and Ti4+ centers grafted onto mesoporous silica have shown activity toward propan-2-ol photo-oxidation under visible light (λ > 400 nm), attributed to charge transfer between the two metal centers yielding Ce4+ and Ti3+.(25) The same group also reported Cu2+-grafted TiO2 as a visible-light-active photocatalyst toward the oxidation of propan-2-ol, attributed to the visible-light-active interfacial charge transfer between Cu2+ and Ti4+,(26) and later combined this approach with W6+ and Ga3+ doping into TiO2 to lower the conduction band relative to that of TiO2 to enhance visible light activity of the system.(27) This is an interesting contrast to the more common method of band gap engineering to obtain visible light absorption by raising the valence band of TiO2 and other oxides, for example, by anion-doping.3,28,29
CaCu3Ti4O12 has a cubic double-perovskite structure, illustrated in Figure 1,(30) with Ca2+ and Cu2+ ordered on the A site and Ti4+ located on the B site; it is a rare example of Cu2+ situated on the A site. The distortion produced by TiO6 octahedral tilting produces a square planar oxide environment suited to the Jahn−Teller distorted Cu2+. The extended structure allows Ti4+ and the open-shell Cu2+ to be incorporated within the same material without requiring doping; both cations occupy distinct ordered sites within the structure. There has been great interest in the properties of CaCu3Ti4O12 since the discovery of the unusually high dielectric constant of this material,(31) the origin of which is still not fully understood: there is some debate as to whether this is intrinsic or extrinsic in nature.32−34 As TiO2 is an efficient UV-active photocatalyst and CuO possesses significant visible light absorption, it was intended that the effective combination of the properties of these oxides in the complex oxide CaCu3Ti4O12 would facilitate visible light photocatalytic activity arising from a simple visible-light-induced charge-transfer transition from a Cu2+ + Ti4+ ground state to a Cu3+ + Ti3+ excited state, with the photogenerated electrons and holes being free to move in the distinct ordered crystallographic sublattices within the extended structure of this complex oxide. As the Cu-based occupied states are higher in energy than the O 2p-based valence band in a closed-shell oxide semiconductor, absorption will create carriers at a lower energy than the UV characteristic of TiO2. Lowering the gap will, of course, change the thermodynamic oxidizing and reducing potential of the generated electron and hole carriers. Cu-doped TiO2(35) and CuO surface-modified TiO2 composites36,37 have been shown to have higher photocatalytic activities toward Rhodamine B degradation and H2 evolution under UV light than TiO2 alone: that these studies are limited to the UV region illustrates the inefficiency of CuO as a photocatalyst. CuO/Cu2O composites have been shown to have greater activity toward the photo-oxidation of methyl orange under visible light than both CuO and Cu2O alone, although no mechanism for light absorption or catalysis was proposed.(38)
Figure 1.

Crystal structure of CaCu3Ti4O12, showing Ca2+ (purple) and chains of orthogonal square planar Cu2+ (green) on the A site and tilted TiO6 (orange) octahedra on the B site. Im3̅, a = 7.4037(5) Å.(30)
The high sinterability of CaCu3Ti4O12, coupled with typically high synthesis temperatures, makes the nanoparticulate synthesis particularly challenging. Here we present the low-temperature synthesis of CaCu3Ti4O12 via a simple sol−gel route and show that this material is active for the photosensitized oxidation of model pollutants methyl orange (MO) dye and 4-chlorophenol (4CP) under visible light (λ > 420 nm) irradiation. The origin of the visible light absorption is clarified by DFT calculations.
Experimental Section
Synthesis of CaCu3Ti4O12
A single-phase ceramic sample of CaCu3Ti4O12 was prepared by intimately mixing stoichiometric quantities of CaCO3 (Puratronic, 99.99%), CuO (Puratronic, 99.99%), and TiO2 (Puratronic, 99.99%) in an agate pestle and mortar. The mixture was then calcined as loose powder at 1000 °C for 12 h in an alumina crucible to decompose the carbonate. The resulting powder was reground and then pelletized using a 10 mm pellet press. The pellet was then calcined at 1100 °C for 2 × 20 h, with regrinding and pelletizing between calcinations.
During the investigation of sol−gel synthesis parameters, the ratio of ethylene glycol and organic acid to the number of metal ions per formula unit was varied (as summarized in Table 1). Typically, for 1 g (1.62 mmol) of CaCu3Ti4O12 synthesized using a 1:1:1 ratio of metal ions per formula unit:ethlyene glycol:organic acid, 1.1757 g (4.86 mmol) of Cu(NO3)2·3H2O (AnalaR, 99.5%) and 0.3842 g (1.62 mmol) of Ca(NO3)2·4H2O (Aldrich, 99%) were dissolved in approximately 40 cm3 water and added to a solution of 3.8306 g (6.48 mmol) of Ti(IV) bis(ammoniumlactato)dihydroxide (Aldrich, 50 wt % in H2O) and 0.8083 g (12.9 mmol) of ethylene glycol (Fluka, 99.5%). To investigate the effect of pH, the pH of this solution was altered to 2, 3, 4, or 5 by the dropwise addition of ammonium hydroxide (Sigma-Aldrich, ACS, concentrated solution): pH 1 was obtained without the addition of base. An aqueous solution (approximately 40 cm3) containing 12.9 mmol of an organic acid (2.7368 g of citric acid (Aldrich, 99.5%), 1.9547 g of l-tartaric acid (Aldrich, 99.5%), or 1.1726 g of oxalic acid (Aldrich, 99%)) was then added to the sol. The final sol was then heated to 80 °C under magnetic stirring for approximately 5 h. The subsequent gel was calcined at 120 and 350 °C for 10 and 2 h, respectively. The resultant black-brown powder was ground before the final calcination at 650 °C for 16 h.
Table 1. Investigation of Sol−Gel Conditions Yielding CaCu3Ti4O12 after Calcination at 650 °Ca.
| particle size/nm |
||||||
|---|---|---|---|---|---|---|
| acid | ratio M:EG:acid | pH | ppt | BET/m2 g−1 | XRD | SA |
| oxalic | 1:1:1 | 1 | yes | 11.2 | 53 | 53 |
| 1:1:1 | 3 | yes | 6.6 | 102 | 89 | |
| 1:1:1 | 4 | yes | 7.7 | 75 | 77 | |
| 1:1:1 | 5 | yes | 6.3 | 67 | 93 | |
| citric | 1:2:2 | 5 | no | 7.2 | 46 | 80 |
| l-tartaric | 1:1:1 | 6 | no | 9.4 | 61 | 63 |
| 1:1:1 | 5 | no | 12.6 | 64 | 47 | |
| 1:1:1 | 4 | no | 7.8 | 63 | 75 | |
| 1:1:1 | 3 | yes | 6.6 | 66 | 89 | |
| 1:1:1 | 2 | yes | 7.9 | 65 | 75 | |
Columns summarize the following parameters from left to right: chosen organic acid, ratio of metal ions per formula unit to molecules of ethylene glycol and molecules of chosen organic acid, the pH of the sol, whether a precipitate (ppt) was observed during sol−gel reaction, BET surface area, XRD particle size as determined by the Scherrer equation, and particle size determined from BET surface area.
Characterization
Samples were analyzed by X-ray power diffraction (XRD) using a Panalytical X’pert Pro diffractometer. Particle size was determined from XRD data using the Scherrer equation. Surface areas were measured by the BET method on a Quantachrome Nova gas sorption analyzer by nitrogen sorption on samples which were previously degassed at 120 °C overnight. The diffuse reflectance spectra of polycrystalline samples were obtained using a Perkin-Elmer Lambda 650 S UV/vis spectrometer equipped with a Labsphere integrating sphere over the spectral range 190−900 nm (6.53−1.38 eV), using BaSO4 reflectance standards. The spectra were analyzed by using the Kubelka−Munk function, F(R) = (1 − R)2/2R = K/S, with K the absorption and S the scattering coefficient, to represent the absorption coefficient of the sample. Zeta potentials of CaCu3Ti4O12 and Pt-CaCu3Ti4O12 over the pH range 4−9 were measured using acetate buffer on a Malvern Zetasizer Nano ZS. MO adsorption isotherms were obtained by sonicating 0.02 g of oxide material in 5 cm3 of MO of varied concentration for 15 min. The concentrations of MO before and after catalyst suspension were measured using a Perkin-Elmer Lambda 650 S UV/vis spectrometer to measure the absorbance of the solution at 465 nm, with the change in concentration giving the amount of dye adsorbed.
Photocatalysis
Pt cocatalyst (1 wt %) was deposited onto samples of CaCu3Ti4O12 via the photoreduction of H2PtCl6. Typically, 0.4 g of CaCu3Ti4O12 was suspended in 400 cm3 of a 1% MeOH aqueous solution to which 0.1048 g of H2PtCl6 (Aldrich, 8 wt % in H2O) was added. The cylindrical Pyrex reactor was then sealed and purged with N2 before being irradiated with two half-cylinder arrangements of 6 × 8 W CoastWave Blacklight UVA lamps, set against a semicircular aluminum reflector for approximately 6 h. During irradiation, the two half-cylinders were pushed together. The platinized material was retrieved by centrifugation at 3000 rpm for 15 min and dried at 60 °C. TEM images confirm that the photoreduction of H2PtCl6 in the presence of CaCu3Ti4O12 (OA 1:1:1, pH 1) deposited Pt particles on the metal oxide surface of ∼2−4 nm in size, as shown in Figure 2.
Figure 2.

TEM image of Pt nanoparticles of 2−4 nm on the surface of Pt-CaCu3Ti4O12, following photoreduction of H2PtCl6 in 1% MeOH solution in the presence of CaCu3Ti4O12 under UV light. The scale bar is 5 nm.
The photosensitized degradations of MO dye (50 cm3, 0.020 g dm−3 in water) and 4CP (50 cm3, 0.025 g dm−3 in water) were each conducted in a 100 cm3 cylindrical Pyrex reactor in the presence of typically 0.1 g of Pt-CaCu3Ti4O12. O2 was bubbled through the system throughout the course of the reaction. The light source used was a 300 W Xe lamp (Oriel) fitted with a water-flow IR filter; a 420 nm cutoff filter was used for visible light experiments. At suitable time intervals, 2 cm3 samples of MO or 4CP solutions were extracted, filtered to remove the catalyst, and their concentration determined before being replaced into the reactor. The concentrations of MO and 4CP were monitored using a Perkin-Elmer Lambda 650 S UV/vis spectrometer to measure the absorbance of the solution at 465 and 280 nm, respectively. In order to confirm that the presence of catalyst material was necessary for reaction to occur, blank reactions in the absence of catalyst were performed for both the MO and 4CP systems. Owing to the heating effects of the light source, the temperature over the course of the reaction was found to rise to a maximum of 43 °C. As such, thermal reactions were performed in the absence of light using an oil bath maintained at 45 °C to confirm that light and not heat was required for the reaction to proceed.
Quantum yield measurements were performed using potassium ferrioxalate actinometry.(39) This technique allows the determination of the photon flux entering a reactor and avoids the need to correct for any influences of light reflections. The same reactor and light source as for the photo-oxidation measurements were used. The light source was fitted with 400 and 450 nm, 10 nm band-pass filters as required. The photon flux incident on the reactor at 400 and 450 nm was measured to be 1.15 × 1020 and 2.10 × 1020 photons h−1, respectively.
Results
Characterization of CaCu3Ti4O12
While there are numerous reports of sol−gel-derived CaCu3Ti4O12, with a focus on the formation of high-density sintered pellets at higher temperatures (typically ∼1100 °C) for dielectric investigations,40−43 and recent reports of lowering the synthesis and sintering temperature by molten salt synthesis,(44) there has been no investigation into maximizing the surface area of high purity samples. Thus, in order to optimize the sol−gel synthesis conditions for high surface area CaCu3Ti4O12, a number of organic acids, pH values, and inorganic reagents were systematically used.
Table 1 summarizes the sol−gel reaction conditions found to yield the highest surface area CaCu3Ti4O12. It was found that nitrate precursors performed better than acetates, due to their greater solubility. Likewise, the water-soluble Ti(IV) bis(ammoniumlactato)dihydroxide produced sols of greater stability than Ti(IV) tert-butoxide, even when ethanol was used as the solvent. Interestingly, the formation of an insoluble precipitate when using oxalic acid could not be prevented by altering the pH of the sol, in contrast to reactions involving l-tartaric or citric acid. This precipitate was likely a metal oxalate complex, similar to that prepared by Thomas et al.,(45) where ethylene glycol was not used. However, in the current study, it was found that the presence of ethylene glycol was influential in the formation of high-purity CaCu3Ti4O12 at 650 °C. The best sol−gel-derived CaCu3Ti4O12 sample in terms of surface area and purity was produced using oxalic acid in a 1:1:1 ratio with ethylene glycol and the number of metal ions per formula unit at pH 1 (OA 1:1:1, pH 1; BET = 11.2 m2 g−1). Complete removal of the organic component after calcination at 650 °C was confirmed by CHN analysis (C:H:N = 0.00:0.16:0.00 for OA 1:1:1, pH 1).
XRD data from the ceramic sample of CaCu3Ti4O12 can be indexed to a unit cell of Im3̅, a = 7.3904(8) Å, and those from the sol−gel-derived sample (OA 1:1:1, pH 1) to a = 7.386(1), which both compare well to the reported structure.(30) A small amount of CuO was found by XRD to be present as an impurity phase in all sol−gel-derived samples after calcination at 650 °C, as shown in Figure 3: Rietveld refinement of XRD data from CaCu3Ti4O12 (OA 1:1:1, pH 1) suggests <2% CuO is present in this sample (Figure S1). Increasing the calcination temperature to 700 °C did not remove CuO but did dramatically decrease the surface area by ∼50%. When an analogous synthesis was conducted with a 3% Cu deficiency, both CuO and TiO2 impurities could be observed by XRD.
Figure 3.

Powder X-ray diffraction patterns of CaCu3Ti4O12 produced via (a) ceramic and (b) sol−gel synthesis indexed to cells of Im3̅, a ≈ 7.39 Å. Asterisk denotes CuO impurity.
Generally, particle sizes derived from BET surface area and XRD data from CaCu3Ti4O12 samples produced for this study were in good agreement, as shown in Table 1, indicating that samples were comprised of approximately spherical particles under little strain and with limited aggregation. SEM images of CaCu3Ti4O12 (OA 1:1:1, pH1: see Figure S2) show agglomerates of ∼50−100 nm sized particles, concordant with the 53 and 50 nm particle sizes calculated from XRD and BET surface area data, respectively.
Optical Properties of CaCu3Ti4O12
The optical properties of a ceramically prepared phase-pure sample of CaCu3Ti4O12 and the related compounds CaTiO3 and CuO were investigated by diffuse reflectance spectroscopy, and results are shown in Figure 4. A plot of [F(R) × E]1/n vs E can be used to determine the band gap energy, with n taking a value depending on the nature of the transition: 1/2 for a direct transition or 2 for an indirect transition. The absorption edge in the spectrum of CuO corresponds to an indirect transition of 1.27 eV (Figure S3), which compares well with values reported elsewhere.46,47 The valence band of CuO comprises strongly hybridized O 2p and Cu 3d orbitals, and the conduction band of Cu 3d orbitals,(48) and as such, the absorption edge of CuO is in the region typical for Cu2+ d−d transitions(49) but is far more intense owing to the hybridized nature of the Cu 3d orbitals. The carrier mobilities in CuO have been shown to be much smaller than those in Si, owing to this primarily Cu 3d nature of both the valence and conduction bands creating relatively flat and localized bands.(48)
Figure 4.

(a) Diffuse reflectance spectra of CaCu3Ti4O12 (black), CuO (red), and CaTiO3 (blue) and linear fittings of the higher energy absorption edge of CaCu3Ti4O12 to (b) an indirect transition of 1.93 eV and (c) a direct transition of 2.21 eV. The CuO absorption edge corresponds to an indirect transition of 1.27 eV, and the CaTiO3 absorption edge to a direct transition of 3.76 eV (Figure S3).
The absorption edge of CaTiO3 can be fitted to a direct transition of 3.76 eV (Figure S3), which compares well with previous reports;16,50 the valence band of CaTiO3 comprises O 2p orbitals, and the conduction band of Ti 3d orbitals.(51) CaTiO3 has orthorhombic symmetry owing to the tilting of TiO6 octahedra.(52) For ternary perovskites, it has been shown that, as the Ti−O−Ti bond angles distort away from 180° and the symmetry is lowered, an induced destabilization of the conduction band minimum decreases the width of the conduction band and therefore increases the band gap relative to that of a cubic structure: this is the case for CaTiO3, which has a significantly greater band gap than cubic SrTiO3.(16)
There are two observable absorption edges in the spectrum of CaCu3Ti4O12, with the most intense absorption edge lying between those of CaTiO3 and CuO. The lower energy absorption edge in the spectrum of CaCu3Ti4O12 is in the region expected for Cu2+ d−d transitions(49) and is seen in samples both with (sol−gel) and without (ceramic) CuO present as an impurity. It can be fitted as an indirect transition at 1.39 eV. Indeed, this absorption edge compares well with the absorption onset of CuO, the band gap of which is indirect and between Cu d-based bands.(48) Both the n = 1/2 and n = 2 plots of the higher energy adsorption edge of the CaCu3Ti4O12 spectrum produced good-quality linear plots, fitted to indirect and direct transitions of 1.93 and 2.21 eV, respectively. The observed reflectance data thus show that, in contrast to the parent oxides with the two-component transition metals in CaCu3Ti4O12 in isolation, the more complex oxide has two distinct band gaps which are both in the visible region. The observed values compare reasonably well with Eg ≥ 1.5 eV reported previously by Homes et al., where the nature of the transition was not addressed,(53) but are significantly smaller than the direct gap of 2.88 eV calculated from optical measurement of a CaCu3Ti4O12 thin film on a LaAlO3 substrate.(54)
Electronic Structure of CaCu3Ti4O12
To elucidate the nature of the band gap of CaCu3Ti4O12, electronic structure calculations were performed. Although such calculations have previously been performed on CaCu3Ti4O12,55−59 they have not gone beyond density functional theory within the basic local spin density (LSDA) or generalized gradient (GGA) approximations, with the exception of one study concerning the effect of Ca−Cu antisite defects.(58) Compared to the experimental measurements discussed above, the gap is grossly underestimated by LSDA and GGA functionals, which give band gaps in the region of 0.2−0.5 eV. He and co-workers(59) suggested a variety of approaches to improve the description of the electronic structure of transition metal oxides of this type, and in this study we have chosen to use the simplest of these, the LSDA+U method,(60) which has been shown to increase the band gap to roughly 2 eV,(58) though to our knowledge, no exact value calculated using LSDA+U has previously been reported.
The LSDA+U method introduces an on-site Coulomb interaction energy, parametrized by U, describing the screened on-site Coulomb interaction between paired electrons in the same orbital and J, describing Hund’s-rule-type intra-atomic exchange. The method attempts to correct for the LSDA’s frequent failure to describe strongly correlated or highly localized electrons, including the d-electrons of transition metals. We use the LSDA+U method as implemented by Dudarev and co-workers,(61) in which the correction is made rotationally invariant and depends only the difference between the two parameters, U − J.
The choice of U and J parameters for a given system is not necessarily straightforward. Screening within bulk materials means that they are often very different from those calculated for isolated atoms or ions. One approach is to assume that J remains unchanged at around 1 eV and vary U until good agreement is found with experiment. Alternatively, U and J can be calculated from first principles by varying the occupancies of the d-shell for one central transition metal atom and fixing the occupancies of the surrounding atoms to model the effect of screening.(62) We have chosen to use parameters calculated in this way for the material CaCuO2: U(Cu) = 7.5 eV, J(Cu) = 0.98 eV.(62) The parameters for Cu2+ were shown to markedly improve the electronic structure of CuO calculated within plane-wave DFT,(63) correctly giving an antiferromagnetic, semiconducting ground state with a band gap of 1.0 eV (experimentally 1.2−1.9 eV) and a local magnetic moment of 0.60 μB (experimentally 0.5−0.68 μB), whereas LSDA calculations give a nonmagnetic, metallic ground state. Since the Ti 3d orbitals are formally unoccupied in CaCu3Ti4O12, we have not introduced the on-site Coulomb energy at the Ti sites.
To perform reasonable electronic structure calculations on CaCu3Ti4O12, it is necessary to take into account the unpaired electrons associated with the Cu2+ atoms by calculating using a spin-polarized electron density. CaCu3Ti4O12 has antiferromagnetic spin ordering below a Néel temperature of 25 K;(64) however, at room temperature this long-range order is absent and the material is paramagnetic. Since our spin-polarized calculations are performed within a supercell, we inevitably introduce magnetic order; nevertheless, we expect the calculated electronic structure of the antiferromagnetically ordered ground state to be a good approximation to that of the paramagnetic state found at the temperature at which experiments were performed. Calculations with antiferromagnetic ordering were performed using the plane-wave-based DFT code, VASP,(65) using the projector-augmented wave method to describe core electrons.(66) The experimental crystal structure(30) was used throughout. The magnetic unit cell was sampled with a 12 × 12 × 12 k-point grid, with a plane-wave cutoff energy of 400 eV; both settings were found to give well-converged results for the electronic structure. The calculations were performed with 240 bands so that the density of states and band structure were converged to 8 eV above the band gap. We have performed calculations using the LSDA functional of Ceperley and Alder, parametrized by Perdew and Zunger,(67) and the LSDA+U method described previously.
The orbital- and site-projected partial density of states and the band structure calculated with the LSDA+U method are shown in Figure 5. Our results using LSDA reproduce those of previous calculations using LSDA and GGA functionals and are shown for comparison in Figure S4.59,68,69 Inclusion of the on-site Coulomb interaction with the LSDA+U method clearly has a large effect on the calculated electronic structure. The band gap of 0.15 eV between the occupied and unoccupied narrow bands in the LSDA calculations is greatly increased. There is now a gap of 1.52 eV from the valence band edge to a narrow band of unoccupied states, which is similar to the lowest unoccupied band in LSDA calculations. In addition, the gap between the valence band and the conduction band has increased to 2.03 eV (we define the conduction band as the wider band of unoccupied states from 2 to 4 eV). A narrow band of occupied states is observed just below the valence band in the LSDA+U calculations but not in the LSDA calculations.
Figure 5.

(a) Total and partial density of states (PDOS) calculated using LSDA+U. The PDOS is projected onto the Ca, Cu, Ti, and O atoms. (b) PDOS projected onto the dxy and the sum of the other four d orbitals of the Cu atom coordinated to O atoms in the xy plane, as shown in the inset figure. Spin-up and spin-down electrons are shown separately above and below the x-axis. (c) Band structure of CaCu3Ti4O12 calculated using the LSDA+U method. Note that, with antiferromagnetic ordering, the separate spin channels have identical band structures. Dots are added to the bands to show the projection of the states onto orbitals centered at different atoms, the radius of the dot is proportional to the extent of overlap. The dots are colored according to projections onto the Ca (green), Cu (blue), Ti (pink), and O (red) atoms.
The character of the occupied states at the valence band edge, the unoccupied states in the narrow band above it, and the occupied states in the narrow band just below the valence band can be understood in terms of simple crystal field theory considerations and are in general agreement with the findings of He and co-workers using a LDA functional.(59) These states have largely Cu dxy, dxz, and dyz and O 2p character (we define the x, y, and z axes as parallel to the lattice vectors of the cubic unit cell). These are the d orbitals with lobes pointing to the surrounding O atoms in the square planar CuO4 group. With d9 Cu2+ cations coordinated in a square planar geometry, these d orbitals are expected to be the highest in energy and hence singly occupied. This is indeed the case in CaCu3Ti4O12, with the added complication that these states are further split by σ-bonding and σ-antibonding interactions with O 2p orbitals.
To further characterize these states, we show the density of states projected onto the d orbitals of the Cu atom in the square planar CuO4 group lying in the xy plane using LSDA in Figure S4 and using LSDA+U in Figure 5b. The σ-bonding and σ-antibonding states involving the Cu dxy orbitals and O 2p orbitals are visible for each electronic spin. In the LSDA calculations, the σ-bonding states form a broad set of peaks at the bottom of the valence band due to extensive covalent hybridization with the other states in the valence band. With the addition of the on-site Coulomb repulsion with the LSDA+U method, these states are pushed down in energy in one spin channel and up in energy in the other. The states pushed down in energy are pushed down below the bottom of the valence band and no longer hybridize with the other states in the valence band. They then form the narrow band of states just below the valence band visible in the total density of states and band structure in Figure 5. Their σ-bonding character is clearly evident in the plot of the electron density arising from these states shown in Figure 6c.
Figure 6.

Plots of electron densities arising from (a) the three occupied Cu(3d)−O(2p) σ-antibonding states at the top of the valence band, (b) the three unoccupied Cu(3d)−O(2p) σ-antibonding states forming the narrow band above the valence band, (c) the three occupied Cu(3d)−O(2p) σ-bonding states just below the valence band, and the unoccupied conduction band showing the contribution from (d) Cu 3d and O 2p orbitals and (e) the Ti 3d orbitals. Densities are plotted in the plane at the base of the unit cell depicted in (f), except for (e), which is plotted in the plane containing Ti atoms, one-fourth of the way up the unit cell. Note that calculations were actually performed in a unit cell with the origin at a Ca atom, but the plots have been shifted by half a unit cell along the y direction to make the CuO4 group central. Plots are made with logarithmic contours which are shown dashed below an arbitrary value.
In both LSDA and LSDA+U calculations, the σ-antibonding states are singly occupied and form the top of the valence band in one spin channel and the narrow band of unoccupied states between the valence and conduction bands in the other spin channel. The character of these states can be seen in the plots of electron density in Figure 6a,b. Since there are six CuO4 within the magnetic unit cell which are largely uncoupled from each other, there will be six sets of σ-bonding and σ-antibonding states, one centered on each Cu atom. Due to the antiferromagnetic ordering, the singly occupied σ-antibonding states are occupied in different spin channels when considering neighboring CuO4 groups (spin densities are shown in Figure S5), with three in one spin channel and three in the other. Above the Néel temperature, we would expect these spins to become disordered but the character and energies of the electronic states to remain similar to those found with antiferromagnetic ordering.
The valence band stretches for 6 eV below the valence band edge. As well as the previously discussed Cu(3d)−O(2p) σ-antibonding states at the top of the band, the valence band consists of fully occupied states with Cu 3d, O 2p, and Ti 3d character, including the remaining four Cu d orbitals which do not have their lobes pointing toward the coordinated O atoms. The conduction band edge lies 2.03 eV above the valence band edge. Although there is some contribution to the unoccupied conduction band from Cu 3d and O 2p states shown in Figure 6d, the large majority of the states in the conduction band have Ti 3d character. The first band of states from 2 to 4 eV are the Ti t2g states, shown in Figure 6e, with mainly dxy, dxz, and dyz character. A second band from 5 to 8 eV mainly consists of the Ti eg states. Above these lie the vacant Ca 4s orbitals.
For comparison with optical experiments, we calculated the separate electronic transitions from the valence band to various bands of unoccupied states. The lowest energy transition from the valence band to the Ti 3d-dominated conduction band is an indirect one of 2.03 eV from R to Γ(0,0,0), with a lowest energy direct transition of 2.20 eV at X. It is also interesting to consider the transition from the valence band to the localized narrow Cu 3d-based band between it and the conduction band. The lowest energy transition between these bands is indirect, with a value of 1.52 eV, from R(0.5,0.5,0.5) to X(0.5,0,0) in the reciprocal unit cell, and the lowest energy direct transition is 1.57 eV at the X point, which is only slightly larger than the indirect transition. It is also worth noting precisely how narrow this Cu 3d band is, with a width of 0.44 eV compared to the 6.33 and 2.03 eV widths of the valence band and conduction band, respectively.
He and co-workers(59) discuss the relative isolation of the CuO4 units within CaCu3Ti4O12 compared to other cuprate structures, which commonly contain extended CuO2 1D chains or 2D planes. In contrast, the CuO4 units within CaCu3Ti4O12 are not really connected, forming 0D groups as shown in Figure 1 (and additionally Figure S6). When moving between two Cu atoms, through an O atom along a Cu−O−Cu pathway, the first Cu−O distance within the CuO4 group is 1.977 Å, which is similar to the Ti−O distance of 1.962 Å. However, the second O−Cu distance is considerably larger, at either 2.785 Å to the nearest-neighbor or 3.262 Å to the next-nearest-neighbor Cu atoms. Furthermore, neighboring CuO4 groups are rotated such that the states formed by overlap of the in-plane Cu 3d orbital and the O 2p orbitals are orthogonal to those on neighboring groups. This structural motif results in electronic states which are localized on the CuO4 groups, and this localization of the states, which are depicted in Figure 6, is reflected in the narrow widths of the bands above and below the valence band. The localization of the σ-antibonding states at the top of the valence band and in the unoccupied narrow band lying between the valence and conduction bands may well lead to a lack of mobility of both holes and electrons following excitation of electrons from one to the other. We do, however, note that the band structure in Figure 5 shows that there are states at the top of the valence band with hybridized Cu 3d/O 2p character which have considerable dispersion in energy, which should aid hole mobility.
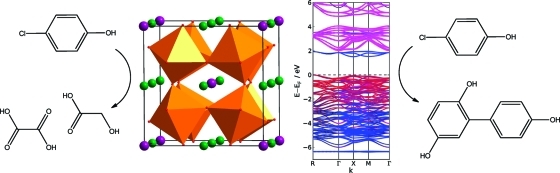
To compare the results of the LSDA+U calculations with experiment directly, we have calculated the frequency-dependent dielectric function (relative permittivity), ε(ω). The interband contribution to the imaginary part of ε(ω), ε′′(ω), was calculated in the self-consistent field approximation as a summation over states, as implemented in VASP,(70) neglecting local field effects. The real part, ε′(ω), was then obtained using a Kramers−Kronig transformation. In order to converge the calculated optical properties, the number of bands included in the calculation was increased to 320. The extinction coefficient, k(ω), was derived from ε(ω)(71) and is related to the Kubelka−Munk function measured in diffuse reflectance spectroscopy. In contrast to similar calculations,(69) we did not need to correct the optical properties due to an underestimated band gap. The calculated values of ε(ω) and k(ω) are shown in Figure 7.
Figure 7.
Calculated (a) dielectric function and (b) extinction coefficient of CaCu3Ti4O12. The extinction coefficient is plotted against wavelength to aid comparison with experimental diffuse reflectance spectra, which is shown in the inset. The corresponding energy scale, in eV, is shown above.
The experimental diffuse reflectance spectrum is reasonably well reproduced in the calculated k(ω) shown in Figure 7b, albeit with a general shift toward higher energies (shorter wavelengths). The first absorption edge in k(ω) appears at a shorter wavelength than in experiment, by 0.2 eV, appearing at an energy of 2.0 eV (608 nm), and by comparison with the density of states and band structure, we attribute it to transitions between the occupied and unoccupied Cu(3d)−O(2p) σ-antibonding states of the top of the valence band and localized narrow band within the gap. The next set of peaks, from 2.9 eV (421 nm) to 4.4 eV (284 nm) in the calculated spectrum, agree with the broad peak from 300 to 500 nm in the experimental spectrum, and we attribute it to transitions from the valence band to the conduction band of unoccupied predominantly Ti 3d orbitals.
The values for the electronic transitions calculated from the electronic structure of CaCu3Ti4O12 are in good agreement with those derived from optical measurement and suggest that the higher energy absorption edge of CaCu3Ti4O12 arises from a direct transition of 2.21 eV from the Cu 3d−O 2p hybridized valence band to the Ti 3d-composed conduction band (calculated as 2.20 eV), while the lower energy indirect transition of 1.39 eV is from the valence band to the localized unoccupied Cu 3d band located within the larger gap (calculated as 1.52 eV).
Photo-oxidation of Methyl Orange
The change in [MO] with irradiation (λ > 420 nm) time in the presence of Pt-CaCu3Ti4O12 is shown in Figure 8. The data sets show that Pt-CaCu3Ti4O12 degraded approximately 8 times more MO in 6 h than bare CaCu3Ti4O12. Dark and thermal reactions were also performed, and the absence of degradation in these control reactions confirmed that the presence of visible light was essential for the oxidation of MO to take place. Likewise, reaction in the absence of CaCu3Ti4O12 confirmed that CaCu3Ti4O12 was required for photo-oxidation to occur. As CuO was present as an impurity phase in all samples, the photocatalytic activity of Pt-CuO, prepared analogously to Pt-CaCu3Ti4O12, for this reaction was also tested, but no activity was observed. The CuO prepared in this way (OA 1:1:1) had a BET surface area of 2.0 m2 g−1, much lower than that reported for the hydrothermally prepared CuO (45.5 m2 g−1), which showed some activity toward MO photo-oxidation.(38) XRD data and SEM images were obtained from samples before and after testing toward the photo-oxidation of MO, and no change in the nature of CaCu3Ti4O12 was observed (Figure S2).
Figure 8.

Normalized change in [MO] with respect to time for (a) P25 + UV light and (c) Pt-CaCu3Ti4O12 + visible light (λ > 420), CaCu3Ti4O12 + visible light, Pt-CaCu3Ti4O12 + heat, and P25 + visible light. Evolution of MO UV/vis spectra with illumination time in the presence of (b) P25 (UV light) and (d) Pt-CaCu3Ti4O12 (visible light).
The UV/vis absorption spectra of MO during the course of the photo-oxidation in the presence of Pt-CaCu3Ti4O12 under visible light are shown in Figure 8d. The photo-oxidation of MO over semiconductor catalysts usually proceeds through a number of steps, including demethlyation, hydroxyl attack of the phenol ring, and cleavage of the azo bond, yielding a colorless solution.72,73 The spectral evolution of MO in the presence of TiO2 under UV light typifies this mechanism (Figure 8b). In contrast, the apparent isosbestic point at ∼374 nm observed in the spectra of MO irradiated under visible light in the presence of Pt-CaCu3Ti4O12 indicates the conversion of MO to a particular and well-defined single species, suggesting that this photo-oxidation process is more selective than the photocatalytic oxidation of MO in the presence of TiO2 and UV light. MS data show four closely related intermediates, with m/z 317, 290, 276, and 261 au (Figure 9; raw data in Figures S7 and S8), which indicate that the isosbestic point observed relates to the stepwise oxidation and removal of the −NMe2 group of MO. The intermediates with m/z 290 and 276 au were also observed in MS studies of MO photo-oxidation by TiO2 under UV light(74) but were not selectively produced in such a stepwise manner; the intermediates with m/z 317 and 261 au were not observed. The MS data from MO photo-oxidation over Pt-CaCu3Ti4O12 under visible light also show products with m/z 366 and 204 au, which were not observed in the UV/vis spectra and indicate direct hydroxyl attack of the phenol ring. While such attack has been indicated by the products of reaction of MO with TiO2 under UV light, this was limited to one hydroxyl group per phenol ring and occurred simultaneously with the removal of one or both methyl groups.(74) The detected intermediates suggest that the photo-oxidation of MO occurs via two pathways in the presence of Pt-CaCu3Ti4O12 under visible light: primarily by the selective stepwise oxidation and removal of the −NMe2 group, evident in the spectral evolution of MO, but also by direct hydroxyl attack of the phenol ring of MO.
Figure 9.

Products of visible light MO photo-oxidation over Pt-CaCu3Ti4O12 detected by MS with m/z values.
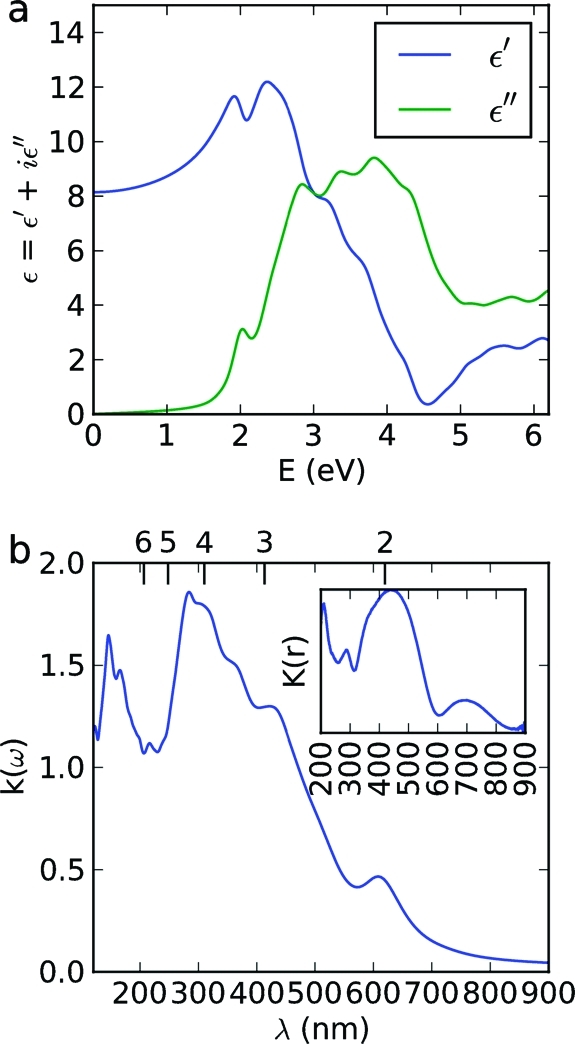
That Pt-CaCu3Ti4O12 displayed higher activity toward photo-oxidation of MO than bare CaCu3Ti4O12 is not unusual; enhanced photocatalytic activity with the use of a Pt cocatalyst is commonly observed, attributed to improved charge separation within the semiconductor material.(75) However, the addition of Pt and other noble metals has also been shown to greatly enhance the adsorption of dye eosin-Y onto TiO2, attributed to multidentate or multilayer complexation to the cocatalyst.(76) Indeed, Figure 10a compares the adsorption isotherms of MO on CaCu3Ti4O12, Pt-CaCu3Ti4O12, and TiO2 (P25): a marked enhancement of dye adsorption onto Pt-CaCu3Ti4O12 compared to bare CaCu3Ti4O12 can be observed. Owing to the influence of Pt loading on pollutant adsorption, the zeta potentials of CaCu3Ti4O12 and Pt-CaCu3Ti4O12 over a pH range 4−9 were measured. CaTiO3 has been shown to have negative zeta potential in this pH range, with a point of zero charge around pH 3.(77) As is clear from Figure 10b, the surface of bare CaCu3Ti4O12 is negatively charged within this pH range. While experimental data of zeta potentials of complex oxides are lacking from the literature, the prediction of the point of zero charge of oxides in water, based on crystal chemistry and electrostatic models, has been demonstrated and includes a theoretical inverse linear dependence on the dielectric constant of materials.(78) As such, the high dielectric constant of CaCu3Ti4O12 leads to a point of zero charge at low pH, leaving the surface negatively charged at higher pH values. Upon Pt deposition, the zeta potentials of CaCu3Ti4O12 are far less negative than those of the bare material—this might explain the enhanced adsorption of the anionic MO. The rate of reaction in such systems typically depends on the amount of adsorbed dye, so enhancing the adsorption of MO could explain the enhanced rate of oxidation.
Figure 10.
(a) MO adsorption isotherms of Pt-CaCu3Ti4O12, bare CaCu3Ti4O12, and TiO2 P25. Curve is a guide for the eye. (b) Zeta potentials of CaCu3Ti4O12 and Pt-CaCu3Ti4O12 at pH 4−9.
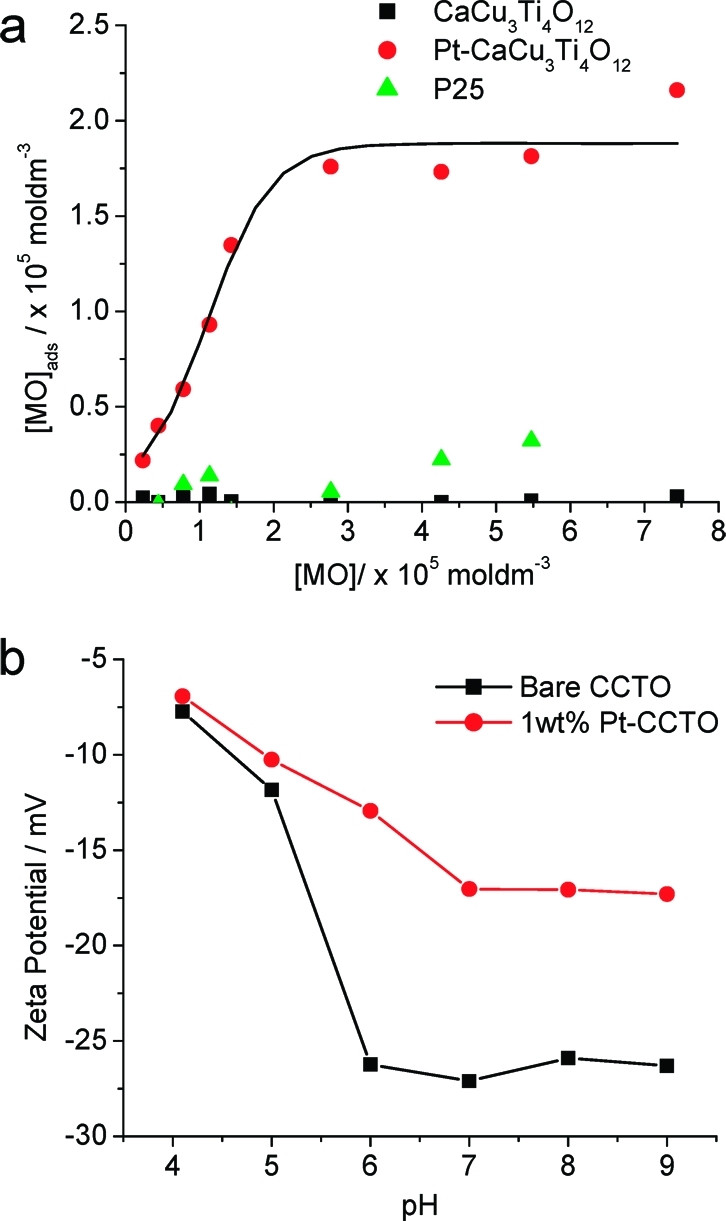
As MO dye strongly absorbs visible light, a dye-sensitized mechanism is possible. The quantum yields of Pt-CaCu3Ti4O12 toward MO photo-oxidation at different wavelengths were measured in order to produce an action spectrum to ascertain whether dye or catalyst was being activated by visible light. Figure 11 shows the action spectrum measured at 400 and 450 nm, directly compared to the absorption spectra of MO and CaCu3Ti4O12. Clearly, the data closely resemble the peak shape of MO adsorption and not the band edge of CaCu3Ti4O12, indicating a dye-sensitized mechanism. Such a mechanism has been observed for MO over TiO2 when irradiated with laser light at 442 nm,(79) but there are a far greater number of examples of visible light MO photo-oxidation where this issue is not addressed.
Figure 11.
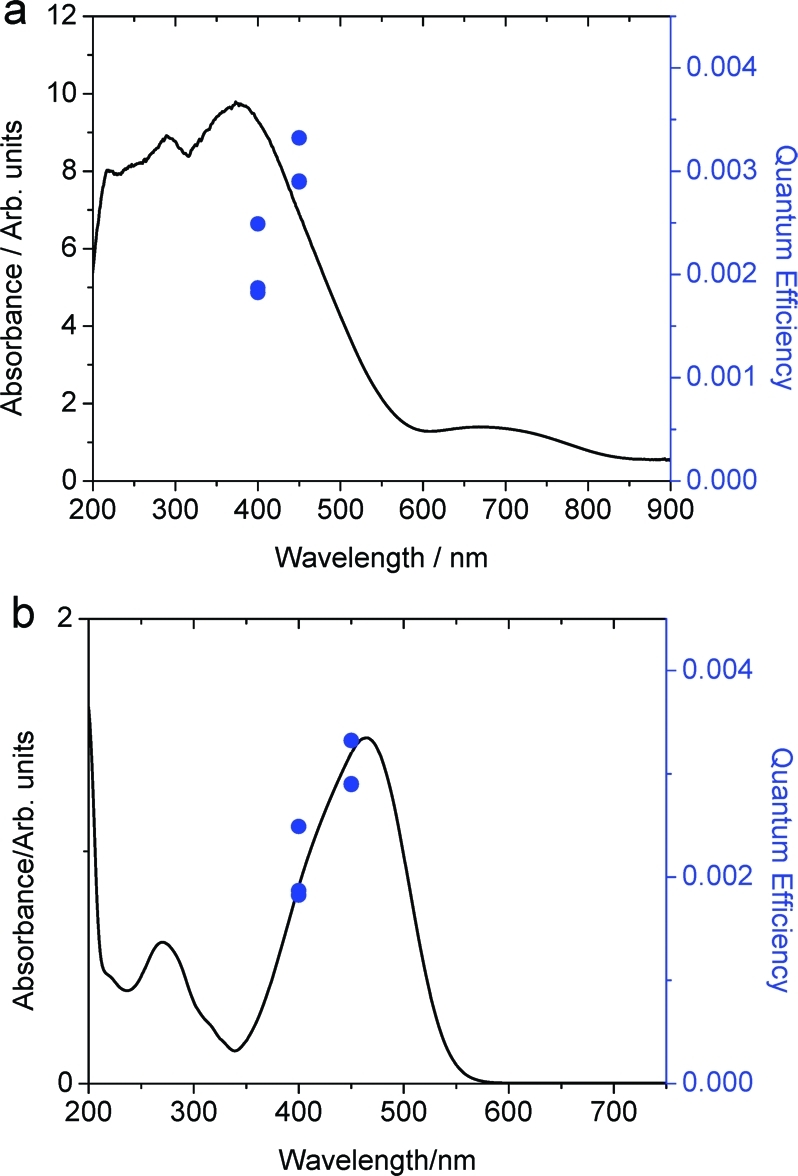
Quantum yields of MO oxidation over Pt-CaCu3Ti4O12 at 400 and 450 nm (10 nm band filters) overlaid with absorption spectra of (a) CaCu3Ti4O12 and (b) MO. Results indicate a dye-sensitized mechanism.
Photo-oxidation of 4-Chlorophenol
The spectral evolution of 4CP in the presence of TiO2 (P25) under UV light is shown in Figure 12. While there is a slight increase in absorbance at ∼250 nm in the P25 case, there is no observable isosbestic point, consistent with an unselective reaction pathway. The key intermediates of 4CP UV photo-oxidation in the presence of P25 as detected by MS are shown in Figure 12b (raw data in Figures S9 and S10) and are in good agreement with previous reports.(80)
Figure 12.

Spectral evolution of 4CP and photo-oxidation products (detected by MS) with UV light irradiation in the presence of 0.1 g of P25 (a,b) and 0.1 g of Pt-CaCu3Ti4O12 (c,d).
Interestingly, P25 showed activity toward the photo-oxidation of 4CP under visible light (Figure 13), despite neither 4CP nor P25 absorbing visible light. If this was due to stray UV light, activity toward MO photo-oxidation would also have been observed, but this was not the case. Contrary to expectations, visible light activity toward photo-oxidation of 4CP has been observed with P25 and attributed to a visible light-absorbing surface complex formed between TiO2 and 4CP, most likely via a phenolate linkage, which has been shown to induce occupied states in the TiO2 band gap.81,82 Indeed, after reaction with 4CP under visible light, P25 appeared yellow in color, as shown in the diffuse reflectance data in Figure S11. The main intermediates detected by MS for visible light photo-oxidation of 4CP in the presence of P25 are shown in Figure 13c (raw data in Figure S12). Under visible light, P25 yielded fewer products than is familiar for the UV reaction, breaking the phenol ring via a more selective route. While the visible-light-induced charge transfer between 4CP and TiO2 has been reported previously, there has been little discussion of products or precise mechanism of degradation. Agrios et al. were unable to detect coupling or other byproducts by GC-MS analysis after 4CP was irradiated with visible light in the presence of P25.(83) The photo-oxidation of trichlorophenol over P25 has been shown to proceed via two distinct pathways: the semiquinone is produced upon band gap excitation, whereas a charge-transfer complex yields the phenoxyl radical at light of energy below TiO2 band gap energies (anatase and rutile).(84)
Figure 13.

Normalized change in [4CP] with respect to time for (a) P25 + 4CP + visible light compared with Pt-CaCu3Ti4O12, (d) 4CP + Pt-CaCu3Ti4O12 + visible light (λ > 420 nm), 4CP + Pt-CaCu3Ti4O12 + heat, 4CP + visible light, 4CP + Pt-CuO + visible light, and 4CP + Pt-CaCu3Ti4O12 + visible light + tert-butOH. Evolution of 4CP UV/vis spectra with illumination time in the presence of (b) P25 (0.1 g) and (e) Pt-CaCu3Ti4O12 (0.20 g). Products of 4CP visible light photo-oxidation over (c) P25 and (f) Pt-CaCu3Ti4O12 detected by MS with m/z values.
In the presence of Pt-CaCu3Ti4O12 under UV light, the spectral evolution of 4CP in Figure 12 shows that the intensities of two additional absorbencies at 245 and 255 nm initially increase with reaction time and then begin to decrease after 270 min. Moreover, an isosbestic point at 232 nm can be observed until these additional absorbencies begin to disappear. This suggests that 4CP is initially selectively transformed and that this intermediate is then further photo-oxidized. The main products for this reaction as detected by MS are shown in Figure 12d: species with m/z values of 75, 89, and 62 au were not observed after the first hour of reaction but were detected after a longer period of 5 h (raw data in Figures S13 and S14). Far fewer intermediates were formed in the presence of Pt-CaCu3Ti4O12 than P25 under UV light, suggesting a more selective mechanism, accordant with the observed isosbestic point in the 4CP spectra. Evidence for the break-down of the phenol ring is apparent in both cases, with this being favored over the formation of dimeric products in the presence of Pt-CaCu3Ti4O12. The UV/vis absorbance spectra of key products hydroquinone (HQ), benzoquinone (BQ), and hydroxyhydroquinone (HHQ) are shown in Figure S15: comparison to the spectrum of photo-oxidized 4CP indicates a greater build-up of BQ in the presence of Pt-CaCu3Ti4O12 than in the presence of P25, leading to the aforementioned emergence of additional absorbencies at ∼250 nm in the spectra of 4CP photo-oxidized over Pt-CaCu3Ti4O12.
The change in [4CP] with respect to visible light irradiation (λ > 420 nm) time in the presence of Pt-CaCu3Ti4O12 is shown in Figure 13. The same blank reactions as performed with MO were repeated for 4CP and confirmed that the presence of Pt-CaCu3Ti4O12 and light was necessary for degradation to occur. It was also observed that increasing the amount of Pt-CaCu3Ti4O12 present resulted in an approximately proportional increase in 4CP photo-oxidation rate, which reached a maximum with 0.2 g (Figure 13 and additionally Figure S16). The decrease in rate when >0.2 g of Pt-CaCu3Ti4O12 material was added can be attributed to light scattering effects.
In contrast with the aforementioned visible light photo-oxidation of 4CP with P25, no color change of catalyst was observed with Pt-CaCu3Ti4O12, possibly owing to the dark red-brown color of this material, and as such the photo-oxidation observed was thought to originate from the visible light absorption of CaCu3Ti4O12. However, this was unable to be confirmed by quantum yield measurements at 400 and 450 nm, owing to the slow rate of reaction (at 450 nm). The quantum yield at 400 nm (±5 nm) was found to be 1.4%. This compares favorably with the quantum yield toward 4CP photo-oxidation of the commercially available, visible-light-active photocatalyst N-doped TiO2, found to be 0.05% at 405 nm (±5 nm).(85)
In order to ascertain whether the activity of Pt-CaCu3Ti4O12 toward the visible light photo-oxidation of 4CP originates from light absorption by CaCu3Ti4O12 or by some 4CP surface complex, reactions were also performed with an excess of tert-butyl alcohol (tert-butOH), an OH• scavenger, to provide insight into the mechanisms of these processes (Figure 13d). As the mechanism of photocatalytic 4CP degradation involves OH•,(15) the rate of degradation over P25 under UV light is decreased when tert-butOH is added; as OH• is not involved in the surface-complex-mediated degradation pathway, 4CP degradation over P25 under visible light is not hindered by the presence of tert-butOH.(81) The activity of Pt-CaCu3Ti4O12 toward 4CP photo-oxidation under visible light decreased in the presence of tert-butOH, suggesting that this process proceeds via a pathway involving OH• generated by band gap absorption by the oxide in the visible region and not a surface complex of 4CP.
The spectral evolution of 4CP in the presence of P25 and Pt-CaCu3Ti4O12 under visible light (λ > 420 nm) is compared in Figure 13. Aside from the different time scales of these reactions, there are subtle yet revealing differences between these sets of 4CP spectra. During both reactions, there is an increase in absorbance at ∼250 nm; however, in the case of Pt-CaCu3Ti4O12, there are isosbestic points to either side of this, at 238 and 265 nm. The small increase in UV/vis absorbance at ∼250 nm is attributed to the presence of BQ in both cases. This indicates that the photo-oxidation of 4CP occurs via a more selective pathway in the presence of Pt-CaCu3Ti4O12 than in the presence of P25. The main intermediates detected by MS for visible light photo-oxidation of 4CP in the presence of Pt-CaCu3Ti4O12 are shown in Figure 13f (raw data in Figure S17). The intermediates detected after visible light 4CP photo-oxidation over Pt-CaCu3Ti4O12 include some of the aromatic species found upon reaction with P25 under visible light, but no aliphatic products associated with further breakdown of the aromatic rings are observed. Pt-CaCu3Ti4O12 favors dimeric aromatic products, whereas P25 forms both dimeric aromatic and aliphatic products under visible light. It is perhaps not surprising to observe these differences, as it has been shown that P25−4CP surface complex initiates a dye-sensitized-type mechanism, whereas CaCu3Ti4O12 absorbs light to photo-oxidize 4CP.
Under both UV and visible light, Pt-CaCu3Ti4O12 photo-oxidizes 4CP more selectively than P25. The dramatic wavelength dependence of products of photo-oxidation of 4CP over Pt-CaCu3Ti4O12, selectively yielding aliphatic and dimeric aromatic products under UV and visible light, respectively, is particularly significant.
In order to assess the catalytic nature of visible light photo-oxidation of 4CP in the presence of Pt-CaCu3Ti4O12, the reaction was run for an extended period of time, after which the pollutant concentration was replenished and the reaction continued. The change in [4CP] with respect to visible light irradiation (λ > 420 nm) time in the presence of Pt-CaCu3Ti4O12 for the first and second runs is shown in Figure 14. The rate of the second run was slower than that of the first due to the presence in the solution of products from the first run, which may compete with the photo-oxidation of 4CP, as shown by the stepwise reactivity revealed from the MS data, and loss of catalyst during the sampling process. Turnover numbers were calculated by estimating the number of active sites accessible at the surface of Pt-CaCu3Ti4O12. It was estimated that 6.8 × 10−6 mol of active sites was present at the surface of 0.1 g of Pt-CaCu3Ti4O12, calculated from BET surface area data and unit cell dimensions by assuming one active site per formula unit (see Supporting Information for details). The total amount of 4CP lost by the end of the second run yielded a turnover number of 0.9 (after allowing for loss of catalyst). However, as has been described, several products are formed during the course of the reaction, with initial products being further oxidized.
Figure 14.
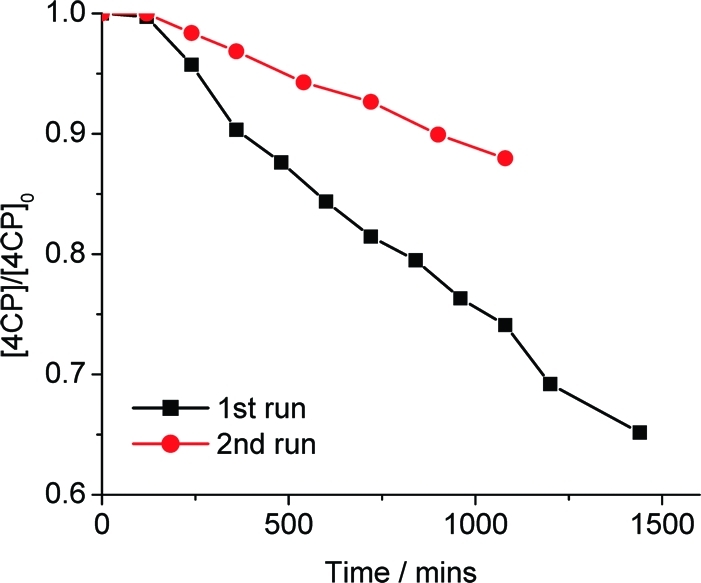
Normalized [4CP] with respect to time for extended and repeat runs in the presence of 0.1 g of Pt-CaCu3Ti4O12 under visible light (λ > 420 nm).
Discussion
Two absorption edges can be observed in the UV/vis diffuse reflectance spectrum of CaCu3Ti4O12, compared to only one in the spectra of each of the parent oxides CaTiO3 and CuO. As has been described, the lower energy absorption edge can be fitted to an indirect transition of 1.39 eV and the higher energy absorption edge to indirect and direct transitions of 1.93 and 2.21 eV, respectively.
DFT calculations, using the LSDA+U method, show that there are three bands in the ground-state electronic structure of CaCu3Ti4O12: the valence band of predominately O 2p character, the Ti 3d-dominated conduction band, and an unoccupied narrow band of localized Cu (3d)−O(2p) σ-antibonding orbitals located within the gap. The lowest energy transition from the valence band to the conduction band is an indirect one of 2.02 eV from R(0.5,0.5,0.5) to Γ(0,0,0), with a lowest energy direct transition of 2.20 eV at X(0.5,0,0). It is this direct transition that would be observed optically due to its higher probability and therefore much stronger absorption compared to that of an indirect transition. This is akin to the calculated electronic structure of anatase TiO2, a well-known UV-active photocatalyst, which has a direct transition at slightly higher energy than the indirect band gap.86,87 The calculated band gap is in much better agreement with the experimentally measured band gap than the small band gaps calculated previously using LSDA and GGA approximations. The calculated position of the narrow band within the gap between the valence band and conduction band is approximately 0.1−0.2 eV higher in energy, in terms of both the calculated band gap and the maximum in the calculated extinction coefficient. The exact position of these states will be dependent on the choice of U and J parameters for Cu within the LSDA+U method. Use of a smaller, arbitrarily chosen U(Cu) would probably result in slightly better agreement with experiment. The calculated direct band gap of 2.20 eV between the valence band and conduction band is in excellent agreement with the experimental value of 2.21 eV. Overall, the good agreement between the calculated and measured band gaps and the reasonable agreement between the calculated extinction coefficient and the experimental diffuse reflectance spectrum suggest that the electronic structure around the band gap is well described by the LSDA+U method. The calculations predict transitions from the valence band edge to an unoccupied narrow band of localized Cu (3d)−O(2p) σ-antibonding orbitals, resulting in the lower energy absorption edge observed in the UV/vis diffuse reflectance spectrum. At energies above 2.21 eV, transitions occur between the valence band and the conduction band, resulting in the higher energy absorption edge observed in the diffuse reflectance spectrum of CaCu3Ti4O12. The use of LSDA+U is only one step toward a better description of the electronic structure of CaCu3Ti4O12. Further improvements would be expected by the use of dynamical mean-field theory or quasiparticle theory as suggested by He and co-workers;(59) however, these improvements would come at the cost of a considerable increase in computational expense and are beyond the scope of the current study. The qualitative agreement with the experimental diffuse reflectance spectrum is reasonable, given the approximations used in the calculation of k(ω). Better agreement would be expected if more computationally expensive techniques were used to calculate ε(ω) and hence k(ω), such as those previously mentioned or the inclusion of local field effects or the use of quasiparticle calculations. In addition, the effect of the scattering coefficient, which contributes to the experimentally measured Kubelka−Munk function along with k(ω), is not simulated in the present calculations.
It is important to consider how the electronic structure of CaCu3Ti4O12 compares to that of TiO2. The electronic structure of TiO2 includes a valence band comprising occupied O 2p orbitals and a conduction band of unoccupied Ti 3d orbitals, separated by a band gap of 3.2 eV (anatase) or 3.0 eV (rutile).(87) The conduction band of CaCu3Ti4O12 comprises primarily unoccupied Ti 3d states with very little contribution from Cu or Ca near the band edge, so it can essentially be thought of as unshifted with respect to the conduction band of TiO2. Perhaps the starkest contrast between the electronic structures of TiO2 and CaCu3Ti4O12 is the band gap energy: CaCu3Ti4O12 has a significantly narrower band gap between the O 2p-based valence band and the Ti 3d-dominated conduction band than is observed in TiO2, resulting in visible light absorption. This difference is largely the influence of the square planar Cu2+ in CaCu3Ti4O12, with occupied Cu 3d states present near the top of the valence band. This hybridization of Cu 3d and O 2p states effectively widens the valence band and narrows the band gap, relative to those of TiO2, and shifts absorbance into the visible region. This is an advantage of the use of complex oxides over doped TiO2 as photocatalyst, with an induced narrowing of the band gap resulting from the addition of delocalized states widening the valence band. However, as has been described above, the electronic structure of CaCu3Ti4O12 also contains an unoccupied narrow localized band of Cu (3d)−O(2p) σ-antibonding orbitals located within the band gap—this is similar to the undesired effect of dn doping directly into TiO2, creating localized states that trap photogenerated electrons and holes and promote recombination. The occurrence of this feature in the electronic structure of an undoped and rigorously ordered complex oxide is perhaps surprising and illustrates the complexities and difficulties of tailoring the electronic structures of materials for any application, not just photocatalysis.
The incorporation of closed-shell Pb2+ into the Aurivillius phase PbBi4Ti4O15 has been shown to shift the absorption properties and photocatalytic activity into the visible region from the UV activity of the parent CaBi4Ti4O15.(24) The shift of the absorption edge (and photocatalytic activity) has been attributed to the hybridization of Pb 6s and Bi 6s with O 2p in the valence band, raising the position of the valence band edge with reference to the calculated electronic structure of the related phase, PbBi2Nb2O9.(88) As discussed above, the presence of open-shell Cu2+ on the ordered A site of CaCu3Ti4O12 is responsible for the visible light absorption and activity of CaCu3Ti4O12. Similarly to the aforementioned Pb2+ materials, this is attributed to hybridization of Cu 3d with O 2p in the valence band edge. The observed red shift of the band edge upon Ca substitution for Pb in CaBi4Ti4O15 (forming PbBi4Ti4O15) is 0.34 eV;(24) the band edge of CaCu3Ti4O12 is red-shifted 1.55 eV compared to that of CaTiO3. That the Cu 3d−O 2p hybridization in CaCu3Ti4O12 induces more visible light adsorption than the combined effects of Pb 6s−O 2p and Bi 6s−O 2p hybridization in the valence band of PbBi4Ti4O15 is of particular note: this illustrates the potential for developing single-phase, visible-light-active photocatalysts that incorporate open-shell cations rather than those that contain only closed-shell cations.
Upon illumination with light of energy equal to or greater than the band gap of CaCu3Ti4O12, an electron is excited from the Cu 3d−O 2p-based valence band to the Ti 3d-based conduction band, generating a hole (h+) and an electron (e−) as Cu3+/O− and Ti3+, respectively. Our calculations have shown that the lowest energy transition between these orbitals is from R(0.5,0.5,0.5) to Γ(0,0,0), i.e., is indirect, but that a direct transition occurs at a slightly higher energy. Owing to the presence of an indirect transition of slightly lower energy than that of the optically observed direct band gap of 2.21 eV, photogenerated e− are able to relax to the conduction band minimum. Recombination with h+ would then be via an indirect transition, thought to be less favorable than the analogous direct process, hindering recombination with h+ in the valence band. However, it is also possible that the photogenerated e− can relax into the localized Cu 3d band located within the band gap, forming Cu+. This electron would then be less able to migrate and react at the surface of CaCu3Ti4O12, and this band may act as an e− trap and facilitate recombination. Excitation of e− from the valence band to the localized Cu 3d−O 2p band is also possible. This situation is similar to that proposed for transition-metal-doped TiO2, where localized defect sites induced by the doping of, for example, Cu2+, are thought to act as trapping sites and recombination centers, thus lowering the efficiency compared to that of the parent TiO2.(13) The band structure of 3d transition-metal-doped TiO2 has a localized 3d-based band located within the band gap.(89) However, it is important to note that, in the case of CaCu3Ti4O12, the localized Cu 3d band is not the result of defects within the parent material but an intrinsic property of this compound that extends throughout the structure, and there is the possibility of band motion of this photogenerated electron to the particle surface. The narrow Cu band means that electrons (Ti3+) in the Ti-derived conduction band are considerably more mobile, and the presence of the Cu 3d states between the broad valence and conduction bands would enhance the recombination rate of photogenerated Ti3+ with valence band holes via Cu+ localized states from the narrow Cu band. Thus, the presence of localized Cu+ would reduce the number of mobile electrons within the conduction band (Ti3+), detrimentally affecting the efficiency of the catalyst. As Cu+ is less reducing than conduction band Ti3+ (Cu+ → Cu2+ + e−, E0 = −0.153 V; Ti3+ → Ti4+ + e−, E0 = −0.498 V), surface Cu+ would also be less reactive than Ti3+. Additionally, the Cu 3d−O 2p states at the valence band edge are not as dispersed as the predominantly O 2p states in the TiO2 valence band, so Cu3+ at the valence band edge may be less mobile than h+ species in TiO2. That the valence band edge of CaCu3Ti4O12 is shifted to higher energy than that of TiO2 means that h+ in CaCu3Ti4O12 is less oxidizing than h+ in TiO2, which may also act to hinder photocatalytic reactions.
MO is a precursor to many azo dyes used globally for coloring textiles. Polluted wastewaters from this industry are a large environmental issue. Current methods of water purification, such as ozonation and chlorination, use strong oxidants that themselves pose problems toward the environment. As such, there has been growing interest in environmentally sound materials that can photo-oxidize and degrade chemicals such as MO, enabling the cleanup of polluted water via a far less aggressive approach. While there are numerous reports of the photo-oxidation of MO by metal oxide materials, relatively little is known about the mechanisms involved. The exception to this is TiO2, perhaps the best-studied photocatalyst. The degradation of MO over TiO2 under UV light is known to proceed via a number of intermediates, which have been identified by HPLC and LCMS techniques.74,79 The main intermediates of MO photo-oxidation have also been reported for reaction over In2S3 under visible light and suggest a pathway similar to that of TiO2 under UV light, although a mechanism was not discussed.(90) As with the photo-oxidation of all other organic pollutants, it is generally thought that the photogenerated hole, h+, plays a major role in the oxidation of MO at the surface of TiO2.(4) The photogenerated electron, e−, reduces O2 dissolved in the solution, which can then also oxidize MO. The form of the reduced O2 and its role in the degradation of organics are still unclear.(4)
That the oxidation of MO over Pt-CaCu3Ti4O12 under visible light is more selective than the well-known UV-induced reaction with P25 must be partly a result of the observed dye-sensitized pathway. The dye-sensitized mechanism of photo-oxidation involves the excitation of a dye molecule (rather than a semiconductor particle) by incident light: an e− is injected from the dye into the conduction band of the semiconductor, leaving the dye in an oxidized state (MO+). In analogy with semiconductor photoexcitation, the conduction band e− can then recombine with MO+ or can react at the semiconductor surface to reduce some species, e.g., dissolved O2. This reaction between O2 and e− would thus prevent recombination between e− and MO+. In the MO−Pt-CaCu3Ti4O12 system, excitation of adsorbed MO injects e− into the conduction band of CaCu3Ti4O12 and reduces Ti4+, forming Ti3+ and MO+. MO+ is most likely a zwitterion with several possible resonance structures. As such, the observed −NMe2 removal proceeds when the positive charge of MO+ is located on the N atom of the −NMe2 group, which can then be subjected to nucleophilic attack by OH−. Addition of OH to the aromatic ring would arise from resonance structures where the positive charge of the zwitterion is located on an aromatic C atom—a milder route of H substitution for OH than direct attack on the aromatic ring. That the removal of −NMe2 is favored over OH substitution onto the aromatic rings reflects the stability of the zwitterion resonance structures. If recombination between the conduction band electron (Ti3+) and MO+ is avoided, Ti3+ can reduce O2. However, the conduction band e− (Ti3+) is also able to relax to the narrow and localized Cu 3d band located within the band gap of CaCu3Ti4O12, forming Cu+ (and Ti4+). Owing to the localized nature of this band, which acts as an electron trapping site, an e− on Cu+ is less able to migrate and react at the surface of CaCu3Ti4O12 than an e− on Ti3+. The selectivity of photo-oxidation products of the dye-sensitized MO−Pt-CaCu3Ti4O12 system, compared to those formed by UV light excitation of P25 and MO, is likely a result of a combination of the different excitation mechanism and the chemical nature of charge carriers.
4CP is used in the manufacture of dyes, drugs, and fungicides. The photo-oxidation and degradation of 4CP is of particular importance, owing to the role of chlorophenols as xenobiotic pollutants, originating largely from industrial chemical synthesis wastewaters.(80) There are several detailed investigations into the mechanism and kinetics of 4CP degradation over TiO2 under UV light,80,91,92 with key intermediates identified by HPLC and MS techniques.80,93 UV light irradiation of TiO2 forms e− and h+: h+ plays a major role in the oxidation of 4CP at the surface,(4) while e− reduces dissolved O2, which can then also react with 4CP. As with MO, the precise mechanism of photo-oxidation of 4CP in the presence of semiconductors other than TiO2 is currently not well understood.
It is well known that the photo-oxidation of 4CP in the presence of TiO2 and UV light proceeds via a number of steps and intermediates, yielding both aliphatic and aromatic species.80,92 That the product selectivity during the photo-oxidation of 4CP over P25 is greater under visible light than under UV light is attributed to the very different pathways of these two reactions, with the visible light absorption by a 4CP-TiO2 surface complex instigating a dye-sensitized path, similar to that described above for MO photo-oxidation over Pt-CaCu3Ti4O12. However, as Pt-CaCu3Ti4O12 photo-oxidizes 4CP via a conventional semiconductor band gap excitation under both UV and visible light, the enhanced selectivity of this catalyst compared with P25, as well as the wavelength-dependent selectivity, cannot be attributed to a change in the reaction pathway. As has been described above, the photo-oxidation of organic pollutants over a semiconductor photocatalyst involves oxidation by photogenerated h+ and reduction of O2 by photogenerated e−. Thus, any mechanistic differences between photo-oxidation reactions of different semiconductors must, in part, reflect the chemical nature of e− and h+. As the conduction band edges of P25 and CaCu3Ti4O12 are of similar energy, conduction band e− (Ti3+) in both materials is able to reduce ambient O2 at the semiconductor surface. As described above, localized Cu+ in CaCu3Ti4O12 may decrease the photocatalytic efficiency by facilitating recombination and reducing the number of mobile Ti3+ in the conduction band and would be less reactive upon surface reaction owing to its lower reduction potential than Ti3+. More importantly, both P25 and CaCu3Ti4O12 are able to oxidize 4CP via reaction of valence band h+. However, as described above, the valence band edge of CaCu3Ti4O12 is at higher energy than that of TiO2 and therefore contains Cu3+ and O− states that are less oxidizing than the O− states within the valence band of TiO2. In addition, the states at the valence band edge of CaCu3Ti4O12 are relatively localized. Thus, h+ in CaCu3Ti4O12 is less reactive (and therefore more selective) than h+ in TiO2.
The dramatic wavelength dependence of products of photo-oxidation of 4CP over Pt-CaCu3Ti4O12, selectively yielding aliphatic and dimeric aromatic products under UV and visible light, respectively, is particularly significant. Considering the electronic structure of CaCu3Ti4O12, it is reasonable to attribute the observed wavelength-dependent selectivity to the aforementioned nature of charge carriers involved. Upon band gap excitation, e− exist as both delocalized Ti3+ and localized Cu+. Under visible light, while band gap excitation is still possible, a greater proportion of e− would exist as localized Cu+ than is the case under UV light. Thus, when Ti3+ is the majority charge carrier (UV light), aliphatic products are formed; when Cu+ is the majority (visible light), dimeric aromatic products are formed.
The ability to control the photo-oxidative selectivity of CaCu3Ti4O12 has implications for the use of heterogeneous photocatalysis in organic transformations and synthesis, where selectivity has already been cited as extremely attractive.(8) Such externally controlled selectively could find applications where specific ratios of products are required, or where the order in which fractions are formed is significant.
The competitive adsorption of photoreaction products begins to retard the destruction of 4CP, with photogenerated charge carriers instead reacting with species such as HQ, BQ, or HHQ. Indeed, BQ has been shown to be an effective electron scavenger over illuminated TiO2 or ZnO, competing with O2 for photogenerated electrons.(94) The equilibrium between HQ and BQ has already been cited as a possible explanation for low photonic efficiencies for the photocatalytic degradation of 4CP and other aromatic compounds.95,96 As such, the calculated turnover number of 0.9 for the destruction of 4CP under visible light over Pt-CaCu3Ti4O12 is likely an underestimate. The formation of BQ from 4CP requires 2 e−, increasing the turnover number to 1.8 (in terms of photogenerated electrons/holes used rather than molecules of 4CP lost) before the continued equilibrium between BQ and HQ is considered.
Conclusion
CaCu3Ti4O12 has two distinct transition metals, open-shell square planar Cu2+ and closed-shell octahedral Ti4+, ordered over the A and B sites of the heavily distorted perovskite structure. The optical absorption behavior is more complex than that of the parent oxides, with two transitions reflecting the distinct cation species and sites within the structure. These gaps are both in the visible region, but the initial goal of a simple visible-light-induced charge-transfer transition between the metals from a Cu2+ + Ti4+ ground state to a Cu3+ + Ti3+ excited state, with the photogenerated electrons and holes being free to move in the distinct A- and B-site-based bands free of disorder, is only partially achieved according to the DFT calculations used to understand the visible light absorption behavior. The higher energy direct gap of 2.21 eV is assigned to transitions from Cu(3d)−O(2p) σ-antibonding top of the valence band to the conduction band, a wide band with largely Ti 3d character. This is effectively moving the conventional valence band−conduction band gap in an oxide semiconductor such as TiO2 into the visible region because the top valence band states have hybridized, dispersive Cu σ*−O 2p character rather than almost pure O 2p as in TiO2. Band gap excitation promotes an electron from the valence band to the conduction band and generates charge carriers Cu3+/O− (h+) and Ti3+ (e−). The lower energy absorption of 1.39 eV arises from transitions from the valence band into a localized Cu 3d-based band within the gap, generating Cu3+/O− (h+) and Cu+ (e−). These states, although giving rise to the low-energy, formally Cu+ electron carriers, permit ready carrier recombination and have less mobility than the broader band states above and below them to permit carrier transport to the surface for reaction. The narrowness of this band thus offsets the low-energy absorption produced by the two distinct metal sites strategy. The electronic structure of CaCu3Ti4O12 can only be understood when the effect of correlations between electronic motions within the narrow 3d Cu bands is taken into account.
In order to exploit this visible light absorption in photocatalysis, relatively high surface area sol−gel-derived CaCu3Ti4O12 (11.2 m2 g−1 compared to less than 1.0 m2 g−1 for ceramic samples) was prepared and found to photo-oxidize model organic pollutants MO and 4CP under visible light (λ > 420 nm). The action spectrum of the photo-oxidation of MO over Pt-CaCu3Ti4O12 suggests that this reaction proceeds via a dye-sensitized pathway due to light absorption by the MO. Pt deposition greatly enhanced the photo-oxidation of MO, an effect generally attributed to improved charge separation within the semiconductor; however, it was also found that the adsorption of MO onto CaCu3Ti4O12 is greatly enhanced by the deposition of a Pt cocatalyst, which could be explained by the relative zeta potentials of the bare and Pt-loaded CaCu3Ti4O12. The photo-oxidation of MO by Pt-CaCu3Ti4O12 under visible light was found to be far more selective than that by P25 under UV light and to occur via a stepwise process, consistent with a dye-sensitized pathway. The photo-oxidation of 4CP by Pt-CaCu3Ti4O12 under visible light has been shown to occur via a mechanism involving OH•, akin to the UV-induced degradation of 4CP over TiO2, and not charge transfer between 4CP and catalyst. The photo-oxidation of 4CP over Pt-CaCu3Ti4O12 was also found to occur by a more selective mechanism than that over P25, further supporting the different mechanism for visible light charge carrier generation involving both ordered metal sublattices revealed by the DFT calculations. The differences in electronic structure give the photogenerated carriers distinct chemical character from those in TiO2 and make the observed phototransformations of MO and 4CP more selective. The role of the Ti is shown by the inactivity of CuO in the evaluated reactions. The products of photo-oxidation of 4CP over Pt-CaCu3Ti4O12 under UV and visible light have been rationalized with reference to the chemical nature of charge carriers compared to those found in TiO2. The relatively poor photocatalytic efficiency compared to P25 under UV light, as well as the observed product selectivity, is attributed to the enhanced localization of valence band holes and Cu+ electrons, the higher energy and therefore lower oxidizing potential of hole states at the valence band edge, and the reduction of the conduction band population (Ti3+) resulting from localized Cu+ acting as an electron/hole trap and facilitating recombination within the electronic structure of CaCu3Ti4O12. The dependence of products of photo-oxidation of 4CP over Pt-CaCu3Ti4O12 on the wavelengths of incident light is of particular note and may have implications particularly for externally controlled organic transformations and synthesis.
This study has demonstrated that utilizing complex oxides containing two ordered sublattices of different metals is an effective method of creating visible light absorption by charge transfer, allowing the creation of electrons and holes in band states unimpeded by disorder. The efficiency of CaCu3Ti4O12 is low (although the quantum yield for 4CP degradation is higher than for a commercial visible light catalyst) because the electron mobility in the narrow σ*-derived band is low due to isolation of the CuO4 units, and because the quasi-localized d states in the gap between the conventional conduction band and valence band facilitate rapid carrier recombination. The development of an efficient visible-light-active photocatalyst requires that the metal sublattices involved in charge transfer must form extended M−O networks with independent broad bands to delocalize the charge carriers. Charge transfer between closed-shell metals would reduce the recombination problem present in CaCu3Ti4O12.
Acknowledgments
We thank the EPSRC for funding under EP/H000925 and a Doctoral Training Award to J.H.C. M.S.D. and R.G.P. are supported by the European Research Council (ERC Grant agreement 227987). We gratefully acknowledge Charlotte Blythe of the Centre for Materials Discovery for assistance with mass spectrometry analysis. We thank members of the EPSRC-funded UK Semiconductor Photochemistry Network for helpful discussions.
Supporting Information Available
Diffuse reflectance data from CuO and CaTiO3, LSDA and LSDA+U PDOS, spin-up and spin-down electron densities, SEM images of CaCu3Ti4O12, raw MS data, [4CP] with respect to visible light irradiation time for different catalyst loadings, UV/vis absorbance spectra of key 4CP photo-oxidation intermediates, diffuse reflectance data from TiO2-4CP, and CaCu3Ti4O12 active site calculation. This material is available free of charge via the Internet at http://pubs.acs.org.
Supplementary Material
References
- Kudo A.; Kato H.; Tsuji I. Chem. Lett. 2004, 33, 1534. [Google Scholar]
- Meyer T. J. Nature 2008, 451, 778. [DOI] [PubMed] [Google Scholar]
- Maeda K.; Domen K. J. Phys. Chem. C 2007, 111, 7851. [Google Scholar]
- Mills A.; LeHunte S. J. Photochem. Photobiol. A−Chemistry 1997, 108, 1. [Google Scholar]
- Hagfeldt A.; Gratzel M. Chem. Rev. 1995, 95, 49. [Google Scholar]
- Hoffmann M. R.; Martin S. T.; Choi W. Y.; Bahnemann D. W. Chem. Rev. 1995, 95, 69. [Google Scholar]
- Fox M. A. Acc. Chem. Res. 1983, 16, 314. [Google Scholar]
- Fox M. A.; Dulay M. T. Chem. Rev. 1993, 93, 341. [Google Scholar]
- Thuillier G.; Herse M.; Labs D.; Foujols T.; Peetermans W.; Gillotay D.; Simon P. C.; Mandel H. Solar Phys. 2003, 214, 1. [Google Scholar]
- Asahi R.; Morikawa T.; Ohwaki T.; Aoki K.; Taga Y. Science 2001, 293, 269. [DOI] [PubMed] [Google Scholar]
- Ohno T.; Mitsui T.; Matsumura M. Chem. Lett. 2003, 32, 364. [Google Scholar]
- Serpone N.; Lawless D.; Disdier J.; Herrmann J. M. Langmuir 1994, 10, 643. [Google Scholar]
- Choi W. Y.; Termin A.; Hoffmann M. R. J. Phys. Chem. 1994, 98, 13669. [Google Scholar]
- Osterloh F. E. Chem. Mater. 2008, 20, 35. [Google Scholar]
- Mills A.; Davies R. H.; Worsley D. Chem. Soc. Rev. 1993, 22, 417. [Google Scholar]
- Eng H. W.; Barnes P. W.; Auer B. M.; Woodward P. M. J. Solid State Chem. 2003, 175, 94. [Google Scholar]
- Domen K.; Ebina Y.; Ikeda S.; Tanaka A.; Konda J. N.; Maruya K. Catal. Today 1996, 28, 167. [Google Scholar]
- Ebina Y.; Sasaki T.; Harada M.; Watanabe M. Chem. Mater. 2002, 14, 4390. [Google Scholar]
- Nowotny M. K.; Sheppard L. R.; Bak T.; Nowotny J. J. Phys. Chem. C 2008, 112, 5275. [Google Scholar]
- Ting V.; Liu Y.; Withers R. L.; Krausz E. J. Solid State Chem. 2004, 177, 979. [Google Scholar]
- Ting V.; Liu Y.; Withers R. L.; Noren L. J. Solid State Chem. 2004, 177, 2295. [Google Scholar]
- Yin J.; Zou Z. G.; Ye J. H. J. Phys. Chem. B 2003, 107, 4936. [Google Scholar]
- Yin J.; Zou Z. G.; Ye J. H. J. Phys. Chem. B 2003, 107, 61. [Google Scholar]
- Kim H. G.; Becker O. S.; Jang J. S.; Ji S. M.; Borse P. H.; Lee J. S. J. Solid State Chem. 2006, 179, 1214. [Google Scholar]
- Nakamura R.; Okamoto A.; Osawa H.; Irie H.; Hashimoto K. J. Am. Chem. Soc. 2007, 129, 9596. [DOI] [PubMed] [Google Scholar]
- Irie H.; Miura S.; Kamiya K.; Hashimoto K. Chem. Phys. Lett. 2008, 457, 202. [Google Scholar]
- Yu H. G.; Irie H.; Hashimoto K. J. Am. Chem. Soc. 2010, 132, 6898. [DOI] [PubMed] [Google Scholar]
- Chen X. B.; Burda C. J. Am. Chem. Soc. 2008, 130, 5018. [DOI] [PubMed] [Google Scholar]
- Maeda K.; Higashi M.; Lu D. L.; Abe R.; Domen K. J. Am. Chem. Soc. 2010, 132, 5858. [DOI] [PubMed] [Google Scholar]
- Subramanian M. A.; Sleight A. W. Solid State Sci. 2002, 4, 347. [Google Scholar]
- Subramanian M. A.; Li D.; Duan N.; Reisner B. A.; Sleight A. W. J. Solid State Chem. 2000, 151, 323. [Google Scholar]
- Homes C. C.; Vogt T.; Shapiro S. M.; Wakimoto S.; Ramirez A. P. Science 2001, 293, 673. [DOI] [PubMed] [Google Scholar]
- Sinclair D. C.; Adams T. B.; Morrison F. D.; West A. R. Appl. Phys. Lett. 2002, 80, 2153. [Google Scholar]
- Lunkenheimer P.; Fichtl R.; Ebbinghaus S. G.; Loidl A. Phys. Rev. B 2004, 70, 172102. [Google Scholar]
- Xin B.; Wang P.; Ding D.; Liu J.; Ren Z.; Fu H. Appl. Surf. Sci. 2008, 254, 2569. [Google Scholar]
- Li G. H.; Dimitrijevic N. M.; Chen L.; Rajh T.; Gray K. A. J. Phys. Chem. C 2008, 112, 19040. [Google Scholar]
- Bandara J.; Udawatta C. P. K.; Rajapakse C. S. K. Photochem. Photobiol. Sci. 2005, 4, 857. [DOI] [PubMed] [Google Scholar]
- Yu H. G.; Yu J. G.; Liu S. W.; Mann S. Chem. Mater. 2007, 19, 4327. [Google Scholar]
- Hatchard C. G.; Parker C. A. Proc. R. Soc. London Ser. a−Math. Phys. Sci. 1956, 235, 518. [Google Scholar]
- Liu L.; Fan H.; Fang P.; Jin L. Solid State Commun. 2007, 142, 573. [Google Scholar]
- Liu J.; Smith R. W.; Mei W.-N. Chem. Mater. 2007, 19, 6020. [Google Scholar]
- Liu L.; Fan H.; Fang P.; Chen X. Mater. Res. Bull. 2008, 43, 1800. [Google Scholar]
- Masingboon C.; Thongbai P.; Maensiri S.; Yamwong T.; Seraphin S. Mater. Chem. Phys. 2008, 109, 262. [Google Scholar]
- Chen K. P.; Zhang X. W. Ceram. Int. 2010, 36, 1523. [Google Scholar]
- Thomas P.; Dwarakanath K.; Varma K. B. R.; Kutty T. R. N. J. Phys. Chem. Solids 2008, 69, 2594. [Google Scholar]
- Koffyberg F. P.; Benko F. A. J. Appl. Phys. 1982, 53, 1173. [Google Scholar]
- Rakhshani A. E.; Barakat F. K. Mater. Lett. 1987, 6, 37. [Google Scholar]
- Wu D. X.; Zhang Q. M.; Tao M. Phys. Rev. B 2006, 73, 235206. [Google Scholar]
- Morpurgo S.; Lojacono M.; Porta P. J. Mater. Chem. 1994, 4, 197. [Google Scholar]
- Mizoguchi H.; Ueda K.; Orita M.; Moon S. C.; Kajihara K.; Hirano M.; Hosono H. Mater. Res. Bull. 2002, 37, 2401. [Google Scholar]
- Ueda K.; Yanagi H.; Hosono H.; Kawazoe H. J. Phys.: Condens. Matter 1999, 11, 3535. [Google Scholar]
- Sasaki S.; Prewitt C. T.; Bass J. D.; Schulze W. A. Acta Crystallogr. Sect. C−Cryst. Struct. Commun. 1987, 43, 1668. [Google Scholar]
- Homes C. C.; Vogt T.; Shapiro S. M.; Wakimoto S.; Subramanian M. A.; Ramirez A. P. Phys. Rev. B 2003, 67, 092106. [Google Scholar]
- Ning T. Y.; Chen C.; Zhou Y. L.; Lu H.; Zhang D. X.; Ming H.; Yang G. Z. Appl. Phys. a−Mater. Sci. Processing 2009, 94, 567. [Google Scholar]
- Li G. L.; Yin Z.; Zhang M. S. Phys. Lett. A 2005, 344, 238. [Google Scholar]
- Li G.-L.; Yin Z.; Zhang M.-S. Mater. Sci. Eng. B−Adv. Funct. Solid-State Mater. 2008, 150, 163. [Google Scholar]
- Delugas P.; Alippi P.; Fiorentini V.; Raineri V. Phys. Rev. B 2010, 81, 081104(R). [Google Scholar]
- Zheng J.-C.; Frenkel A. I.; Wu L.; Hanson J.; Ku W.; Bozin E. S.; Billinge S. J. L.; Zhu Y. Phys. Rev. B 2010, 81, 144203. [Google Scholar]
- He L.; Neaton J. B.; Cohen M. H.; Vanderbilt D.; Homes C. C. Phys. Rev. B 2002, 65, 214112. [Google Scholar]
- Anisimov V. I.; Zaanen J.; Andersen O. K. Phys. Rev. B 1991, 44, 943. [DOI] [PubMed] [Google Scholar]
- Dudarev S. L.; Botton G. A.; Savrasov S. Y.; Humphreys C. J.; Sutton A. P. Phys. Rev. B 1998, 57, 1505. [Google Scholar]
- Anisimov V. I.; Zaanen J.; Andersen O. K. Phys. Rev. B 1991, 44, 943. [DOI] [PubMed] [Google Scholar]
- Wu D.; Zhang Q.; Tao M. Phys. Rev. B 2006, 73, 235206. [Google Scholar]
- Koitzsch A.; Blumberg G.; Gozar A.; Dennis B.; Ramirez A. P.; Trebst S.; Wakimoto S. Phys. Rev. B 2002, 65, 052406. [Google Scholar]
- Kresse G.; Furthmüller J. Phys. Rev. B 1996, 54, 11169. [DOI] [PubMed] [Google Scholar]
- Kresse G.; Joubert D. Phys. Rev. B 1999, 59, 1758. [Google Scholar]
- Perdew J. P.; Zunger A. Phys. Rev. B 1981, 23, 5048. [Google Scholar]
- Li G.-L.; Yin Z.; Zhang M.-S. Phys. Lett. A 2005, 344, 238. [Google Scholar]
- Li G.-L.; Yin Z.; Zhang M.-S. Mater. Sci. Eng.: B 2008, 150, 163. [Google Scholar]
- Gajdoš M.; Hummer K.; Kresse G.; Furthmüller J.; Bechstedt F. Phys. Rev. B 2006, 73, 045112. [Google Scholar]
- Ravindran P.; Delin A.; Johansson B.; Eriksson O.; Wills J. M. Phys. Rev. B 1999, 59, 1776. [Google Scholar]
- Brown G. T.; Darwent J. R. J. Phys. Chem. 1984, 88, 4955. [Google Scholar]
- Rajeshwar K.; Osugi M. E.; Chanmanee W.; Chenthamarakshan C. R.; Zanoni M. V. B.; Kajitvichyanukul P.; Krishnan-Ayer R. J. Photochem. Photobiol. C−Photochem. Rev. 2008, 9, 171. [Google Scholar]
- Baiocchi C.; Brussino M. C.; Pramauro E.; Prevot A. B.; Palmisano L.; Marci G. Int. J. Mass Spectrom. 2002, 214, 247. [Google Scholar]
- Subramanian V.; Wolf E.; Kamat P. V. J. Phys. Chem. B 2001, 105, 11439. [Google Scholar]
- Jin Z.; Zhang X.; Lu G.; Li S. J. Mol. Catal. A: Chem. 2006, 259, 275. [Google Scholar]
- Coreno J.; Coreno O. J. Biomed. Mater. Res., Part A 2005, 75A, 478. [DOI] [PubMed] [Google Scholar]
- Sverjensky D. A. Geochim. Cosmochim. Acta 1994, 58, 3123. [DOI] [PubMed] [Google Scholar]
- Galindo C.; Jacques P.; Kalt A. J. Photochem. Photobiol. A−Chemistry 2000, 130, 35. [Google Scholar]
- Theurich J.; Lindner M.; Bahnemann D. W. Langmuir 1996, 12, 6368. [Google Scholar]
- Kim S.; Choi W. J. Phys. Chem. B 2005, 109, 5143. [DOI] [PubMed] [Google Scholar]
- Orlov A.; Watson D. J.; Williams F. J.; Tikhov W. M.; Lambert R. M. Langmuir 2007, 23, 9551. [DOI] [PubMed] [Google Scholar]
- Agrios A. G.; Gray K. A.; Weitz E. Langmuir 2004, 20, 5911. [DOI] [PubMed] [Google Scholar]
- Hurum D. C.; Gray K. A.; Rajh T.; Thurnauer M. C. J. Phys. Chem. B 2004, 108, 16483. [DOI] [PubMed] [Google Scholar]
- Emeline A. V.; Zhang X.; Jin M.; Murakami T.; Fujishima A. J. Photochem. Photobiol. A−Chemistry 2009, 207, 13. [Google Scholar]
- Asahi R.; Taga Y.; Mannstadt W.; Freeman A. J. Phys. Rev. B 2000, 61, 7459. [Google Scholar]
- Mo S. D.; Ching W. Y. Phys. Rev. B 1995, 51, 13023. [DOI] [PubMed] [Google Scholar]
- Kim H. G.; Hwang D. W.; Lee J. S. J. Am. Chem. Soc. 2004, 126, 8912. [DOI] [PubMed] [Google Scholar]
- Umebayashi T.; Yamaki T.; Itoh H.; Asai K. J. Phys. Chem. Solids 2002, 63, 1909. [Google Scholar]
- He Y.; Li D.; Xiao G.; Chen W.; Chen Y.; Sun M.; Huang H.; Fu X. J. Phys. Chem. C 2009, 113, 5254. [Google Scholar]
- Alsayyed G.; Doliveira J. C.; Pichat P. J. Photochem. Photobiol. A−Chemistry 1991, 58, 99. [Google Scholar]
- Mills A.; Morris S. J. Photochem. Photobiol. A−Chemistry 1993, 71, 75. [Google Scholar]
- Durand A. P. Y.; Brown R. G. Chemosphere 1995, 31, 3595. [Google Scholar]
- Richard C. New J. Chem. 1994, 18, 443. [Google Scholar]
- Augugliaro V.; Palmisano L.; Sclafani A.; Minero C.; Pelizzetti E. Toxicol. Environ. Chem. 1988, 16, 89. [Google Scholar]
- Okamoto K.; Yamamoto Y.; Tanaka H.; Tanaka M.; Itaya A. Bull. Chem. Soc. Jpn. 1985, 58, 2015. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.