

Abstract
The endoplasmic reticulum (ER) represents the entry point into the secretory pathway and from here newly synthesized proteins and lipids are delivered to the Golgi. The selective cargo export from the ER is mediated by COPII-assembly at specific sites of the ER, the so-called transitional ER (tER). The peripheral membrane protein Sec16, first identified in yeast, localizes to transitional ER and plays a key role in organization of these sites. Sec16 defines the tER and is thought to act as a scaffold for the COPII coat assembly. In humans two isoforms of Sec16 are present, the larger Sec16A and the smaller Sec16B. Nevertheless, the functional differences between the two isoforms are ill-defined. Here we describe characterization of the localization and dynamics of Sec16B relative to Sec16A, provide evidence that Sec16B is likely a minor or perhaps specialized form of Sec16, and that it is not functionally redundant with Sec16A.
The organization of COPII dependent budding is very different between some of the key species. In mammalian cells and the yeast Pichia pastoris budding of COPII-coated vesicles occurs at specific sites of the ER, the transitional ER1,2. These subdomains are regions of rough ER, which are devoid of ribosomes and show characteristic COPII positive buds2,3. The tER membrane together with the first post-ER membrane structures, which are prior to fusion with (or to become) the ERGIC make up the so-called ER exit site (ERES, reviewed in4). Mammalian cells have several hundred ERES distributed throughout the cytoplasm which tend to accumulate in the juxtanuclear region1. Time-lapse imaging reveals them to be relatively long-lived structures5,6.
COPII vesicle budding in vivo requires the peripheral membrane protein Sec16p. SEC16 gene was first identified in Saccharomyces cerevisiae7; SEC16 mutations result in a lack of 50 nm vesicles in these mutants8. Sec16p localizes to the ER membrane and has three main functional domains namely the central conserved domain (CCD), the N-terminal domain and the C-terminal domain, which are separated by clusters of proline rich regions9. Protein interaction assays showed that Sec16p interacts with multiple COPII subunits; Sec24p and Sec31p bind Sec16p at the CCD, whereas Sec23p interacts with the C-terminus9,10,11. These results suggest a key involvement of Sec16p in the formation of COPII-coated vesicles, possibly by acting as a scaffold for the assembly of the COPII coat. In vitro, Sec16p facilitates the recruitment of Sec23/24p and Sec13/31p to synthetic liposomes12. Additionally, Sec16p stabilizes the Sec23/24p-Sar1p-GTP complex on liposomes, possibly preventing its premature disassembly after GTP-hydrolysis.
Database search of the human genome using the CCD of the yeast Sec16p identified two mammalian orthologues of yeast Sec16p – a longer Sec16A and a shorter Sec16B13,14,15. Overexpression and siRNA depletion showed the involvement of mammalian Sec16A in ERES organization. In both cases, a disorganization of the ERES was observed as the number ERES in the cell periphery as well as in the juxtanuclear region decreased13,14. In addition to that, depletion of Sec16A resulted in a delay and overexpression in an inhibition of ER-to-Golgi transport13,14,15.
Pull-down assays revealed a physical interaction of Sec16A with the COPII subunits Sec23 and Sec13, which is in line with the idea that Sec16 functions as a platform for COPII assembly14,15. Fluorescence recovery after photobleaching (FRAP) analysis showed that YFP-Sec16A rapidly cycles on and off the membrane13, although there is a significant steady-state pool of Sec16A in the cytoplasm, FRAP analysis revealed that Sec16A has a greater half-life (∼ 6 sec) than other COPII components such as Sec23A or Sec24D (both ∼ 4 sec)16,17. Moreover, Sec16A has a greater immobile pool (around 40%) on the ER membrane compared to Sec23A (around 20%), suggesting that recycling of Sec16A is slower than that of other COPII components16. These data are consistent with the idea that Sec16A provides a platform for the assembly of the COPII.
Recently, laser scanning confocal microscopy and electron microscopy was used to define the steady-state localization of human Sec16A in more detail16. Confocal microscopy and immunogold labelling of ultrathin cryosections showed that Sec16A localizes to sites adjacent to Sec31A. Electron tomography showed that Sec16A localized to concave cup-shaped structures, but it remains unclear if Sec16A is involved in generating this curvature or sensing curved membranes.16. Consistent with its role in organizing COPII assembly, the localization of Sec16A to ERES was shown to be independent from other COPII components as Sec16A remained associated with the tER membrane in the absence of Sec23/24 and Sec13/3116. Whether Sec16A plays a regulatory or structural role in COPII assembly remains to be determined; these possibilities are of course not mutually exclusive. Consistent with a role in regulating assembly Sec16p in P. pastoris is found at significantly lower abundance when compared to COPII proteins18. It is not fully understood whether Sec16A becomes incorporated into vesicles or is only exchanged between a membrane bound pool and a cytosolic pool. S. cerevisiae Sec16p for example was shown to be incorporated into budded vesicles in vitro and electron micrographs revealed Sec16A labelled structures that were consistent with COPII vesicles9,16. It would therefore be possible, that Sec16 is incorporated transiently into COPII-coated vesicles16.
The Drosophila melanogaster orthologue of Sec16, dSec1619 shares 23% amino acid identity with human Sec16A with the highest conservation in the C-terminal domain and the CCD. The peripheral membrane protein dSec16 also localizes to ERES and in agreement with the findings for human Sec16, depletion of dSec16 resulted to a disruption of ERES as shown by immunofluorescence and electron microscopy. Conversely, an ER-to-plasma membrane transport assay using plasma membrane marker Delta and the Golgi marker Fringe-GFP revealed that upon depletion of dSec16 ER export was inhibited19. As shown for human Sec16A, dSec16 localized to cup-shaped structures on the ER, which was determined by immuno-EM. Moreover, investigation of dSec23 depleted cells by immuno-EM revealed that dSec16 still localized to the tER in the absence of dSec23. These results suggest that dSec16 localizes upstream of other COPII subunits confirming the results obtained for human Sec16A16,19. Taken together, Sec16 seems to play an important role in the organization of ERES and seems to act as a scaffold for the assembly of the COPII coat. Sec16 orthologues have been identified in all species tested so far, including for example Mus musculus (mouse), Gallus gallus (chicken) and Danio rerio (zebrafish) underlining the importance of Sec16 in ER-to-Golgi transport15. Many species contain two isoforms (including mammals) while some only have one isoform, as for example C. elegans and Drosophila. However, the distinct functions of the two Sec16 isoforms remain elusive.
The role of the second mammalian ortholog Sec16B is less well defined15. The 117 kD protein has been previously described as a regucalcin gene promoter region-related protein(RGPR-p117, reviewed in20). A GFP-tagged version of Sec16B was shown to localize to ERES and siRNA depletion of Sec16B resulted in a disruption of ERES as seen for Sec16A21,22. Moreover, Sec16B is required for export from the ER as cells depleted of Sec16B showed an inhibition of GalNAc-T2-GFP transport from the ER15. Sec16A and Sec16B were shown to co-immunoprecipitate and thus likely function in a heteromeric complex15. Here we have used a combination of approaches that reveal Sec16B to be a minor isoform in terms of expression profile and also provide evidence that Sec16B cannot compensate for loss of Sec16A function in cells. Our data point to an as yet undefined specialized role for Sec16B in human cells.
Results
Characterization of anti-Sec16B antibodies
In order to identify endogenous Sec16B and further investigate its functions, we sought to generate specific antibodies against human Sec16B. We generated and affinity purified three different anti-peptide antibodies, anti-Sec16B (996–1010), anti-Sec16B (883–897), and anti-Sec16B (393–408). The peptides were designed against unique sequences of Sec16B, so that no cross-reaction of the antibodies with Sec16A could occur. In order to determine if the antibodies are able to detect endogenous Sec16B by western blotting, several cell types were lysed and the proteins separated by SDS-PAGE. Moreover, the three antibodies were tested for specificity on HeLa cells overexpressing GFP-Sec16B . The results are shown in Figure 1A–C. All three antibodies detected over-expressed GFP-Sec16B, which has a molecular weight of ∼ 144 kD (Figure 1A, B and C, lane 1 #). Immunoblotting with anti-Sec16B (393–408) detected multiple protein bands, with some very faint bands at around 100 kD (Figure 1A). Immunoblotting with anti-Sec16B (883–897) revealed a number of protein bands, including three bands at around 100 kD (Figure 1B). None of the bands detected in HeLa cells were lost upon siRNA-mediated suppression of Sec16B (data not shown). The antibody anti-Sec16B (996–1010) detected a single band at ∼ 60 kD (Figure 1C). This protein does not have the molecular weight expected for Sec16B (∼ 117 kD) and was also unaffected by siRNA depletion of Sec16B in HeLa cells (data not shown).
Figure 1. Sec16B expression is undetectable in a panel of cell lines.

HeLa cells were transfected with GFP-Sec16B, lysed 16 hours after transfection. Lysates were also produced from HepG2, RPE-1, NHDF, SH-SY5Y, C13NJ, and HeLa cells. The cell lysates were separated by SDS-PAGE and immunoblotted with the antibodies (A) anti-Sec16B (393–408), (B) anti-Sec16B (883–897), and (C) anti-Sec16B (996–1010). All three antibodies detected GFP-Sec16B (A, B and C lane 1 #). No endogenous Sec16B could be detected in any of the cell types. The bands at ∼ 100 kD recognized by anti-Sec16B (883–897) and anti-Sec16B (393–408) are non-specific bands (A and B, arrows) since they could not be depleted using siRNA (not shown). (D–F) HeLa cells were transfected with GFP-Sec16B, methanol fixed and stained with the antibodies (D) anti-Sec16B (393–408), (E) anti-Sec16B (883–897), and (F) anti-Sec16B (996–1010). The antibodies anti-Sec16B (883–897) and anti-Sec16B (996–1010) both detected overexpressed GFP-Sec16B and clearly labelled GFP-Sec16B marked ERES (E and F, zoom, arrows). Scale bar = 20 µm. (G–J) NHDF and C13NJ cells were methanol fixed and double labelled with the antibodies anti-Sec16B (883–897) or anti-Sec16B (996–1010) (green) and anti-Sec31A or anti-Sec24C (red). (G) Anti-Sec16B (883–897) detected some puncta in NHDF cells, these did not localize in proximity of Sec31A (zoom, arrows). (H) Anti-Sec16B (996–1010) recognized small puncta in NHDF cells. These were distinct from Sec24C (zoom, arrows). (I, J) Anti-Sec16B (883–897) and anti-Sec16B (996–1010) labelling was cytosolic in C13NJ cells. Scale bar = 20 µm.
To test the specificity of the antibodies on over-expressed Sec16B by immunofluorescence, HeLa cells were transfected with GFP-Sec16B, methanol fixed 16 hours after transfection and stained with the antibodies anti-Sec16B (393–408), anti-Sec16B (996–1010), and anti-Sec16B (883–897). The antibody anti-Sec16B (393–408), however, was unable to detect GFP-Sec16B by immunofluorescence (Figure 1D). Two of the three antibodies, anti-Sec16B (883–897) and anti-Sec16B (996–1010), recognized over-expressed GFP-Sec16B. Defined labelling of GFP-Sec16B marked ERES with the antibodies was observed (Figure 1E and 1F, arrows). These antibodies were further tested on different cell types, NHDF and C13NJ cells (Figure 1G–J). Cells were methanol fixed and double labelled with the antibodies anti-Sec16B (883–897) and anti-Sec31A or anti-Sec16B (996–1010) and anti-Sec24C. The antibodies anti-Sec16B (883–897) and anti-Sec16B (996–1010) both detected some puncta in NHDF cells (Figure 1G, 1H). However, these did not localize adjacent to Sec31A or Sec24C and are therefore not ERES (Figure 1G, 1H, arrows). Therefore, these puncta do not represent ERES and are more likely accumulated antibody or non-specific staining. In C13NJ cells both antibodies showed unspecific cytosolic staining (Figure 1I and J). Similar results were obtained in SH-SY5Y cells. Both antibodies showed an overall diffuse staining and anti-Sec16B (883–897) also detected puncta, but these were distinct from Sec31A labelling indicating that they are not bona fide ERES (data not shown). We conclude that the three antibodies tested – anti-Sec16B (996–1010), anti-Sec16B (393–408) and anti-Sec16B (883–897) – did not detect endogenous Sec16B by immunofluorescence or western blotting. Thus, either those cell types examined do not express Sec16B, or the antibodies are not of sufficient avidity to detect a very low level of endogenous expression of Sec16B.
Effects of siRNA depletion of endogenous Sec16B
Depletion of endogenous Sec16B was previously described to result in a disruption of ERES and the Golgi as well as a block of ER-to-Golgi transport15, similar to the phenotype observed in Sec16A depleted cells13,14,15. We depleted Sec16B in HeLa cells using siRNA in order to verify the previously described phenotype15. Here we used the same siRNA sequence (termed here Sec16B “G”). Using immunoblotting or transfection of cells expressing GFP-Sec16B, we confirmed that isoform specificity of these siRNAs. RPE-1 cells expressing GFP-Sec16B were transfected with the siRNA Sec16B “G”, and Sec16B UTR, which targets Sec16B in the 3′ untranslated region. Figure 2 shows that these siRNAs are effective against GFP-Sec16B in stably transfected RPE-1 cells. Sec16B UTR is ineffective in this assay at it targets a sequence within the non-coding region that was not included in the construct used to generate this cell line. We then sought to determine the effect of depleting Sec16B on early secretory pathway function. In addition to depletion of Sec16B alone, Sec16A and Sec16B were depleted simultaneously by adding both siRNAs Sec16B “G” and Sec16A UTR to the cells (Figure 3A–D). 72 hours after transfection the cells were methanol fixed and double stained with an anti-Sec24C and anti-ERGIC-53 antibody. When compared to the control GL2 (Figure 3A), Sec16A depleted cells showed a reduction of Sec24C labelled ERES (Figure 3B). Sec16A depletion also induced a redistribution of ERGIC-53 to an overall ER-like pattern (Figure 3B) compared to the control cells (Figure 3A). By contrast, transfection with duplexes targeting Sec16B had no effect on Sec24C or ERGIC-53 (Figure 3C). Significantly, targeting both, Sec16A and Sec16B (Figure 3D), resulted in an indistinguishable phenotype to that observed on suppression of Sec16A alone (Figure 3B). Cells showed a reduced number of ERES and an ER-like distribution of ERGIC-53 (Figure 3D).
Figure 2. Validation of Sec16B siRNAs.

RPE1 cells stably expressing GFP-Sec16B were transfected with siRNA duplexes targeting Sec16B (Sec16B “G” as used in21, Sec16B UTR targeting the 3′ untranslated region, Sec16B #1 and Sec16B #2 targeting the coding sequence). GFP-Sec16B is knocked down efficiently with all siRNAs except Sec16B UTR which only targets the endogenous Sec16B.
Figure 3. Depletion of Sec16B in HeLa cells does not affect the localization of Sec24C or ERGIC-53, nor can GFP-Sec16B functionally replace Sec16A. (A–D) Cells transfected with (A) a control siRNA duplex or those targeting (B) Sec16A, (C) Sec16B or (D) both isoforms as indicated were methanol fixed and processed for immunofluorescence to detect Sec24C and ERGIC-53.

Boxed regions show 2x enlargements. Note the loss of peripheral puncta labelled with Sec24C or ERGIC-53 when cells are depleted of Sec16A but not when depleted of Sec16B. Bars = 20 µm. (E) Depletion of Sec16A in HeLa and HeLa GFP-Sec16B cells. HeLa or HeLa GFP-Sec16B cells were transfected with siRNA duplexes as follows: GL2 (non-targeting control), lamin A/C (positive control), Sec16A UTR (targeting the 3′ untranslated sequence of Sec16A13). Cells were lysed 72 hours after transfection. Efficient depletion of Sec16A could be detected in HeLa control cells and HeLa GFP-Sec16B cells (top row, lane 3 and 6). A slight reduction of GFP-Sec16B can be observed in HeLa GFP-Sec16B cells depleted of Sec16A (second row, lane 6). Lamin A/C knockdown was efficient (third row, lane 2 and 5). α-tubulin was used as a loading control (bottom row). (F–I) GFP-Sec16B HeLa cells were methanol fixed 72 hours after transfection with the non-targeted control GL2 and Sec16A UTR. (F, G) Cells were stained for Sec31A. In Sec16A depleted cells (G) a reduction of Sec31A positive ERES could be observed compared to controls (F, note boxed 2x enlargements). (H, I) Immunolabelling revealed that ERGIC-53 was relocalized to the ER in Sec16A depleted cells (I) compared to controls (H). Expression of GFP-Sec16B appeared lower in Sec16A depleted cells (G and I, bottom panels, zoom). Scale bars = 20 µm. Single optical Z-sections are shown.
Subsequently, we set out to investigate whether GFP-Sec16B could compensate for loss of function of Sec16A. As we could not detect Sec16B expression in cell lines, we generated two cell lines stably expressing GFP-Sec16B for further characterization. GFP-Sec16B was amplified by PCR and cloned into the lentiviral expression vector pLVX-puro as described in23. HeLa and RPE-1 cells were subsequently transduced with the virus and stably expressing cells were selected with puromycin. Immunoblotting with an anti-GFP antibody revealed that in both cell lines GFP-Sec16B was expressed as a correct fusion protein having a molecular weight of ∼ 144 kD (data not shown). In order to determine the correct cellular localization of GFP-Sec16B, HeLa GFP-Sec16B cells and RPE-1 GFP-Sec16B cells were first imaged live. In both cell lines GFP-Sec16B localized to puncta which were distributed throughout the cell in an ERES-like pattern that showed mainly short range movements (data not shown) consistent with other ERES markers13,24. HeLa GFP-Sec16B cells were then transfected with siRNA duplexes targeting Sec16A as well as two control siRNAs. Immunoblotting of GFP-Sec16B cells transfected with control siRNA duplexes for 72 hours showed no effect on Sec16A expression (Figure 3E). Sec16A was effectively suppressed with the siRNA used and this was not affected by the presence of GFP-Sec16B. Of note, GFP-Sec16B expression was slightly reduced following suppression of Sec16A. There is no significant homology between the duplex used to target Sec16A here and the sequence of Sec16B, so we believe this reflects the fact that Sec16A is required to stabilize GFP-Sec16B. Immunofluorescence using these cells showed that suppression of Sec16A resulted in a loss of peripheral ERES (Sec31A labelling, Figure 3G compared to Figure 3F). Compared to control cells (Figure 3H), a redistribution of ERGIC-53 to a more ER-like distribution was observed in Sec16A depleted cells (note enhanced nuclear envelope labelling and reduction in number of peripheral puncta, Figure 3I). Thus, depletion of Sec16A in HeLa GFP-Sec16B cells had the same effect on other components of the secretory pathway as in wild type HeLa cells indicating that loss of Sec16A was not compensated by the presence of GFP-Sec16B. Thus, neither endogenous nor overexpressed GFP-Sec16B can compensate for loss of Sec16A.
Localization of GFP-Sec16B and COPII proteins analyzed by high resolution light microscopy
HeLa GFP-Sec16B cells were processed for immunofluorescence to determine the degree of colocalization of GFP-Sec16B with ERGIC markers. We showed previously that Sec16A localizes to the tER clearly distinct from the ERGIC23. Cells were imaged at the highest possible resolution allowed by conventional laser scanning microscopy ensuring that the Nyquist sampling theorem was satisfied and no pixels were saturated. Figure 4A shows that, GFP-Sec16B clearly colocalizes with Sec16A and localizes offset from markers of the ERGIC, ERGIC-53 (Figure 4B) and β′-COPI (Figure 4C). These results show that GFP-Sec16B localizes to ERES, most likely the tER, in both HeLa and RPE-1 cells.
Figure 4. Localization of GFP-Sec16B relative to other ERES and ERGIC components.

(A) GFP-Sec16B shows a high degree of overlap with endogenous Sec16A. (B, C) GFP-Sec16B localizes offset from (B) ERGIC-53 and (C) COPI which label the ERGIC. Bars = 20 µm, 200 nm in zoom a, 500 nm in zoom b and c. (D) Live cell imaging showed a minor population of stably transfected cells show a nuclear localization of GFP-Sec16B. GFP-Sec16B could be found throughout the nucleus showing some brighter puncta. (E, F) In methanol fixed cells punctate structures within the nucleus could be observed. Labelling with an anti-coilin (E) and anti-PML (F) antibody showed that the puncta were distinct from coilin and PML. Scale bar = 20 µm (10 µm in zooms). Single optical Z-sections are shown.
During the analysis of the stable GFP-Sec16B cell lines a very minor population of GFP-Sec16B expressing cells were observed to show GFP fluorescence in the nucleus. When RPE-1 GFP-Sec16B cells were imaged, in a subset of cells GFP-Sec16B could be found diffusely distributed throughout the nucleus with localization to some brighter puncta (Figure 4D, arrows). That this distribution is evident during live cell imaging (Figure 4D) shows that this is not an artefact of fixation. Methanol fixation of RPE-1 GFP-Sec16B cells extracted the diffuse staining revealing a punctate localization of GFP-Sec16B within the nucleus (Figure 4E, F). When RPE-1 GFP-Sec16B cells were fixed with paraformaldehyde (PFA) the nuclear staining resembled that observed in living cells showing a few brighter punctae in an overall diffuse stained nucleus (data not shown). This is likely to be because PFA fixation does not extract the soluble fraction of GFP-Sec16B. Immunostaining for the protein Coilin, the main component of the nuclear localized Cajal bodies25 and the promyelocytic leukemia protein (PML), a transcription factor present in PML bodies within the nucleus26, showed no colocalization of the GFP-Sec16B labelled punctae with Coilin (Figure 4E) or PML (Figure 4F). In order to quantify the number of cells with nuclear localization of GFP-Sec16B, cells with typical ERES localization and nuclear localization of GFP-Sec16B were counted. Nuclear localization of GFP-Sec16B was found in less than 0.1% of cells (n>600).
Requirements for tER localization of Sec16B
We and others have defined the minimal region of Sec16A required for efficient targeting of the protein to the tER as comprising the central conserved domain (CCD) along with an adjacent upstream sequence23,27. To further investigate the targeting requirements of Sec16B, the complete tER localization domains as well as four fragments of each of these domains were N-terminally tagged with GFP (Figure 5). The complete tER localization domain constructs were designed according to the published sequences15 and only minor variations were undertaken: four additional amino acids upstream of the original tER localization domain of Sec16A and one amino acid downstream of the tER localization domain of Sec16B were included for the constructs used here. These additional amino acids show a high conservation among various species and could therefore be involved in the targeting process. Figure 5A shows a schematic of the constructs investigated here. GFP-tagged constructs were transfected into HeLa cells and imaged in living as well as fixed cells. Immunoblotting confirmed that the constructs resulted in expression of protein of the expected size without significant degradation (data not shown). The control GFP-Sec16B showed a distinct ERES pattern when imaged in living and fixed cells (Figure 5B and C, first panel). The GFP-tagged construct containing the complete tER localization domain GFP-Sec16B (34–235), however, was distributed throughout the cytoplasm and the nucleus (Figure 5B and C). A similar localization was observed for the four fragments of the tER localization domain of Sec16B – GFP-Sec16B (34–109), GFP-Sec16B (34–159), GFP-Sec16B (159–235) and GFP-Sec16B (109–235). In very few cells a diffuse punctate pattern could be observed after fixation, for example, for the construct GFP-Sec16B (109–235) (Figure 5C, #). These puncta did not resemble ERES and did not label with Sec24C or Sec31A (data not shown) and are therefore non-specific and could be fixation artefacts, or represent aggregated protein. In summary, none of the GFP-tagged tER localization domain constructs designed here localized to ERES and we conclude that the requirement for accurate localization of Sec16B to ERES are the same as those for Sec16A23, namely the CCD and immediate upstream sequence.
Figure 5. Requirements for localization of GFP-Sec16B to ERES.

(A) Schematic of deletion constructs used. Full-length GFP-Sec16B was used as a control. (B) GFP-tagged tER localization domain constructs of Sec16B were transfected into HeLa cells. Live cell imaging showed that only full-length GFP-Sec16B localized to ERES. None of the deletion constructs localized to ERES, all deletion constructs were cytosolic and nuclear localized. (C) Following methanol fixation, GFP-tagged tER localization constructs were primarily cytosolic and the nuclear pool was mainly extracted. Very few cells (<5%) showed a diffuse punctate pattern (see # for example). Scale bar = 20 µm.
Identification of interaction partners of Sec16B
In order to identify other interaction partners of Sec16B, HeLa GFP-Sec16B cells and HeLa control cells transfected with pEGFP-C2 were lysed and a large scale immunoprecipitation carried out. GFP-Sec16B and the proteins bound to Sec16B were extracted from whole cell lysate using the GFP-Trap®. Subsequently, the beads fraction was separated by SDS-PAGE on a 5% and 15% gel and the proteins which were bound to GFP-Sec16B visualized with SYPRO® Ruby (Figure 6A and B). GFP-Sec16B could be determined by its size of ∼ 144 kD on the 5% gel (Figure 6B, #). By comparison with bands present in the GFP control, three distinct bands of the 15% gel were chosen for further identification by mass spectrometry (Figure 6A, arrows). The three proteins were extracted from the 15% gel, digested and the peptides analyzed by MS/MS spectrometry. The proteins were then identified by blast search against a human database. Band 1 was determined to be the heat shock 70 kD protein 8 (HSPA8) or heat shock cognate 70 (Hsc70). Bands 2 and 3 were both identified as Sec13. The interaction with Sec13 was validated by immunoblotting (Figure 6C). Again, as a control, HeLa cells transfected with pEGFP-C2 were used. 1% of input and unbound fraction as well as 20% of the beads were separated by SDS-PAGE. Incubation with an anti-GFP antibody showed that GFP as well as GFP-Sec16B (∼ 144 kD) efficiently bound to the GFP-Trap® beads (Figure 6C, top row, lane 2 and 5, #). Immunoblotting for various COPII components revealed that GFP-Sec16B binds to Sec13 (Figure 6C, second row, lane 5) and Sec16A (Figure 7C, bottom row, lane 5), but not to Sec31A (Figure 6C, third row, lane 5). These results are consistent with the idea that Sec16B functions in ER export through interaction with Sec16A and Sec13. Lack of detectable interaction with Sec31A is also consistent with the idea that Sec16B forms a direct complex with Sec13 through its central conserved domain28. This domain forms an ACE (ancestral coat element) which interacts with Sec13 in a similar way to the integration of Sec13 into a complex with Sec31 and separately with nuclear pore complex29. These findings are supported by the relatively high level of conservation of the central conserved domains (ACE domains) of Sec16A and Sec16B.
Figure 6. GFP-Sec16B interacts with Sec16A and Sec13.
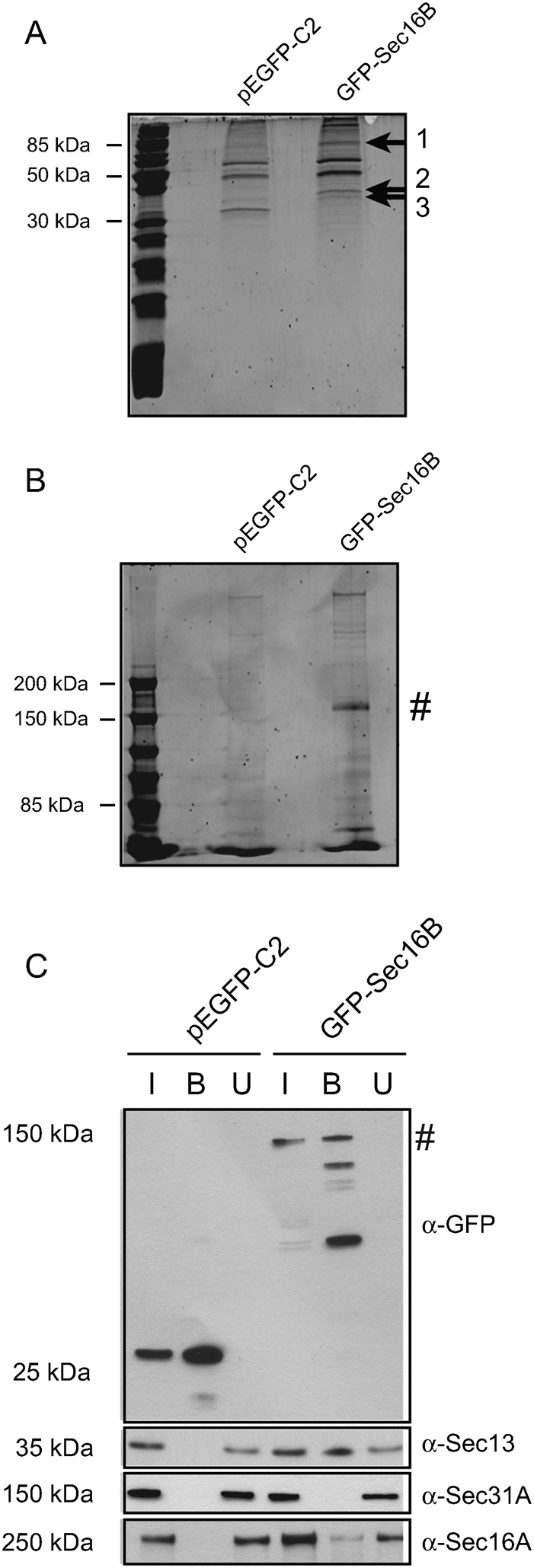
GFP-Sec16B and proteins bound to Sec16B were extracted from whole cell lysate using GFP-Trap®. The beads fraction was loaded on a 15% (A) and 5% (B) gel and subjected to SDS-PAGE to separate the proteins bound to GFP-Sec16B. The proteins were stained using SYPRO® Ruby. Three distinct protein bands were extracted from the 15% gel (1, 2 and 3, arrows), the proteins digested and analyzed by MS/MS spectrometry. The proteins were then identified by blast search against a human database. GFP-Sec16B migrated at ∼ 144 kD (B, #). (C) Identification of interaction partners of GFP-Sec16B by immunoprecipitation and immunoblotting. HeLa GFP-Sec16B cells and HeLa control cells expressing GFP were lysed. GFP and GFP-Sec16B were subsequently bound to GFP-Trap® beads. 1% of input and unbound fraction and 20% of beads were loaded. Probing with an anti-GFP antibody revealed some degradation of GFP-Sec16B during the sample analysis. Western blotting revealed that GFP-Sec16B (∼ 144 kD, #) and the GFP control were bound efficiently to the GFP-Trap® beads (top row, lane 2 and 5). GFP-Sec16B bound to Sec13 (second row, lane 5) and Sec16A (bottom row, lane 5), but not to Sec31A (third row, lane 5).
Discussion
Analysis of various cell types by immunoblotting and immunofluorescence revealed that Sec16B protein is not detectable with any of the antibodies tested. Specificity of the antibodies, however, was proven by detection of over-expressed GFP-Sec16B. Consistent with this lack of detection, Q-PCR using mRNA from different cell types showed that Sec16B expression is very low indeed in comparison to that of Sec16A (data not shown). Thus, it seems possible that Sec16B possesses a more specialized role within the cell and its function is only required at times of higher cargo load. Additionally, Sec16B could only be needed for transport of certain cargo molecules or at a specific cellular state. It is also possible that the expression of Sec16B is strictly regulated.
Due to the inability to detect endogenous Sec16B, further characterization of Sec16B was performed using GFP-Sec16B. High resolution imaging revealed that GFP-Sec16B is found almost completely overlapping with Sec16A and, reminiscent of the localization of Sec16A itself, was found offset from rather than overlapping with COPI and ERGIC markers. Significantly, and in contrast to work published previously, our data following depletion of Sec16A in HeLa GFP-Sec16B cells suggested that Sec16A and Sec16B have non-redundant functions. Silencing of Sec16A in HeLa cells stably expressing GFP-Sec16B cells had the same effect as in control cells showing that the presence of GFP-Sec16B does not compensate for the loss of Sec16A. This apparent discrepancy from previously published data15 could reflect a requirement for Sec16B that is only reflected in some assays and not others. This also supports the notion of a specialized function of Sec16B that we have yet to define.
Our data support the idea that the CCD and immediate upstream sequence are both required for effective targeting of Sec16B to the tER. This is analogous to data obtained for the Sec16A isoform23 and is in line with recent findings by Rabouille and colleagues (2008) who showed that the minimal targeting region in Drosophila dSec16 consists of a non-conserved region upstream of the CCD, which includes several charged residues, as well as the CCD itself19. Previous data implicated a smaller fragment of Sec16B (GFP-Sec16B (34–234)) in tER targeting; we only observed this at low expression level and in a minor population of cells; cell type and expression level differences could account for this. Overall, our data support a role for the CCD as well as upstream sequence in robust targeting of Sec16B to tER.
In summary, these data provide strong evidence for a function of Sec16B at ERES. While we have been unable to characterize the endogenous protein, our data points to a specialized function for Sec16B linked to but perhaps distinct from that of Sec16A. It is possible that Sec16B possesses a dedicated role within the cell and perhaps its function is only required at times of higher cargo load. Sec16B could only be needed for transport of certain cargo molecules or at a specific cellular state; it is also possible that the expression of Sec16B is strictly regulated. GFP-Sec16B was also shown to interact with Sec16A and the COPII coat component Sec13 supporting the hypothesis that Sec16B functions in ER export. Fluorescence recovery after photobleaching also indicates that Sec16B (half-life of recovery of 5.7 seconds and 56% immobile fraction22) is very similar in its dynamics of membrane association to Sec16A (half-life of recovery of 5.8 seconds and 43% immobile fraction23) showing both a highly dynamic pool as well as substantial immobile component. Our own (unpublished) observations are in line with these findings. This is perhaps not surprising given the data showing that these two proteins form a complex.
We also show that GFP-Sec16B interacts with the COPII coat component Sec13 supporting further the hypothesis that Sec16B functions in ER export. Additionally, mass spectrometry revealed heat shock 70 kD protein 8, also known as heat shock cognate 70, as a potential interaction partner of Sec16B. Despite its function in protein folding, the ATPase Hsc70 is also involved in vesicle transport where it is required for the uncoating of clathrin-coated vesicles30,31. An interaction of Hsc70 with Sec16B could potentially be interesting and would require further verification as well as investigation. However, a true interaction between the two proteins is questionable as Hsc70 is known to be a common contaminant in immunoprecipitation assays using GFP-Trap® as it binds non-specifically to the Sepharose matrix of the beads32. We have no data to indicate that the binding of Hsc70 to Sec16B is anything other than a false positive result.
Sec16B was first described as regucalcin gene promoter region-related protein (RGPR-p117) (reviewed in20). Regucalcin is a Ca2+-binding protein that has been shown to exhibit multiple roles within the cell as for example in maintaining intracellular Ca2+ homeostasis, inhibition of protein kinases, protein phosphatases, protein synthesis as well as RNA and DNA synthesis (reviewed in33). RGPR-p117 was shown to bind to the regucalcin promoter in a yeast one hybrid screen and overexpression of RGPR-p117 in NRK cells resulted in an increase of regucalcin mRNA levels. Moreover, HA-tagged RGPR-p117 was found to localize to the nucleus as well as the cytoplasm and to exhibit a putative leucine zipper motif, supporting the idea that the protein functions as transcription factor (reviewed in20). Conversely, recombinant RGPR-p117 could not bind to the regucalcin promoter region in vitro.20,34. Interestingly, here we found that in very few cells stably expressing GFP-Sec16B a nuclear localization of GFP-Sec16B was observed. Together with the data described above suggesting that RGPR-p117/Sec16B functions as a transcription factor, one could speculate that Sec16B might have additional functions to those in ER-to-Golgi transport. Such an additional function as a transcription factor has been proposed for Sec1235,36. Sec12 was also identified as a putative prolactin regulatory element binding (PREB) transcription factor36. Balch and co-workers suggested that Sec12/PREB could be released from the ER membrane in order to act as a transcription factor in a similar fashion as observed for the membrane bound transcription factors SREBPs35. Such a regulated release from the ER could also be imagined for Sec16B. It cannot be excluded that GFP-Sec16B is simply proteolytically processed and that the degradation products enter the nucleus.
The data shown here, however, provides strong evidence for a function of Sec16B in ER-to-Golgi transport. Sec16B was shown to interact with Sec13 and Sec16A, two proteins known to play key roles in ER export. The steady-state localization of Sec16B in relation to other early secretory pathway markers is similar to that observed for Sec16A16 underlining a probable function of Sec16B in ER export. Analysis of protein and mRNA expression hints at some form of regulation of Sec16B expression and the possibility of a specialized role of Sec16B in protein transport.
Methods
Production of polyclonal antibodies
Peptide antibodies against Sec16B were raised against the following sequences:
anti-Sec16B (393–408) (sheep) raised against aa 393–408 (CKKLEKYKRQPPVANL)
anti-Sec16B (883–897) (rabbit) raised against aa 883–897 (CSDEADKNSPRNTAQR)
anti-Sec16B (996–1010) (sheep) raised against aa 996–1010 (CGVGGLSGPESVSFEL)
The rabbit anti-Sec16B antibody was produced by Eurogentec (Belgium). The two antibodies raised in sheep were produced by conjugation of peptides to keyhole limpet haemocyanin (KLH). Conjugates were used for immunization by the Scottish National Blood Transfusion Service (Penicuick, Scotland). Affinity purification was done using SulfoLink® coupling gel (Pierce). Peak fractions from the elution were combined.
Antibodies used
Antibodies used in this study were as follows. Anti-Sec24C and anti-β′-COP rabbit polyclonal antibodies generated in the Stephens lab and described previously37; anti-giantin and anti-GFP (Covance, Cambridge UK), anti-ERGIC-53 (Sigma-Aldrich, Poole, UK), anti-Sec31A (Transduction labs, Cambridge, UK). Anti-coilin (Sigma-Aldrich) and anti-PML (Santa Cruz Biotechnology) were both kindly provided by Harry Mellor, anti-Sec13 antibodies were generous gifts from Wanjin Hong (IMCB, Singapore) and Beatriz Fontoura (Southwestern Medical Center, Dallas, TX), anti-Sec16A (anti-KIAA0310, Bethyl Laboratories). Horseradish peroxidase (HRP)-conjugated secondary antibodies were from Jackson Laboratories (Suffolk, UK). Donkey anti-rabbit, anti-mouse, and anti-sheep secondary antibodies conjugated to Alexa Fluor dyes were from Molecular Probes (Invitrogen).
Cell lines used
Mammalian cell lines and primary cells were kindly provided by the following people: HeLa, hTERT-RPE-1 human retinal pigment epithelial cells, and HEK 293T human embryonic kidney cell line (ATCC), HepG2 human hepatocellular liver carcinoma cell line (Giles Cory, University of Exeter), NHDF normal human dermal fibroblasts (ATCC), NRK normal rat kidney cell line (Jon Lane, University of Bristol), C13NJ human microglial cell line and SH-SY5Y human neuroblastoma cell line (both from Jeremy Henley, University of Bristol). HeLa cells, HEK 293T, SH-SY5Y and C13NJ cells were grown in DMEM supplemented with 10% fetal calf serum (FCS) and 4 mM glutamine. HepG2 were grown in MEM + GlutaMAX™-I (Gibco) containing 10% FCS. RPE-1 cells were grown in DMEM/F12 + GlutaMAX™-I (Gibco) containing 10% FCS.
Microscopy
Immunofluorescence and microscopy of both living and fixed cells (including fluorescence recovery after photobleaching) were carried out essentially as described previously23.
Cloning and expression of GFP-Sec16B
GFP-Sec16B was generated by PCR using primers designed according to the sequences of Sec16B (GenBank accession AB063357) and cloned into pLVX-Puro (Clontech). Virus was produced and cells transduced according to the manufacturer's recommended protocols. Stably expressing cells were selected using puromycin. Clones were not generated – all experiments were on mixed populations. Transient transfections were achieved using Lipofectamine 2000 (Invitrogen, Paisley, UK) according to the manufacturer's instructions). Truncation mutants were generated using PCR and verified by DNA sequencing (MWG Eurofins).
RNA interference
Transfection of siRNA duplexes was done using a modified calcium phosphate method as described previously38. siRNA duplexes were synthesized by Eurofins/MWG Biotech with the following sequences with the exception of the two from Ambion as indicated. GL2 5′-CGU ACG CGG AAU ACU UCG A-3′; lamin A/C: 5′-CUG GAC UUC CAG AAG AAC A-3′; Sec16A UTR: 5′-GCA GCU CUG GAA CUU AGU A-3′; Sec16B #1∶5′-UGG AGG UUA UUC UUA AUG A-3′; Sec16B #2∶5′-AUC UUC GAG UAG UGU CAG A-3′; Sec16B “G”: 5′-UAG UGA AUU UCU CCA CGA UCU GCG C-3′ (as described in21); Sec16B UTR∶5′-UGA GUG CCU GAU AUU GUU A-3′; Sec16B 1: 5′-CGA AGG UGG UUA UCG CAA U-3′; ( #s40166, Ambion Applied Biosystems, UK); Sec16B 2: 5′-GGA GGA AUA UGC UUA UGG A-3′; (#s40165, Ambion Applied Biosystems, UK).
Q-PCR
The primers for the Q-PCR of Sec16A and Sec16B were designed so that a Q-PCR product of ∼ 250 bp, ideally spanning two exons, is amplified. Primer sequences were as follows: Forward primer for Q-PCR of RNA Polymerase 2: 5′-TAT CTG CAC TGC CAA GAC TGA-3′; Reverse primer for Q-PCR of RNA Polymerase 2: 5′-CCA CAA TGC TCA TGC CTT CTT TCA-3′; Forward primer for Q-PCR of Sec16A: 5′-CAC TCC CTG GCT CTG AAC TC-3′; Reverse primer for Q-PCR of Sec16A: 5′-AAT CCT CCC TAG CCT TGA GC-3′; Forward primer for Q-PCR of Sec16B 5′-TCA GCC TGT GTC TGG AGT TG-3′; Reverse primer for Q-PCR of Sec16B: 5′-CGG TAA TCT TCG TGC CAG AT-3′. Q-PCR was carried out as described in39.
Protein interactions and mass spectrometry
Cells were lysed and GFP-tagged proteins isolated using GFP-Trap® beads (Chromotek) according the manufacturers protocol. For a large scale immunoprecipitation, HeLa GFP-Sec16B cells were plated on two 15 cm tissue culture dishes, the cells lysed in 300 µl lysis buffer and the lysate of the two plates pooled together. As a negative control, HeLa cells transfected with pEGFP-C2 were used. The cleared lysate was subsequently incubated for 2 h at 4°C with 30 µl of equilibrated GFP-Trap® beads to allow binding of GFP fusion protein. The beads where then washed four times 5 min with 500 µl of dilution buffer and resuspended in 150 µl 2x sample buffer. The beads were boiled for 10 min at 95°C and 20 µl of sample separated by SDS-PAGE on a 5% and 15% gel. Proteins were subsequently stained using SYPRO® Ruby and protein bands visualized using the Typhoon™ scanner 9400 (GE Healthcare). Proteins, which only bound to the protein of interest but not to the GFP control, were subsequently determined. These proteins were extracted from the gel, prepared for and analyzed by MS/MS spectrometry by Kate Heesom (Proteomics Facility, University of Bristol).
Author Contributions
A.B. performed all experiments, K.H. performed proteomics experiments and analysis, and A.B. and D.J.S. wrote the manuscript. All authors reviewed the manuscript.
Acknowledgments
The authors would like to thank the following for funding: the BBSRC and University of Bristol for a PhD studentship to AB and the MRC for a Non-Clinical Senior Research Fellowship to DJS. We are also extremely grateful to Pete Watson, Anna Townley, Jade Cheng, and Virginie Betin for their contributions to aspects of this work, and to current members of the Stephens lab for critical comments on the manuscript. We would also like to thank the Kazusa consortium, Harry Mellor, and Jon Lane for sharing reagents with us, and Francis Barr, Jeremy Henley, Giles Cory, and Jon Lane for cell lines.
References
- Bannykh S. I., Rowe T. & Balch W. E. The organization of endoplasmic reticulum export complexes. J. Cell. Biol. 135, 19–35 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palade G. Intracellular aspects of the process of protein synthesis. Science 189, 347–358 (1975). [DOI] [PubMed] [Google Scholar]
- Orci L. et al.. Mammalian Sec23p homologue is restricted to the endoplasmic reticulum transitional cytoplasm. Proc. Natl. Acad. Sci USA 88, 8611–8615 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budnik A. & Stephens D. J. ER exit sites—localization and control of COPII vesicle formation. FEBS Letters 583, 3796–3803 (2009). [DOI] [PubMed] [Google Scholar]
- Stephens D. J., Lin-Marq N., Pagano A., Pepperkok R. & Paccaud J. P. COPI-coated ER-to-Golgi transport complexes segregate from COPII in close proximity to ER exit sites. J. Cell. Sci. 113, 2177–2185 (2000). [DOI] [PubMed] [Google Scholar]
- Hammond A. T. & Glick B. S. Dynamics of transitional endoplasmic reticulum sites in vertebrate cells. Mol. Biol. Cell 11, 3013–3030 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novick P., Field C. & Schekman R. Identification of 23 complementation groups required for post-translational events in the yeast secretory pathway.[see comment]. Cell 21, 205–215 (1980). [DOI] [PubMed] [Google Scholar]
- Kaiser C. A. & Schekman R. Distinct sets of SEC genes govern transport vesicle formation and fusion early in the secretory pathway. Cell 61, 723–733 (1990). [DOI] [PubMed] [Google Scholar]
- Espenshade P., Gimeno R. E., Holzmacher E., Teung P. & Kaiser C. A. Yeast SEC16 gene encodes a multidomain vesicle coat protein that interacts with Sec23p. J Cell Biol 131, 311–324 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaywitz D. A., Espenshade P. J., Gimeno R. E. & Kaiser C. A. COPII subunit interactions in the assembly of the vesicle coat. J. Biol. Chem. 272, 25413–25416 (1997). [DOI] [PubMed] [Google Scholar]
- Gimeno R. E., Espenshade P. & Kaiser C. A. COPII coat subunit interactions: Sec24p and Sec23p bind to adjacent regions of Sec16p. Mol. Biol. Cell 7, 1815–1823 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supek F., Madden D. T., Hamamoto S., Orci L. & Schekman R. Sec16p potentiates the action of COPII proteins to bud transport vesicles. J. Cell. Biol. 158, 1029–1038 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson P., Townley A. K., Koka P., Palmer K. J. & Stephens D. J. Sec16 defines endoplasmic reticulum exit sites and is required for secretory cargo export in mammalian cells. Traffic 7, 1678–1687 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iinuma T. et al.. Mammalian Sec16/p250 plays a role in membrane traffic from the endoplasmic reticulum. J. Biol. Chem. 282, 17632–17639 (2007). [DOI] [PubMed] [Google Scholar]
- Bhattacharyya D. & Glick B. S. Two mammalian Sec16 homologues have nonredundant functions in endoplasmic reticulum (ER) export and transitional ER organization. Mol. Biol. Cell 18, 839–849 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes H. et al.. Organisation of human ER-exit sites: requirements for the localisation of Sec16 to transitional ER. J. Cell Sci. 122, 2924–2934 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forster R. et al.. Secretory cargo regulates the turnover of COPII subunits at single ER exit sites. Curr. Biol. 16, 173–179 (2006). [DOI] [PubMed] [Google Scholar]
- Connerly P. L. et al.. Sec16 is a determinant of transitional ER organization. Curr. Biol. 15, 1439–1447 (2005). [DOI] [PubMed] [Google Scholar]
- Ivan V. et al.. Drosophila Sec16 mediates the biogenesis of tER sites upstream of Sar1 through an arginine-rich motif. Mol. Biol. Cell 19, 4352–4365 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi M. Novel protein RGPR-p117: its role as the regucalcin gene transcription factor. Mol. Cell. Biochem. 327, 53–63 (2009). [DOI] [PubMed] [Google Scholar]
- Bhattacharyya D. & Glick B. S. Two mammalian Sec16 homologues have nonredundant functions in endoplasmic reticulum (ER) export and transitional ER organization. Mol. Biol. Cell 18, 839–849 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witte K. et al.. TFG-1 function in protein secretion and oncogenesis. Nat. Cell Biol. 13, 550–558 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes H. et al.. Organisation of human ER-exit sites: requirements for the localisation of Sec16 to transitional ER. J. Cell Sci. 122, 2924–2934 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens D. J., Lin-Marq N., Pagano A., Pepperkok R. & Paccaud J. P. COPI-coated ER-to-Golgi transport complexes segregate from COPII in close proximity to ER exit sites. J. Cell Sci. 113, 2177–2185 (2000). [DOI] [PubMed] [Google Scholar]
- Morris G. E. The Cajal body. Biochim. Biophys. Acta – Mol. Cell Res. 1783, 2108–2115 (2008). [DOI] [PubMed] [Google Scholar]
- Lallemand-Breitenbach V. & de The H. PML nuclear bodies. Cold Spring Harb. Persp. Biol. 2, a000661 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivan V. et al.. Drosophila Sec16 mediates the biogenesis of tER sites upstream of Sar1 through an arginine-rich motif. Mol. Biol. Cell 19, 4352–4365 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whittle J. R. & Schwartz T. U. Structure of the Sec13-Sec16 edge element, a template for assembly of the COPII vesicle coat. J. Cell Biol. 190, 347–361 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leksa N. C. & Schwartz T. U. Membrane-coating lattice scaffolds in the nuclear pore and vesicle coats: Commonalities, differences, challenges. Nucleus 1, 314–318 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappell T. G. et al.. Uncoating ATPase is a member of the 70 kilodalton family of stress proteins. Cell 45, 3–13 (1986). [DOI] [PubMed] [Google Scholar]
- Mayer M. P. & Bukau B. Hsp70 chaperones: cellular functions and molecular mechanism. Cell. Mol. Life Sci. 62, 670–684 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinkle-Mulcahy L. et al.. Identifying specific protein interaction partners using quantitative mass spectrometry and bead proteomes. J. Cell Biol 183, 223–239 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi M. Role of regucalcin in calcium signaling. Life Sciences 66, 1769–1780 (2000). [DOI] [PubMed] [Google Scholar]
- Yamaguchi M., Misawa H. & Ma Z. J. Novel protein RGPR-p117: the gene expression in physiologic state and the binding activity to regucalcin gene promoter region in rat liver. J. Cell Biochem. 88, 1092–1100 (2003). [DOI] [PubMed] [Google Scholar]
- Weissman J. T., Plutner H. & Balch W. E. The mammalian guanine nucleotide exchange factor mSec12 is essential for activation of the Sar1 GTPase directing endoplasmic reticulum export. Traffic 2, 465–475 (2001). [DOI] [PubMed] [Google Scholar]
- Fliss M. S., Hinkle P. M. & Bancroft C. Expression cloning and characterization of PREB (prolactin regulatory element binding), a novel WD motif DNA-binding protein with a capacity to regulate prolactin promoter activity. Mol. Endocrinol. 13, 644–657 (1999). [DOI] [PubMed] [Google Scholar]
- Townley A. K. et al.. Efficient coupling of Sec23-Sec24 to Sec13-Sec31 drives COPII-dependent collagen secretion and is essential for normal craniofacial development. J. Cell Sci. 121, 3025–3034 (2008). [DOI] [PubMed] [Google Scholar]
- Watson P. & Stephens D. J. Microtubule plus-end loading of p150(Glued) is mediated by EB1 and CLIP-170 but is not required for intracellular membrane traffic in mammalian cells. J. Cell Sci. 119, 2758–2767 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer K. J., Hughes H. & Stephens D. J. Specificity of cytoplasmic dynein subunits in discrete membrane-trafficking steps. Mol. Biol. Cell 20, 2885–2899 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]