

Abstract
Thalamic and extrathalamic nicotinic α4β2 receptors found in the brain have been implicated in Alzheimer’s disease, Parkinson’s disease, substance abuse and other disorders. We report here the development of 3-(2-(S)-azetidinylmethoxy)-5-(3′-fluoropropyl)pyridine (nifzetidine) as a new putative high affinity antagonist for nicotinic α4β2 receptors. Nifzetidine in rat brain homogenate assays containing α4β2 sites labeled with 3H-cytisine exhibited a binding affinity, Ki = 0.67 nM. The fluorine-18 analog, 3-(2-(S)-azetidinylmethoxy)-5-(3′-18F-fluoropropyl)pyridine (18F-nifzetidine) was synthesized in 20–40% yield and apparent specific activity was estimated to be above 2 Ci/μmol. Rat brain slices indicated selective binding of 18F-nifzetidine to thalamus, subiculum, striata, cortex and other regions consistent with α4β2 receptor distribution. This selective binding was displaced >85% by 150 μM nicotine. PET imaging studies of 18F-nifzetidine in anesthetized rhesus monkey showed slow uptake in the various brain regions. Retention of 18F-nifzetidine was maximal in the thalamus and lateral geniculate followed by regions of the temporal and frontal cortex. Cerebellum showed the least uptake. Thalamus to cerebellum ratio was about 2.3 at 180 min post-injection and continued to rise. 18F-Nifzetidine shows promise as a new PET imaging agent for α4β2 nAChR. However, the slow kinetics suggests a need for >3 hr PET studies for quantitative studies of the α4β2 nAChRs.
Keywords: Nicotine, Autoradiography, Monkey PET, Fluorine-18
1. Introduction
Development of imaging agents for the α4β2 neuronal nicotinic acetylcholine receptor (nAChR) has attracted much attention (1). These receptors consist of five subunits, each of which contains four transmembrane domains. The α4β2 nAChR subtype has high affinity for acetylcholine and nicotine and has been implicated in a number of disorders such as Alzheimer’s disease, Parkinson’s disease, schizophrenia and tobacco dependence (2–5).
A number of imaging agents with 3-pyridyl ethers have been investigated in an effort to optimize in vivo imaging properties (6–8). Human studies have been carried out with 2-18FA85380, 6-18FA85380 and 5-123I-A85380 for imaging α4β2 nAChRs (9–12). However, due to the slow binding kinetics of the three pyridylethers currently in human use, several efforts have been made to improve and hasten the binding kinetics (1).
Our goal has been to develop antagonists and agonists for imaging α4β2 nAChRs. The moderate affinity helps in accelerating the in vivo binding kinetics as was observed with the agonist 18F-nifene, 2 (13). Replacing halogen or hydrogen at the meta-position in 3-pyridyl ethers with an alkyl group such as propyl or butyl group, reduces agonist character and increases the antagonist character (14,15). On the basis of this finding we developed nifrolidine, 3 as an antagonist based PET imaging agent (16). Initially, we had chosen to develop a moderate affinity compound by using the pyrrolidine ring in 18F-nifrolidine, rather than the azetidine ring (16).
In an effort to increase target to nontarget ratios of nifrolidine, we considered replacing the pyrrolidine ring in nifrolidine with an azetidine ring. Azetidine rings have higher affinity at the α4β2 receptor site as evidenced by 2-18FA85380, 6-18FA85380 and 5-123I-A85380. We anticipated that inclusion of the azetidine ring would result in a more potent compound than nifrolidine.
We have now introduced a 3′-fluoropropyl group at the meta-position in 3-pyridyl ether linked to the azetidine ring. We report here the synthesis of 3-(2-(S)-azetidinylmethoxy)-5-(3′-fluoropropyl)pyridine (abbreviated name: nifzetidine, 4), in vitro binding affinity at the nAChRs, radiolabeling with fluorine-18 to provide 3-(2-(S)-azetidinylmethoxy)-5-(3′-18F-fluoropropyl)pyridine (18F-nifzetidine), in vitro autoradiographic studies in rat brain slices and PET studies in rhesus monkeys with 18F-nifzetidine.
2. Materials and Methods
All chemicals (except 1-azetidinecarboxylic acid, 2-(hydroxymethyl)-1,1-dimethylethyl ester (S), 5, ChemPacific Corporation, Baltimore, MD) and solvents were obtained from Aldrich Chemical Co. and Fisher Scientific. Electrospray mass spectra were obtained on a Model 7250 mass spectrometer (Micromass LCT). Proton NMR spectra were recorded on Bruker Instruments OMEGA 500- MHz. High specific activity 18F-fluoride was produced in the MC-17 cyclotron or the CTI RDS-112 cyclotron using oxygen-18 enriched water in a 18O(p, n)18F nuclear reaction. All chemical reactions used high specific activity 18F-fluoride. Chromatographic separations were carried out on semi-preparative reverse-phase columns using the Gilson high performance liquid chromatography (HPLC) systems. Flourine-18 radioactivity was counted in a Capintec dose calibrator while low level counting was carried out in a well-counter (Cobra quantum, Packard Instruments Co., Boston, MA). Radioactive thin layer chromatograms were obtained by scanning in a Bioscan system 200 Imaging scanner (Bioscan, Inc., Washington, DC). Rat brain slices were obtained on a Leica 1850 cryotome. Fluorine-18 autoradiographic studies were carried out by exposing tissue samples on storage phosphor screens. The apposed phosphor screens were read by Cyclone Storage Phosphor System (Packard Instruments Co., Boston, MA). Monkey PET were carried out using a high-resolution ECAT HR+ scanner. All animals were approved by the Institutional Animal Care and Use Committee of University of California-Irvine and Wright State University, Dayton, Ohio.
2.1. 3-Bromo-5-(1-tert-butoxycarbonyl-2-(S)-azetidinylmethoxy)pyridine (7)
In THF (12 mL) solution of 5 (0.98 g, 5.28 mmol), 3-bromo-5-hydroxypyridine 6 (0.92 g, 5.36 mmol) and triphenylphosphine (1.71 g, 6.5 mmol) under nitrogen was cooled to 0°C and treated drop wise with diisopropyl azodicarboxylate (1.27 mL, 6.5 mmol) in THF (2 mL). The reaction mixture was stirred for 1 h at 0°C, and then allowed to warm to room temperature and continued stirring overnight. The mixture was concentrated in vacuo; the residue was treated with pentane/EtOAc (200 mL, 9:1) and stirred at 0°C until crystalline solid was obtained. The reaction mixture was filtered and the white solid was washed with pentane/EtOAc (9:1), (2 × 10 mL). The combined filtrate was concentrated in vacuo and the residue was purified by flash chromatography on silica gel (20×300 mm), eluting with 40% EtOAc in hexane. Column fractions were combined and concentrated in vacuo to give 3-bromo-5-(1-tert-butoxycarbonyl-2-(S)-azetidinylmethoxy)pyridine 7 (1.04 g, 58%) as a pale yellow gummy oil. NMR (500 MHz; CDCl3) δ 8.28 (m, 2H), 7.43 (dd, J = 1.8, 2.6 Hz, 1H), 4.33 (m, 1H), 4.13 (m, 2H), 3.89 (m, 2H), 2.33 (m, 2H), 1.42 (s, 9H). MS, m/z 343, 345 (35%, [M + H]+), 365, 367 (80%, [M + Na]+).
2.2. 3-Allyl-5-(1-tert-butoxycarbonyl-2-(S)-azetidinylmethoxy)pyridine (8)
3-Bromo-5-(1-tert-butoxycarbonyl-2-(S)-azetidinylmethoxy)pyridine 7 (0.80 g, 2.33 mmol) in anhydrous toluene (25 mL) was treated with tetrakis(triphenylphosphine)palladium (0) (100 mg, 0.08 mmol) followed by addition of allyltri-n-butyltin (0.92 g, 2.77 mmol). The reaction mixture was refluxed under nitrogen atmosphere for 20 hours. The reaction mixture was then cooled to room temperature and filtered. The filtrate was evaporated to dryness. The residue was purified by silica column chromatography (25 × 300 mm) eluting with 40% EtOAc in hexane to furnish 3-allyl-5-(1-tert-butoxycarbonyl-2-(S)-azetidinylmethoxy)pyridine 8 (0.49 g, 70%) as a colorless oil. NMR (500 MHz; CDCl3) δ 8.19 (d, J = 2.6 Hz, 1H), 8.08 (s, 1H), 7.07 (s, 1H), 5.94 (m, 1H, olefinic H), 5.13 (m, 2H, olefinic H), 4.31 (m, 1H), 4.12 (dd, J = 9.9, 2.8 Hz, 2H), 3.90 (m, 2H), 3.37 (d, J = 6.5 Hz, 2H), 2.32 (m, 2H), 1.42 (s, 9H). MS, m/z 305 (100%, [M + H]+), 327 (80%, [M + Na]+).
2.3. 3-(1-tert-Butoxycarbonyl-2-(S)-azetidinylmethoxy)-5-(3′-hydroxypropyl)pyridine (9)
The olefin 8 (0.47 g, 1.54 mmol) in THF (5 mL) was treated with borane-tetrahydrofuran complex (1.5 M, 3.5 mL) by drop wise addition at 0°C and stirring under nitrogen atmosphere. The reaction mixture was then stirred at room temperature overnight and was then treated with NaOH (3 N, 3 mL) followed by addition of 30% H2O2 (200 μL) at 0°C. Subsequently, this mixture was stirred at room temperature for 12 hours and then concentrated to dryness. The residue was poured into saturated ammonium chloride solution and extracted with ethyl acetate. The organic layer was dried over MgSO4 and the solvent evaporated in vacuo. The residue was purified by preparative TLC (15% MeOH in DCM) to furnish alcohol 9 (0.33 g, 67% yield). The alcohol was obtained in 70% yield. NMR (500 MHz; CDCl3) δ 8.29 (s, 1H), 8.15 (s, 1H), 7.45 (s, 1H), 4.42 (bm, 1H), 4.21 (m, 2H), 3.93 (m, 2H), 3.73 (t, J = 6.0 Hz, 2H), 2.82 (t, J = 7.0 Hz, 2H), 2.42 (m, 2H), 1.95-1.92 (m, 2H), 1.47 (s, 9H). MS, m/z 323 (100%, [M + H]+), 345 (75%, [M + Na]+).
2.4. 3-(1-tert-Butoxycarbonyl-2-(S)-azetidinylmethoxy)-5-(3′(4-methylbenzenesulfonyloxy)-propyl)pyridine (10)
To a solution of alcohol 9 (0.30 g, 0.93 mmol) and triethylamine (0.40 mL, 2.80 mmol) in anhydrous dichloromethane (3.0 mL), p-toluenesulfonyl chloride (0.186 g, 0.97 mmol) was added and the reaction mixture was stirred at room temperature for 18 hrs. The reaction mixture was then diluted with water and extracted with dichloromethane. The dichloromethane layer was washed with water, dried and concentrated. The residue was chromatographed over silica gel and eluted with ethyl acetate and hexane (1:1) gave tosylate precursor 10 (88 mg, 20% yield). (500 MHz; CDCl3) δ 8.18 (s, 1H), 7.99 (s, 1H), 7.78 (d, J = 8.0 Hz, 2H), 7.35 (d, J = 8.0 Hz, 2H), 7.34 (m, 1H), 4.31 (m, 1H), 4.13 (m, 2H), 4.04 (t, J = 6 Hz, 2H), 3.89 (m, 2H), 2.66 (t, J = 7.7 Hz, 2H), 2.45 (s, 3H), 2.34 (m, 2H), 1.98–1.89 (m, 2H), 1.41 (s, 9H). MS, m/z 477 (25%, [M + H]+), 499 (65%, [M + Na]+).
2.5. 3-(1-tert-Butoxycarbonyl-2-(S)-azetidinylmethoxy)-5-(3′-fluoropropyl)pyridine (11; Method 1)
The tosylate 10 (0.23 g, 0.48 mmol) was dissolved in THF (2 mL). The solution was treated with tetrabutylammonium fluoride (TBAF), 1.0M solution in THF (1 mL, 1 mmol) and heated at 60 °C for 2 hr and cooled. The reaction mixture was concentrated to dryness in vacuo and the residue was then purified by flash column chromatography (silica gel DCM/MeOH, 95:5) providing 11 (95 mg, 60% yield) as a colorless oil. (500 MHz; CDCl3) δ 8.20 (d, J = 2.4 Hz, 1H), 8.10 (s, 1H), 7.11 (s, 1H), 4.50 (t of d, J = 47 Hz, 5.7 Hz, CH2-F, 2H), 4.32 (brm, 1H), 4.14 (dd, J = 9 Hz, 2.8 Hz, 2H), 3.89 (m, 2H), 2.75 (t, J = 7.5 Hz, 2H), 2.35 (m, 2H), 2.01 (m, 2H), 1.41 (s, 9H). MS, m/z 325 (38%, [M + H]+), 347 (100%, [M + Na]+).
2.6. 3-(1-tert-Butoxycarbonyl-2-(S)-azetidinylmethoxy)-5-(3′-fluoropropyl)pyridine (11; Method 2)
To a solution of N-BOC alcohol 9 (0.19 g, 0.60 mmol) and triethylamine (250 μL) in dichloromethane (1 mL) was added drop wise a solution of DAST (75 μL) in dicloromethane (0.5 mL) at 0°C under nitrogen atmosphere. The reaction mixture was warmed to ambient temperature and kept stirring at room temperature for 16 hrs. The reaction mixture was neutralized with saturated sodium bicarbonate solution. The aqueous phase was extracted with dichloromethane, and the extracts were washed with water, dried (anhydrous magnesium sulfate), and evaporated to dryness. Column chromatography of the residue over silica gel and elution with dichloromethane:methanol (95:5) gave N-BOC fluoro compound 11 (52 mg, 26%). (500 MHz; CDCl3) δ 8.22 (d, J = 2.4 Hz, 1H), 8.14 (s, 1H), 7.11 (s, 1H), 4.56 (t of d, J = 47 Hz, 5.7 Hz, CH2-F, 2H), 4.35 (brm, 1H), 4.17 (dd, J = 9 Hz, 2.8 Hz, 2H), 3.88 (m, 2H), 2.75 (t, J = 7.5 Hz, 2H), 2.34 (m, 2H), 2.04 (m, 2H), 1.42 (s, 9H). MS, m/z 325 (38%, [M + H]+), 347 (100%, [M + Na]+).
2.7. 3-(2-(S)-Azetidinylmethoxy)-5-(3′-fluoropropyl)pyridine (4)
Deprotection of N-BOC fluorinated derivative 11 was carried out by treatment with trifluoroacetic acid (TFA). The N-BOC derivative 11 (84 mg, 0.25 mmol) was dissolved in dichloromethane (4 mL) and the solution was cooled at 0 °C. The reaction solution was treated with TFA (0.6 mL) with stirring. The reaction solution was stirred at 0 °C for one hour followed by room temperature for 30 min. The reaction mixture was concentrated to dryness. The residue neutralized with 1N NaOH to pH 11 and the aqueous layer was extracted with dichloromethane, dried (MgSO4), filtered, and concentrated to give crude nifzetidine 4. The product 4 was dissolved in diethyl ether (2 mL) and cooled to 0 °C. The solution was treated with 1M HCl in ether (2 mL) and the temperature was brought into room temperature. The solution was concentrated to dryness. The residue was triturated with diethyl ether to give light brown solid compound 4.2HCl was obtained (28 mg, 50%). NMR (CDCl3), 500 MHz) δ ppm: 8.21 (d, J = 2.7 Hz, 1H, ArH), 8.17 (d, J = 2.69 Hz, 1H, ArH), 7.17 (s, 1H ArH), 4.45 (t of d, J = 47 Hz, 5.8 Hz, CH2-F, 2H), 4.27 (m, 1H), 4.03 (m, 2H), 3.67 (m, 2H), 2.72 (t, J = 7.5 Hz, 2H), 2.59 (m, 2H), 1.98 (m, 2H). MS, m/z 225 (100%, [M + H]+), 247 (5%, [M + Na]+).
2.8. In Vitro Binding Affinity
Rat brain homogenate assays using 3H-cytisine were carried out to measure binding affinity of the compounds to α4β2 receptors using previously described procedures (17). The cerebrum of male Sprague-Dawley rats was isolated and homogenized using Tekmar tissumizer (15 secs, half maxima speed) in the incubation buffer 1:100 (wt:vol). This was then centrifuged at 40,000g for 10 mins and the supernatant discarded. The pellet was then resuspended in the same volume of buffer, homogenized and centrifuged again at 40,000g for 10 mins. The supernatant was then removed and the final pellet was taken up in the incubation buffer described below to a concentration of 120 – 150 mg/mL of tissue (the protein concentration in these homogenate equals 60 μg/mg tissue). Binding assays were carried out in 50 mM Tris buffer at pH 7.0 containing 120 mM NaCl, 5 mM KCl, 1mM MgCl2 and 2.5 mM CaCl2 in the presence of 1 nM 3H-cytisine at 2 °C for 75 min incubation. The total assay volume was 0.25 mL. Different concentrations of the assay compound, nifzetidine in 0.025 mL (final concentration range 10−10 to 10−3M) was taken in 0.1 mL of buffer along with 0.025 mL of 1 nM 3H-cytisine. Nonspecific binding was measured using 300 μM nicotine. To start the incubation, 0.1 mL of the rat brain homogenate was added into each assay tube. The incubation was terminated by rapid vacuum filtration through Whatman GF/C filter paper (presoaked in 0.1% polyethyleneimine for 60 mins) using Brandel tissue harvester. The filters were washed three times with cold buffer and the filters were suspended in 10 mL of Bio-Safe II scintillation fluid and counted for 10 mins in the scintillation counter. Data was analyzed using following procedure: (a) the non-specific binding of 3H-cytisine was subtracted for all samples; (b) the specific binding was normalized to 100% (no competitive ligand) and (c) the binding isotherms were fit to the Hill equation (KELL BioSoft software (v 6), Cambridge, U.K.). The dissociation constant, Kd for the 3H-cytisine to nicotine α4β2 receptor of 0.59 nM was used (18).
2.9. Radiosynthesis
The radiosynthesis of 18F-nifzetidine was conducted in the chemistry-processing computer unit (CPCU). Hydrogen 18F-fluoride in H218O from the MC-17 cyclotron was passed through light QMA sep-pak (Waters Corp.), preconditioned with 3 mL of potassium carbonate, 140 mg/mL, followed by 3 mL of anhydrous acetonitrile. The trapped 18F in QMA was eluted with 1 mL of Kryptofix 2.2.2 (Aldrich)/potassium carbonate solution (stock solution of 360 mg Kryptofix and 75 mg potassium carbonate in 24 mL acetonitrile and 1 mL water) and transferred to the reaction vessel in the CPCU. The SYNTH1 program was used for the synthesis that involved an initial drying step of the 18F-fluoride, Kryptofix 2.2.2., and K2CO3 mixture at 120°C for 10 min. The 18F solution was further dried with acetonitrile (2 mL) at 120 °C for 7 min. Dried 18F was treated with the precursor 10 (2 mg) in acetonitrile (0.5 mL). This solution was heated at 96 °C for 30 min and cooled.
Radiosynthesis of 18F-nifzetidine took two steps. As described above, the first step was the nuclophilic displacement of tosylate group by 18F ion. The crude product N-BOC-18F-nifzetidine was transferred out of CPCU using dichloromethane (5 mL). The dichloromethane containing N-BOC-18F-nifzetidine was evaporated in vacuo for the deprotection step. Purity of an aliquot of this intermediate N-BOC-18F-nifzetidine was performed by HPLC using an Alltech C18 column (10 μm, 250×10 mm) and UV detector (254 nm), mobile phase: 60% acetonitrile-40% 0.1 M ammonium formate, flow rate 2.5 mL/min, r.t. = 25 min (Fig-3).
Figure-3.

Reaction scheme showing the radiosynthesis of 18F-N-BOC-nifzetidine and 18F-nifzetidine. (A) Radioactivity and UV trace at 254 nm of HPLC analysis of 18F-N-BOC-nifezetidine intermediate using C18 reverse-phase semi-preparative column eluted with 60% acetonitrile- 40% 0.1M ammonium formate at a flow rate of 2.5 mL/min. Retention of 18F- N-BOC-nifzetidine was found to be 24.5 min. (B). Radioactivity and UV trace at 254 nm of HPLC purification of 18F-nifzetidine after two-step radiosynthesis using C18 reverse-phase semi-preparative column eluted with 60% acetonitrile- 40% 0.1M ammonium formate at a flow rate of 2.5 mL/min. Retention of 18F-nifzetidine was found to be 17.5 min.
The above N-BOC-18F-nifzetidine intermediate was taken in dichloromethane (1 mL) and trifluroacetic acid (0.2 mL). The solution was heated at 80°C for 30 min and evaporated to dryness. The residue was neutralized with 10% NaHCO3 solution to pH 6–7. Semipreparative HPLC purification was performed using an Alltech C18 column (10 μm, 250×10 mm) and UV detector (254 nm), mobile phase: 60% acetonitrile-40% 0.1 M ammonium formate, flow rate 2.5 mL/min, r.t. = 17 min. The collected fraction was taken to near dryness in vacuo. The final formulation was carried out using approx. 5 mL of saline (0.9% NaCl INJ) followed by filtration through a membrane filter (0.22 μm) into a sterile dose vial.
2.10. In Vitro Autoradiographic Studies
Horizontal brain slices (20 μm thick) were obtained from male Sprague-Dawley rats. Sets of brain slices were preincubated in buffer (50 mmol/L Tris HCl containing 120 mmol/L NaCl, 5mmol/L KCl, 2.5 mmol/L CaCl2. 1 mmol/L MgCl2, pH 7.4) for 10 min. Subsequently, the slices were incubated with 18F-nifzetidine (4 μCi/cc) at 37°C for 60 min. Nonspecific binding was measured in the presence of 15 μmol and 150 μmol of nicotine. After incubation, slides were washed twice (1 min each) with ice-cold Tris HCl buffer, pH 7.4, followed by a quick rinse in cold deionized water. The slides were then air dried and apposed to phosphor screens overnight and read by the Cyclone Phosphor Imaging System (Packard Instruments Co). The amount of bound 18F-nifzetidine in the autoradiograms was evaluated in various brain regions (as digital lights units (DLU]/mm2) using the OptiQuant acquisition and analysis program (Packard Instruments Co.
2.11. PET Imaging
Rhesus monkeys (10 kg) were anesthetized using ketamine (10 mg/kg) and xylazine (0.5 mg/kg) and was subsequently maintained on 1–1.5% isoflurane. Two intravenous catheters were placed, one on each arm, for the purpose of administration of 18F-nifzetidine and for obtaining blood samples during the study. The head of the animal was placed in the gantry of an ECAT EXACT HR+ PET scanner and positioned in place with adhesive tape as described previously (16). A transmission scan using a 68Ge/68Ga rod source was acquired before administration of 18F-nifzetidine to correct for tissue attenuation of the coincident radiation. A dynamic sequence of scans for a total of approximately 180 min was acquired in the 3- dimensional mode immediately after administering approximately 3.5–4 mCi (mass injected, <0.2 μg of nifzetidine) of 18F-nifzetidine intravenously. Data in the final form were expressed in units of the percentage injected dose per milliliter (%ID/ml) or kilobecquerels per milliliter (kBq/mL). Areas showing maximal 18F-nifzetidine binding in the mediodorsal thalamus, ventrolateral thalamus, temporal cortex, occipital cortex, frontal cortex, and cerebellum were delineated in the images. Time-activity curves were obtained for all these regions.
3. Results
3.1. Chemistry
We followed similar chemistry reported previously for nifrolidine starting from 3,5-dibromopyridine (16). The synthesis route to prepare nifzetidine 4 is shown in Fig-2. Starting with 3-bromo-5-methoxypyridine (available from Aldrich Chemical Co.), 3-bromo-5-pyridinol 6 was prepared in 80% yield by refluxing with 48% hydrogen bromide (16). The 3-bromo-5-pyridyl ether compound 7 was synthesized in 58% yield by etherification of 6 with 1-azetidinecarboxylic acid, 2-(hydroxymethyl)-1,1-dimethylethyl ester (S) 5 under Mitsunobu conditions (19). The coupling between 5 and 6 took place in the presence of triphenylphosphine and diisopropyl azodicarboxylate (DIAD). The DIAD is more stable and lower in cost compared to diethyl azodicarboxylate (DEAD). The hydrated DIAD adduct can be easily eliminated as a crystalline salt, simplifying isolation of the product from the reaction mixture. The next step was how to introduce 3′-fluoropropyl group at 5 positions of 3-pyridyl ethers. The bromo compound 7 was heated under reflux with allyltributyltin in toluene in the presence of catalytic amount of tetrakis(triphenylphosphine)palladium (20) catalyst; the reaction went smoothly to give allylpyridyl ether derivative 8 in 70% yield. The olefin 8 was treated with BH3.THF and subsequently oxidized with NaOH/H2O2 to furnish the alcohol in 35% yield. We have improved the yield of alcohol 9 from 35% to 67% by stirring the reaction mixture for longer time (12 hrs) before and after adding NaOH/H2O2. The key intermediate alcohol 9 was reacted with p-toluenesulfonyl chloride in the presence of pyridine gave a complex mixture. On the other hand, with triethylamine the reaction was cleaner and gave pure tosylate labeling precursor 10 in 20–30% isolated yield. Synthesis of nifzetidine was carried out either from the alcohol or tosylate precursor. Reaction of alcohol 9 with diethylaminosulfur trifluoride (21) (DAST) was investigated, but the N-BOC-fluoride 11 was produced only as a minor component of a complex mixture. Alternatively, the compound 11 was prepared in good yield from 10 by the reaction with TBAF.THF (22) suggesting it to be a superior fluorination approach than DAST for these derivatives. Removal of the N-BOC group was achieved by treating with TFA at room temperature to provide 4 in 50% yield. Final product 4, nifzetidine, was used as a hydrochloride salt for in vitro binding assays. The tosylate precursor 10 and salt of Nifzetidine.2HCl 4 were stored at 0 °C to −20 °C.
Figure-2.

Synthesis scheme for 3-(2-(S)-Azetidinylmethoxy)-5-(3′-fluoropropyl)pyridine, 4 and labeling tosylate precursor, 10.
3.2. In Vitro Binding Affinity
In vitro binding affinity of nifzetidine to nicotine α4β2 receptor was determined using 3H-cytisine in rat brain homogenate. The agonist 3H-cytisine binds to α4β2 receptor sub-type and displacement of 3H-cytisine binding in the rat brain homogenate suggest that the nifzetidine does indeed have the specificity to the α4β2 receptor sub-type. Nifzetidine bound to single binding site with a Ki value of 0.67 nM.
3.3. Radiochemistry
Our initial validation studies confirmed N-BOC-18F-nifzetidine to be the major radiochemical product (Fig-3A). The final radiolabeled product 18F-nifzetidine was obtained in >99% purity, assessed by HPLC. Radiochemical yields of 18F-nifzetidine were 20–40% range decay corrected, and specific was approx. 2000 Ci/mmol. For routine studies, to reduce the production time only one final step HPLC purification (Fig-3B) was done after completion of N-BOC deprotection and the radiosynthesis took approximately 2 hrs.
3.4. In Vitro Autoradiographic Studies
As seen in Fig-4A, in vitro autoradiography in horizontal rat brain slices indicated selective high binding of 18F-nifzetidine in the thalamus (TH) as well as extrathalamus regions such as cortex (CO), striata (STR), subiculum (SUB), cerebellum (CB) and other regions consistent with α4β2 receptor distribution. The anteroventral thalamic nucleus (AVT) exhibited the highest amount of binding. Subiculum also exhibited significant amounts of 18F-nifzetidine binding, which is known to contain α4β2 receptors sites. Cerebellum showed some binding and was used as a reference region. Displacement experiments were carried out with different concentrations of nicotine. Nicotine at 15 μM ( Fig-4B) was able to partially displace the selective binding in various brain regions while 150 μM nicotine (Fig-4C) in these brain regions displaced >85% of 18F-nifzetidine (Fig-4D). The ratios of various brain regions with respect cerebellum were: TH/CER = 12; SUB/CER = 5; STR/CER = 4; COR/CER = 3.7.
Figure-4.

In vitro autoradiographic studies of 18F-nifzetidine in rat brain slices. Binding of 18F-nifzetidine in 20 μm horizontal slices (4 μCi/cc at 37 °C. Horizontal brain slices of rat brain showing binding of 18F-nifzetidine (red = highest binding and white = lowest binding). (A). total binding (CO: frontal cortex; STR: striatum; AVT: anterioventral thalamus; TH: thalamus; SUB: subiculum; CB: cerebellum). (B). binding in the presence of 15 μM nicotine showing partial displacement of 18F-nifzetidine. (C). binding in the presence of 150 μM nicotine showing >80 displacement of 18F-nifzetidine. (D). Plot showing amount of 18F-nifzetidine binding in brain regions.
3.5. Monkey PET Imaging
After intravenous administration of 18F-nifzetidine, vital signs of the monkeys did not exhibit any unusual derivation from baseline values. PET imaging study revealed a maximal uptake of 0.01%ID/cc in thalamic regions. The results indicated a significant amount of binding in the thalamic regions in all three sections axial, sagittal and coronal planes (Fig-5a-c). Maximal brain uptake of 18F-nifzetidine was in thalamus (Fig-5f) and lateral geniculate (Fig-5h,i). Regions outside the thalamus (extrathalamic) were also observed. Frontal cortex (Fig-5c), striatum (Fig-5d), temporal cortex (Fig-5i) showed significant amounts of binding. Cerebellum showed the lowest amount of binding (Fig-5c).
Figure-5.
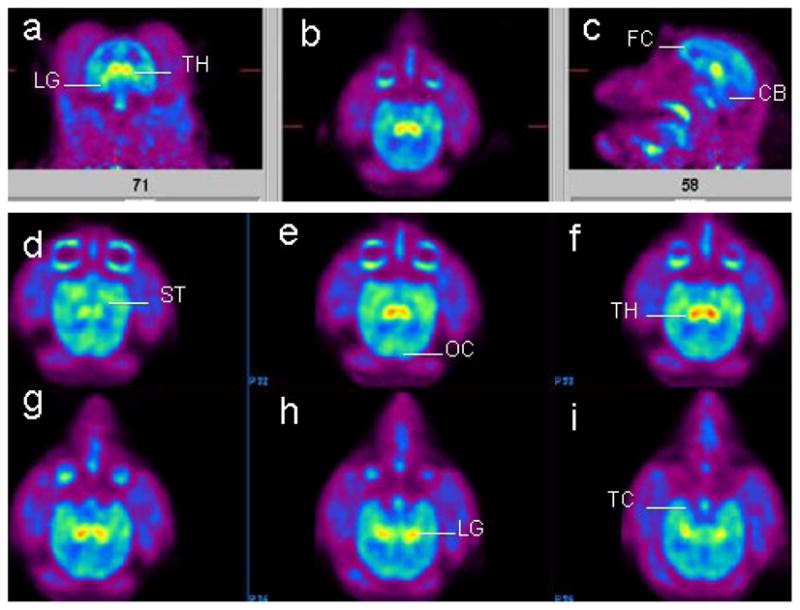
Distribution of 18F-nifzetidine in the rhesus monkey brain. Summed PET images (100–200 mins) showing binding of 18F-nifzetidine in select brain slices (a-c: orthogonal planes; d-i: horizontal planes). Brain regions include thalamus (TH), lateral geniculate (LG), frontal cortex (FC), cerebellum (CB), striatum (ST), temporal cortex (TC), occipital cortex (OC).
The 18F-nifzetidine uptake was very slow in various brain regions of rhesus monkey (Fig-6A). Highest uptake of 18F-nifzetidine was seen in the thalamus and lower uptake in cortical areas such as frontal cortex. Cerebellum had the least amount of uptake as seen in Fig-6A and was used as the reference region. A high level of specific binding was achieved at 2 hours after administration, with a thalamus to cerebellum ratio of 2.4 at 3 hrs postinjection. Ratios of 18F-nifzetidine continued to rise beyond 3 hrs in the thalamus, indicating slow kinetics. Lateral geniculate to cerebellum ratios was >2 at 3 hrs postinjection. Ratios of striatal and cortical regions with respect to cerebellum were 1.3–1.6 (Fig-6B).
Figure-6.

(A). Time-activity curves of the binding of 18F-nifzetidine in select areas of the monkey brain corresponding to the regions identified in Fig-5 (AMT/VLT: anteriomedial thalamus/ventrolateral thalamus; CG: cingulate gyrus; subi: subiculum) (B). Ratio plot of brain regions to cerebellum.
4. Discussion
A series of 3-pyridyl ethers with subnanomolar affinity for the major α4β2 subtype of central nAChRs were synthesized by Abbott laboratories. The 3-pyridyl ethers selectively bind to α4β2 receptors unlike epibatidine analogs which bind also to α3β2 and α3β4 receptors. Several agonist-based radioligands have been explored in the literature for imaging α4β2 receptors (13, 23). Here we focused our attention towards antagonist-based imaging agents with faster binding kinetics. By placing alkyl groups at 5 positions of 3-pyridyl ethers, agonist character is dramatically decreased (15,16). Based on this approach we previously developed antagonist 18F-nifrolidine (16), which exhibited reasonable binding kinetics and selectivity for α4β2 receptors. In an attempt to increase binding affinity and enhance target-to-nontarget ratios, we focused our attention towards nifzetidene which carries azetidine ring. The structure of nifzetidine is similar to that of azetidinylether halogenated analog 2-FA85380 except for an “n-3′-fluoropropyl” at 5-position and lacking the ring fluorine. We accomplished synthesis of nifzetidine by modifying the synthesis of the pyridylethers (16). We used a similar approach for the synthesis of nifzetidine using 1-azetidinecarboxylic acid, 2-(hydroxymethyl)-1,1-dimethylethyl ester (S) for the Mitsunobu coupling. We successfully introduced 3′-fluoropropyl group at 5-position via allylation of 5-bromo-3-pyridyl ether compound with an allylating agent allyltri-n-butyltin, hydroboration of the corresponding double bond and conversion of alcohol to fluoride. The corresponding alcohol was the key intermediate for making the precursor for 18F-nifzetidine. The synthetic approach for preparation of the unlabeled nifzetidine was carried out with diethylaminosulfur trifluoride (DAST) as the fluorinating agent. This was the first approach to make unlabeled nifzetidine directly from the intermediate alcohol 9. But the DAST reaction did not give satisfactory results. Finally, Nifzetidine was prepared from tosylate 10 by reacting with TBAF followed by deprotection at room temperature. The reaction yields were good to moderate. Nifzetidine was prepared as HCl salt for in vitro binding studies.
The binding affinity of nifzetidine is comparable to our previously reported nifrolidine and nifene (Table-1). The four membered ring structure in nifzetidine was expected to significantly influence the binding affinity, since azetidinyl derivatives (e.g., 2-18FA85380) have high affinity (<100 pM, 15–19). However, nifzetidine was found only to be marginally better than nifrolidine in brain homogenate binding assays suggesting that other factors may be involved in this class of fluoropropyl derivatives.
Table I.
Binding Affinity at Nicotinic α4β2 Receptors
| Drug | Kia, nM |
|---|---|
| Nicotine | 2.83 |
| Nifene | 0.50 |
| Nifrolidine | 0.80 |
| Nifzetidine | 0.67 |
Binding assay with 3H-cytisine in rat brain homogenates using reported methods (Pabreza et al., 1991);
Ki calculated from IC50 using Cheng-Prusoff equation (1973) and Kd = 0.59 nM for 3H-cytisine.
Rat brain autoradiographic studies with 18F-nifzetidine were similar to our previous observations with 18F-nifrolidine (16). Nicotine displaced binding of 18F-nifzetidine in a dose dependent manner in all brain regions (Fig-4). Brain uptake of 18F-nifzetidine in the monkey brain was slow, although eventual distribution of the radiotracer was consistent with the distribution of α4β2 receptors. This slow uptake is seen in the time-activity curves for the various brain regions as shown in Fig-6A. The time-activity curves showed the maximum binding in the thalamus. Thalamus to cerebellum ratio was about 1.7 for 18F-nifrolidine at 2 hours post-injection whereas thalamus to cerebellum ratio was about 2.1 for 18F-nifzetidine at 2 hours post-injection (Fig-6B). The kinetic profile of 18F-nifzetidine was slower in all brain regions compared to 18F-nifrolidine and pseudoequilibrium did not occur during the 180 min period of the PET scan. This behavior of 18F-nifzetidine seems to be similar to the slow kinetics observed with other azetidine-based radiotracers, such as 18F-2FA85380 (24).
On the other hand 18F-nifene showed fast in vivo kinetics, rapid uptake and cleared rapidly from various brain regions (13). 18F-Nifene has a unique “3,4-dehydropyrrolidine” ring system which provides for its unique imaging properties. In order to take advantage of this unique ring system we have replaced the “pyrrolidine” ring in nifrolidine (16) with “3,4-dehydropyrrolidine” and have developed 18F-nifrolene (25, 26; manuscript in preparation).
5. Conclusions
18F-Nifzetidine is a high affinity antagonist and has potential as a PET agent for imaging the α4β2 nAChR. Uptake was seen in thalamus and extrathalamic regions which is consistent with α4β2 receptor distribution. In vivo imaging studies showed that 18F-nifzetidine has slow kinetics which may limit its use as PET radioligand for quantitative studies of the α4β2 nAChR with PET.
Figure-1.

Chemical Structures of α4β2 radioligands: (1). 2-F-A85380 ; (2). Nifene; (3). Nifrolidine; (4). Nifzetidine (this work).
Acknowledgments
The project was supported by grant number R01AG029479 from the National Institute on Aging. We like to thank the reviewers for their valuable comments.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Horti AG, Gao Y, Kuwabara H, Dannals RF. Development of radioligands with optimized imaging properties for quantification of nicotinic acetylcholine receptors by positron emission tomography. Life Sci. 2010;86:575–584. doi: 10.1016/j.lfs.2009.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Levin ED. Nicotine receptor subtypes and cognitive function. J Neurobiol. 2002;53:633–640. doi: 10.1002/neu.10151. [DOI] [PubMed] [Google Scholar]
- 3.Paterson D, Nordberg A. Neuronal nicotinic receptors in the human brain. Prog Neurobiol. 2000;61:75–111. doi: 10.1016/s0301-0082(99)00045-3. [DOI] [PubMed] [Google Scholar]
- 4.Benowitz NL. Pharmacology of nicotine: addiction and therapeutics. Annu Rev Pharmacol Toxicol. 1996;36:597–613. doi: 10.1146/annurev.pa.36.040196.003121. [DOI] [PubMed] [Google Scholar]
- 5.Benwell ME, Balfour DJ, Anderson JM. Evidence that tobacco smoking increases the density of [3H]nicotine binding sites in human brain. J Neurochem. 1988;50:1243–1247. doi: 10.1111/j.1471-4159.1988.tb10600.x. [DOI] [PubMed] [Google Scholar]
- 6.Brown L, Chefer S, Pavlova O, Vaupel DB, Koren AO, Kimes AS, Horti AG, Mukhin AG. Evaluation of 5-(2-(4-pyridinyl)vinyl)-6-chloro-3-(1-methyl-2-(S)-pyrrolidinylmethoxy)pyridine and its analogues as PET radioligands for imaging nicotine acetylcholine receptors. J Neurochem. 2004;91:600–612. doi: 10.1111/j.1471-4159.2004.02762.x. [DOI] [PubMed] [Google Scholar]
- 7.Iida Y, Ogawa M, Ueda M, Tominaga A, Kawashima H, Magata Y, Nishiyama S, Tsukada H, Mukai T, Saji H. Evaluation of 5–11C-methyl-A-85380 as an imaging agent as an imaging agent for PET investigations of brain nicotinic acetylcholine receptors. J Nuc Med. 2004;45:878–884. [PubMed] [Google Scholar]
- 8.Roger G, Lagnel B, Rouden J, Besret L, Valette H, Demphel S, Gopisetti JM, Coulon C, Ottaviani M, Wrenn LA, Letchworth SR, Bohme GA, Benavides J, Lasne M-C, Bottaender M, Dolle F. Synthesis of a [2-pyridinyl-18F]-labelled fluoro derivative of (−)-cytisine as a candidate radioligand for brain nicotinic α4β2 receptor imaging with PET. Bioorg Med Chem. 2003;11:5333–5343. doi: 10.1016/j.bmc.2003.09.042. [DOI] [PubMed] [Google Scholar]
- 9.Fujita M, Ichise M, van Dyck CH, Zoghbi SS, Tamagnan G, Mukhin AG, Bozkurt A, Seneca N, Tipre D, DeNucci CC, Iida H, Vaupel DB, Horti AG, Koren AO, Kimes AS, London ED, Seibyl JP, Baldwin RM, Innis RB. Quantification of nicotinic acetylcholine receptors in human brain using 123I-5-IA85380 SPET. Eur J Nucl Med. 2003;30:1620–1629. doi: 10.1007/s00259-003-1320-0. [DOI] [PubMed] [Google Scholar]
- 10.Kimes AS, Horti AG, London ED, Chefer SI, Contoreggi C, Ernst M, Friello P, Koren AO, Kurian V, Matochik JA, Pavlova O, Vaupel DB, Mukhin AG. 2-[18F]F-A-85380: PET imaging of brain nicotinic acetylcholine receptors and whole body distribution in humans. FASEB J. 2003;17:1331–1333. doi: 10.1096/fj.02-0492fje. [DOI] [PubMed] [Google Scholar]
- 11.Bottlaender M, Valette H, Roumenov D, Dolle F, Coulon C, Ottaviani M, Hinnen F, Ricard M. Biodistribution and radiation dosimetry of 18F-fluoro-A-85380 in healthy volunteers. J Nucl Med. 2003;44:596–601. [PubMed] [Google Scholar]
- 12.Ding Y-S, Fowler J, Logan J, Wang GJ, Telang F, Garza V, Biegon A, Pareto D, Rooney W, Shea C, Alexoff D, Volkow ND, Vocci F. 6-[18F]Fluoro-A-85380, a new PET tracer for the nicotinic acetylcholine receptor: studies in the human brain and in vivo demonstration of specific binding in the white matter. Synapse. 2004;53:184–189. doi: 10.1002/syn.20051. [DOI] [PubMed] [Google Scholar]
- 13.Pichika R, Easwaramoorthy B, Collins D, Christian BT, Shi B, Narayanan TK, Potkin SG, Mukherjee J. Nicotinic α4β2 receptor imaging agents Part II. Synthesis and biological evaluation of 2-[18F]fluoro-3-[2-((S)-3-pyrrolinyl)methoxy]pyridine (18F-nifene) in rodents and imaging by PET in nonhuman primate. Nucl. Med Biol. 2006;33:294–304. doi: 10.1016/j.nucmedbio.2005.12.017. [DOI] [PubMed] [Google Scholar]
- 14.Lin N-H, Li Y, He Y, Holladay MW, Kuntzweiler T, Anderson DJ, Campbell JE, Arneric SP. Synthesis and structure-activity relationships of 5-substituted pyridine analogs of 3-[2-((S)-pyrrolidinyl)methoxy]pyridine, A-84543: A potent nicotine receptor ligand. Bioorg Med Chem Lett. 2001;11:631–633. doi: 10.1016/s0960-894x(01)00030-0. [DOI] [PubMed] [Google Scholar]
- 15.Lin N-H, Gunn DE, Li Y, H Y, Bai H, Ryther KB, Kuntzweiler T, Donnelly-Roberts DL, Anderson DJ, Campbell JE, Sullivan JP, Armeric SP, Holladay MW. Synthesis and structure-activity relationships of pyridine-modified analogs of 3-[2-((S)-pyrrolidinyl)pyridine, A-84543, A potent nicotinic acetylcholine receptor agonist. Bioorg Med Chem. 1998;8:249–254. doi: 10.1016/s0960-894x(98)00019-5. [DOI] [PubMed] [Google Scholar]
- 16.Chattopadhyay S, Xue B, Collins D, Pichika R, Bagnera R, Leslie F, Christian BT, Shi B, Narayanan T, Potkin S, Mukherjee J. Synthesis and Evaluation of Nicotine a4b2 receptor radioligand, 5-(3′-18F-fluoropropyl)-3-(2-(S)-pyrrolidinylmethoxy)pyridine, in rodents and PET in nonhuman primate. J Nucl Med. 2005;46:130–140. [PubMed] [Google Scholar]
- 17.Pabreza LA, Dhawan S, Kellar KJ. [3H]Cytisine binding to nicotinic cholinergic receptors in brain. Mol Pharmacol. 1991;39:9–12. [PubMed] [Google Scholar]
- 18.Smulders CJGM, Awart R, Bermudez I, van Kleef RGDM, Groot-Kormelink PJ, Vijverberg HPM. Cholinergic drugs potentiate human nicotinic A4B2 acetylcholine receptors by a competitive mechanism. Eur J Pharmacol. 2005;509:97–108. doi: 10.1016/j.ejphar.2004.12.037. [DOI] [PubMed] [Google Scholar]
- 19.Hughes DL. Organic Reactions. Vol. 42. New York: John Wiley & Sons, Inc; 1992. pp. 335–656. [Google Scholar]
- 20.Kosugi M, Sasazawa K, Shimizu Y, Migita T. Chem Letts. 1977:301. [Google Scholar]
- 21.Middleton WJ, Bingham EM. Diethylaminosulfur trifluoride. Organic Synth. 1977:57. [Google Scholar]
- 22.Cox P, Terpinski J, Lawrynowicz W. Anhydrous tetrabutylammonium fluoride: a mild but highly efficient source of nucleophilic fluoride ion. J Org Chem. 1984;49:3216. [Google Scholar]
- 23.Sihver W, Nordberg A, Langstrom B, Mukhin AG, Koren AO, Kimes AS, London ED. Development of ligands for in vivo imaging of cerebral nicotinic receptors. Behav Brain Res. 2000;113:143–157. doi: 10.1016/s0166-4328(00)00209-6. [DOI] [PubMed] [Google Scholar]
- 24.Chefer SI, London ED, Koren AO, Pavlova OA, Kurian V, Kimes AS, Horti AG, Mukhin AG. Graphical analysis of 2-[18F]FA binding to nicotinic acetylcholine receptors in rhesus monkey brain. Synapse. 2003;48:25–34. doi: 10.1002/syn.10180. [DOI] [PubMed] [Google Scholar]
- 25.Pichika R, Easwramoorthy B, Collins D, Potkin S, Mukherjee J. 18F-Nifrolene: A new PET imaging agent for a4b2 nAChR nicotinic receptors. J Nucl Med. 2006;47 (Suppl):27P. [Google Scholar]
- 26.Vu K, Sevrioukov E, Constantinescu C, Pan ML, Pichika R, Mukherjee J. 18F-Nifrolene MicroPET studies of nicotinic a4b2 receptors. J Nucl Med. 2010;51 (Suppl 2):219. [Google Scholar]